
Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis


1
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China
2
Research Group PLASMANT, Department of Chemistry, University of Antwerp, Universiteitsplein 1, BE-2610 Wilrijk-Antwerp, Belgium
*
Author to whom correspondence should be addressed.
†
These authors contributed equally.
Catalysts 2021, 11(5), 590; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050590
Submission received: 12 April 2021
/
Revised: 28 April 2021
/
Accepted: 29 April 2021
/
Published: 1 May 2021
(This article belongs to the Special Issue Feature Papers to Celebrate “Environmental Catalysis”—Trends & Outlook)
Abstract
:Direct oxidation of methane to methanol (DOMTM) is attractive for the increasing industrial demand of feedstock. In this review, the latest advances in heterogeneous catalysis and plasma catalysis for DOMTM are summarized, with the aim to pinpoint the differences between both, and to provide some insights into their reaction mechanisms, as well as the implications for future development of highly selective catalysts for DOMTM.
1. Introduction
Methane (CH4), as one of the most important molecules in C1 chemistry, is widely present in natural gas, shale gas, coalbed gas, combustible ice, etc. Noticeably, natural gas, which consists of approximately 70% to 90% CH4, will be part of the energy system for decades [1]. Unfortunately, most of these CH4 reserves are located in remote areas, indicating the need of transportation for utilization of CH4 [1,2]. However, due to a very low boiling point (−161.6 °C at a pressure of 1 atm) and high flammability, compression of CH4(gas) into CH4(liquid) for transportation requires huge amounts of energy, making it economically infeasible [3]. In addition, another important issue is the rising global emission of CH4, mainly as by-product of oil production, and its global warming potential is ca. 30 times that of CO2 [1]. The International Energy Outlook 2019 (IEO2019) estimated that 140 billion cubic meters (bcm) were flared and 60 bcm released into the atmosphere in 2018, more than the annual LNG (Liquefied Natural Gas) imports of Japan and China combined. This enormous source of emissions accounts for 40% of the total indirect emissions from global oil supply. Therefore, the conversion of CH4 to value-added chemicals has attracted intensive interests from both academic and industrial communities.
In general, as shown in Figure 1, the conversion of CH4 into value-added chemicals can be classified into indirect and direct routes [4]. As implemented in industry, indirect routes are, actually, initiated through a steam reforming (SR) and/or auto-thermal reforming (ATR) process to produce syngas (mixture of CO and H2), and then a variety of products such as olefins, gasoline, and diesel, as well as oxygenates, can be obtained using the well-established technology of Fischer-Tropsch synthesis (FTS) promoted by Fe-based or Co-based catalysts [5,6]. Alternatively, using Cu-Zn-Al-based catalysts, syngas can also be converted into methanol (CH3OH), which has been used as feedstock to produce light olefins, gasoline and aromatics through industrial technologies of methanol-to-olefins (MTO), methanol-to-gasoline (MTG) and methanol-to-aromatics (MTA) conversion, respectively [6]. Although the above indirect routes are carried out in industry to produce value-added chemicals from CH4, the syngas production by SR and ATR is energy-intensive and costly, motivating researchers to develop direct routes (not syngas-based) [7].
Direct routes (Figure 1), including CH4 dehydroaromatization, CH4 coupling to hydrocarbons (both oxidative and non-oxidative coupling), CH4 pyrolysis (high temperature pyrolysis and catalytic pyrolysis) and direct oxidation of CH4 to methanol/formaldehyde, have also been developed and are still being improved [2,3,4,5,6,7,8]. However, CH4, a molecule with tetrahedral geometry and four equivalent C–H bonds, is inert and difficult to activate and convert. The absence of a dipole moment and a rather small polarizability (2.84 × 10−40 C2·m2·J−1) imply that CH4 needs a relatively high local electric field to be polarized and to allow electrophilic or nucleophilic attack [2]. CH4 exhibits the highest C–H bond strength among all alkanes, with the first bond dissociation energy (BDE) of 493.3 kJ mol−1 (5.1 eV), meaning that CH4 is the least reactive alkane [2]. Therefore, most of the direct routes must be operated at ultra-high temperature (973~1223 K), except for direct oxidation to methanol/formaldehyde, which can be realized at relatively low temperature (300~700 K), indicating low cost and high feasibility in industry [2,3]. For that reason, direct oxidation has attracted more attention, and in this review, we mainly focus on recent progress of direct oxidation of methane to methanol (DOMTM).
CH4 + 0.5 O2 → CH3OH ΔH (298K) = −126.4 kJ·mol−1
DOMTM by O2 (1) has been considered as a dream reaction in chemical industry and a holy grail in catalytic chemistry [9,10], and it has attracted intensive interests from both academic and industrial communities for more than 100 years. DOMTM has been studied by homogeneous catalysis, in which noble metals (Pt and Pd) are typically used as the central atoms of the complex catalysts, and the reaction is usually carried out in strong acid media (sulfuric and trifluoroacetic acid) [11,12,13]. Alternatively, DOMTM can also be realized by heterogeneous catalysis. In the 1980s, a Mo-based catalyst was developed for CH4 oxidation. The Mo=O species was considered to be the active site for the oxidation of CH4 to CH3OH, and at that time, Mo was considered to be the most active metal catalyst for this reaction [14,15]. However, the biggest drawback of a Mo-based catalyst is that Mo can be easily lost at high temperature through volatilization, which was not conducive for industrial application [16]. Compared with a Mo-based catalyst, a V-based catalyst is more stable, but the CH4 conversion was too low (less than 10%) [17,18]. At the beginning of this century, inspired by the active sites of double iron and double copper in methane monooxygenase, Fe-based and Cu-based zeolite catalysts have been used in DOMTM [19,20,21,22], as well as supported noble metals, such as Au, Pd and Rh [23,24]. Detail information for the developing process of DOMTM can be obtained in the review paper by Bokhoven [3].
Although many studies about DOMTM have been reported, it has to overcome two challenges, caused by thermodynamics and kinetics, respectively [3,9]. The first is how to improve the CH3OH selectivity. Thermodynamically, CH3OH is not the favorable product, as CO and CO2 are more stable than CH3OH. Specifically, as shown in Figure 2A, a low temperature (<890 K) favors the production of CO2 and H2O, while a high temperature (>890 K) favors CO and H2. In other words, due to the higher reactivity of CH3OH than the feedstock CH4, the catalytic sites, capable of oxidizing CH4 into CH3OH, can also further oxidize CH3OH into CO or CO2 before CH3OH desorbs from the catalyst surface. Figure 2B indeed illustrates that CH3OH and other oxygenates and hydrocarbons are only formed with much lower selectivity.
The second challenge is how to reduce the kinetic energy barrier (Ea) of DOMTM by O2 at ambient conditions. The Ea of DOMTM by O2 is much higher than for DOMTM using N2O or H2O2 as oxidants, because both N2O and H2O2 can more easily release an oxygen atom, as the main species to trigger the oxidation of CH4 to CH3OH. Therefore, when using O2 as oxidant, high temperature and high activity catalysts are needed to overcome the Ea of DOMTM, which unfortunately leads to deep oxidation. However, for reaction (1) the entropy is reduced (ΔS < 0) while it is exothermic (ΔH < 0). Therefore, CH3OH production is favorable at low temperature, because a low temperature can result in a negative ΔG value (i.e., a spontaneous process), leading to a contradiction between the dynamics and thermodynamics of the DOMTM process.
Recently, several innovative studies by heterogeneous catalysis and plasma catalysis have been performed to overcome the above-mentioned challenges, which we will discuss in this review.
2. Heterogeneous Catalysis
Heterogeneous catalysis is more suitable for industrial application than homogeneous catalysis, being more convenient for product and catalyst recovery. A number of excellent reviews have been recently published on DOMTM, adding to the classic reviews in the field [3,4,5,7,8,9]. The collective desire on these articles was emphasized to find a breakthrough, pushing DOMTM technology closer to commercialization. Generally, seeking for efficient catalytic systems with a controllable reaction kinetics process is the key of CH4 conversion, and the dissociation of the first C–H bond was regarded as the rate-determining step on various CH4 conversion reactions. Basically, as shown in Figure 3, the mechanism of C–H bond cleavage of CH4 at low temperature can be mainly classified into two categories [9]. The first mechanism involves H abstraction from CH4 to form •CH3 radicals by electrophilic oxygen atoms, while the other mechanism includes the formation of metal-CH3 (M-C) σ–bond as a reaction intermediate which can directly cleave the C–H bond with concomitant coordination of the CH3 group [9].
Except seeking advanced catalytic systems that enable efficient C–H activation, the insight obtained in the different roles of oxidants in DOMTM will be also of benefit to the field. On the one hand, strong oxidants (e.g., N2O and H2O2) can easily break the C–H bond, but they will also enhance the over-oxidation of intermediate products adsorbed on the catalyst surface to thermodynamically more stable CO or CO2, which is not conducive to the generation of CH3OH or its derivatives. On the other hand, these strong oxidants with better oxidation properties have higher raw material prices, leading to higher economic costs. In this section, we summarize the research progress of DOMTM by different oxidants (Table 1) in heterogeneous catalysis, with emphasis on the introduction of the catalytic systems with O2 as oxidant.
2.1. Nitrous Oxide as Oxidant
Nitrous oxide (N2O) is widely used as oxidant in DOMTM (2), mainly based on Fe-modified zeolites, which is particularly attractive from the perspective of natural catalysts (i.e., methane monooxygenase (MMO) enzymes), activating CH4 to produce CH3OH under mild conditions. The unique properties of MMO are related to the Fe-containing sites capable of generating extremely active oxygen species that can insert into non-activated C–H bonds of CH4 under ambient conditions [42]. Great interest has been paid to Fe-based zeolites, which may contain various iron species, especially activated by N2O. Early results showed that Fe/ZSM-5 catalyst can decompose N2O effectively through a Fe/O highly active site on its surface at low temperature (<300 °C) and realize partial oxidation of benzene to phenol [43,44]. This active site on the Fe/ZSM-5 catalyst is called the α-Fe or α-O site, and it was found that DOMTM can be achieved at low temperature [45]. The nature of the active site with exceptional reactivity is difficult to prove spectroscopically mainly due to the presence of inactive spectator iron species.
CH4 + N2O → CH3OH + N2 ΔH (298K) = −159.0 kJ·mol−1
The properties of α-oxygen and the state of iron constituting α-sites were studied in many experimental [46,47] and theoretical works [48,49] and discussed in review papers [3,50]. Despite great research efforts have been made, the spectroscopic insight into the structure of actual active site was illustrated only recently on Fe-BEA zeolite [46]. By magnetic circular dichroism (MCD), a mononuclear, high-spin, square planar Fe(ii) site was identified and this high-reactive site was previously regarded as an α-Fe(II) site by Panov and co-workers. Furthermore, the α-O site constrained by the zeolite lattice, considered as the reactive intermediate, is, actually, a mononuclear, high-spin Fe(iv)=O species [46]. From the DR-UV–Vis spectra, an intense band at 40,000 cm−1 and three weak bands at 15,900, 9000, and 5000 cm−1 were observed for the inactivated Fe-BEA (Figure 4a). After N2O activation and CH4 reaction, the peak at 16,900 cm−1, along with a weak feature at around 5000 cm−1 present after N2O activation, disappear upon reaction with CH4, which corresponds to the process of the α-O site which forms on α-Fe (II) active site. The 5000–13,000 cm−1 region of the CH4-reacted spectrum overlaps with that of Fe(II)-BEA, indicating that features in this region originate from inactive iron species. Furthermore, the MCD spectra show a band at 15,100 cm−1 that is corresponding to the 15,900 cm−1 absorption band of the α-Fe(II) site from DR-UV–Vis, which is sensitive to a magnetic field and temperature. Additionally, DFT-optimized cluster models with high-energy ligand-field band were predicted by CASPT2, and α-Fe(II) site can be assigned to a high-spin, square planar Fe(II) site with four anionic Si–O–Al ligands (Figure 5), which can be further verified by DFT-calculated Mössbauer parameters.
An Fe-FER (Ferrierite) catalyst was also studied, in which the formation of active oxygen species from N2O decomposition can react with CH4 to form CH3O groups and valuable products at moderate temperatures. The emergence of methoxy group bands in the FTIR spectrum with introduction of CH4 on Fe-FER catalysts indicates the active surface species leading to the formation of selective products [51]. As shown in Figure 6, the amount of active α-oxygen sites and number of Bronsted acid sites on the Fe-modified zeolites, e.g., Fe/ZSM-5 and Fe/FER, are crucial factors to alter CH4 conversion and product distribution as well [52]. The Fe/ZSM-5 zeolite, with a large number of strong acid sites, could produce a higher selectivity to unsaturated C2-C3 hydrocarbons and dimethyl ether (DME), while the Fe/FER zeolite, with a large number of weak Bronsted acid sites, gave rise to oxygenates such as CH3OH and DME [52]. The formed coke precursors seem to be more dominant on the Fe/ZSM-5 zeolite with stronger acid sites due to a facile reaction pathway through the MTO reaction. The amount of coke deposition on acid sites and α-oxygen sites was well correlated with the catalytic performance, and thus a possible reaction pathway (Figure 6) was proposed by the authors, based on the product distribution with respect to acid sites and α-oxygen sites [52]. The general consensus of DOMTM reaction pathway over α-O sites follows a radical-based H atom abstraction mechanism (cf. Figure 3), although some steps based on this mechanism are still debated. Briefly, oxygen atoms are introduced by N2O decomposition before subsequent CH4 addition. The abstracted hydrogen atom from CH4 by the α-O results in an FeⅢ-O-H fragment and a CH3 radical, and produced CH3 radical may then either react with a further α-O to form FeⅢ-O-CH3 or form FeⅡ-O(H)-CH3 with near fragment, which may then desorb and eventually form CH3OH [48,53].
The remarkable activity of the α-O site can be partially attributed to confinement effects with the zeolite channels [48,54], which are thought to increase the local concentration of the molecular orbitals within the micropores, resulting in increased interactions between confined reactants and enabling unusual transition states to be accessed. Furthermore, the confined molecules within zeolites have also been shown to induce dipoles and multi-poles, potentially strengthen or weaken some C−C bonds, which may lead to modifications of the HOMO/LUMO energy levels.
Periodic structure and cluster modeling of α-O sites in the SSZ-13 zeolite at different levels of theory has been performed and the results showed that the confinement effect of zeolite channels may reduce the energy barrier of DOMTM by over 50% [48]. This confinement effect is mainly electrostatic in nature, which could stabilize all transition states, the reaction intermediate and products. DFT calculations on a periodic system of MO+/ZSM-5 zeolite (M=Fe, Co, Ni and Cu) indicate that the confinement effect is attributed to the nanopores of ZSM-5 zeolites, in which interaction between CH4 and MO+ species was confined [55], resulting in a significant destabilization of CH4 adsorption and further lowering of the activation energy for the C–H bond dissociation. In addition, mono-nuclear α-Fe2+ in an extra-lattice site within Fe-beta zeolite (BEA) also showed that the reactive intermediate was a high spin Fe4+=O species and the confinement of the zeolite lattice promotes the reactivity [46]. Therefore, by preventing geometric distortion, the confined α-O sites can activate CH4 and cleave the strongest aliphatic C–H bond at room temperature to form CH3OH.
Variant zeolite topology has thus been considered an important factor in rationally tuning of the active site properties for DOMTM. The single-site α-Fe in the CHA topology zeolite was demonstrated to be active to form a highly reactive α-O, capable of activating CH4 at room temperature to form CH3OH, which subsequently desorb from catalyst surface by on-line steaming at 200 °C [26]. It has been found that the topology’s 6MR geometry of CHA zeolite has great effect on the structure, the ligand field, and consequently the spectroscopy of the α-Fe site, by comparing α-Fe in Fe/BEA and Fe/CHA. At higher Fe loading (>0.26 wt%; Si:Al = 12.5), Fe2O3 was identified other than α-Fe sites in Fe/CHA zeolite. Therefore, the level of introducing Fe into CHA zeolite is a crucial criterion to improve reaction reactivity with increased active site density [26]. Additionally, indicated by nitrous oxide titration, the method of introducing appropriate extra-framework Al in Fe/MOR catalysts is in favor of increasing iron ions in tetrahedral or octahedral coordination, which can promote the formation of more α-sites in Fe-containing zeolites [56].
The DMTOM process was studied in a so-called quasi-catalytic mode over Fe/ZSM-5 zeolite with N2O as an oxidant, in which the reaction intermediates or products just migrate from catalytically active centers to other sites of the surface, rather than desorption into the gas phase, mainly due to the activation energy of surface diffusion of adsorbed species is much smaller than desorption energy of the species into the gas phase [25,57]. As shown in Figure 7, the produced CH3OH over α-O sites moved to neighboring sites without desorption into the gas phase in the quasi-catalytic mode, and the adsorbed species could further be converted to coke or extracted from surface to get methanol and dimethyl ether (DME). The reaction could switch to conventional catalytic process at above 200 °C, with products direct desorption into gas phase. Low CH4 conversion (0.19%) is indeed a huge limitation for DOMTM reaction, but this approach provides new insight and a possibility to identify the potential intermediates [57].
In addition to Fe-modified zeolites, some other catalysts have been also studied for DOMTM by using N2O as an oxidant. Liu et al. investigated the catalytic performance of a Mo-based catalyst in partial oxidation of CH4 to CH3OH using N2O as oxidant. However, only 3% CH4 conversion was achieved with 78% oxygenates selectivity (CH3OH and HCHO) [58]. Cu/SSZ-13 catalyst activated by N2O was also reported for DOMTM at low temperatures and ambient pressure, with highest CH3OH productivity of 55 μmol CH3OH g−1 h−1, which was more than twice the rate of Cu-mordenite and more than four times the rate of Cu-ZSM-5 [59]. Higher partial pressures of CH4 and H2O with lower N2O partial pressure was thought to further improve CH3OH selectivity [59]. Recently, the performance of N2O and O2 oxidants were compared by DFT calculation on the CoN3–Graphene catalyst, and the results shows that DOMTM reaction can proceed via a two-step pathway, with N2O as an oxygen donor [60]. CoN3–Graphene catalyst exhibited higher catalytic activity for the adsorption of gas reactants, which can be used as an efficient catalyst to fabricate effective C/N catalysts on methane oxidation by N2O.
N2O, a relatively stable greenhouse gas (GHGs), mainly from agricultural production and other anthropogenic activities, like waste management, or the combustion of fossil fuels and biomass, which could be correlated to stratospheric ozone destruction by atmospheric photochemical reactions [61]. Indeed, the co-conversion of CH4 and N2O is of great interest in reducing the anthropogenic forcing of the climate system, as a win-win strategy for both ozone and climate. However, the utilization of N2O as an oxidant confronts an insurmountable hurdle associated with its low availability which cannot satisfy the large demand for industrial scale CH4 valorization.
2.2. Hydrogen Peroxide as an Oxidant
As an important industrial oxidant, hydrogen peroxide (H2O2) is widely used in papermaking, sewage treatment, metallurgy, medical and health care, and other fields [62]. The benefit of H2O2 oxidant is the clean decomposition to water as an environmentally benign byproduct. Noticeably, H2O2 has been considered as a highly efficient oxidant in DOMTM (3). Many studies realized the DOMTM process under milder conditions compared to N2O oxidant, mainly employing noble metal catalysts.
CH4 + H2O2 → CH3OH + H2O ΔH (298K) = −223.9 kJ·mol−1
Singly dispersed noble metal atoms anchored on oxides could offer a distinctly different electronic state in contrast to continuously packed metal atoms on the surface of a metal nanoparticle, and thus could exhibit a distinct catalytic activity or/and selectivity [63,64]. Pd1O4 single-sites anchored on the internal surface of micropores of ZSM-5 exhibited excellent performance for CH4 activation [65]. Under the assistance of H2O2, CH4 was partially oxidized into CH3OH over Pd1O4 site at low temperature (50–95 °C), but extra H2O2 resulted in further oxidation of CH3OH to CHOOH. The CH3OH selectivity remained at 86.4%, while the CH3OH productivity at 95 °C was about 2.78 molecules per Pd1O4 site per second when CuO was used as a co-catalyst. The experiment of varied amounts of Pd on Pd/ZSM-5 catalysts exhibited quite similar yields of the products, although their loadings of Pd were quite different (from 0.01 to 2.0 wt.%), suggesting that the excess Pd, present in the external surface of the zeolite, did not alter the activity, as well as the selectivity of CH3OH. Furthermore, DFT calculations showed that CH3OH is the thermodynamically stable product over Pd1O4, which was consistent with the experimental result that CH3OH production was highly favorable compared to the formation of a byproduct, methylperoxide [65]. Kwon and co-workers reported an atomically dispersed Rh/ZrO2 catalyst using aqueous H2O2 as an oxidant for the selective oxidation of CH4, where the property of Rh active sites significantly affected the CH4 oxidation [30]. Single atomic Rh could make CH3OH with the highest productivity, whereas Rh nanoparticles on SiO2 produced only CO2 without the formation of C1 oxygenates (Figure 8A). The amount of oxygenated products showed only a little difference up to the fifth cycle (Figure 8B). When replacing the oxidant with O2, C2H6 was observed as main product in gas-phase for direct CH4 oxidation, as shown in Figure 8C [30]. Additionally, combing EXAFS, XANES, HAADF-STEM/EDS images and CO-adsorption using DRIFTS measurements, the model of single-atom Rh/ZrO2 catalyst was proposed, based on which DFT calculations were carried out. The results showed that CH3 intermediates can be energetically stabilized on the catalyst, which was further verified by DRIFTS measurements. The active single-site Rh1O5 anchored in microporous aluminosilicates (Rh/ZSM-5) in solution can realize the oxidation of CH4 to CH3COOH (acetic acid) and CH3OH below 150 °C [66]. An isotope experiment confirmed that the C atoms of the methyl and carboxyl groups in CH3COOH were derived from CH4 and CO, respectively. Noticeably, high pressure of CO is detrimental to the production of CH3COOH and finally poisoned the active sites [66]. The conversion of CH4 to oxygenates on Rh/ZSM-5 may occur via M–CH3 functionalization. Firstly, CH4 is activated in the presence of O2 on isolated Rh+ cations under mild conditions to produce Rh–CH3. The formed Rh–CH3 can then be functionalized via two independent reaction pathways: oxygen insertion to produce CH3OH, or CO insertion to produce CH3COOH. After a hydrolysis step, the whole catalytic cycle can be completed [24]. However, the in-depth mechanism responsible for this activation is still not clear and further investigation is indispensable to guide the design of novel and more efficient CH4 oxidation catalysts.
Interestingly, Au-Pd nanoparticles (NP) are highly effective catalysts for the direct synthesis of H2O2, and the hydroperoxy species (HOO) is effective for the oxidation reaction [67]. Thus, Au-Pd supported nanoparticles (AuPd/TiO2) are active for the oxidation of CH4, giving a high selectivity for CH3OH formation under mild aqueous conditions with H2O2 as oxidant [28]. A similar productivity, but with improved CH3OH selectivity, was observed when using the in situ generated H2O2 by adding H2/O2 gases mixture, compared to the experiments performed with pre-synthesized H2O2. Additionally, both methyl (•CH3) and hydroxyl (•OH) radicals were observed by electron paramagnetic resonance (EPR) spectroscopy, which suggests that the CH4 reaction proceeds through a radical mechanism, in contrast to the reaction mechanism previously proposed for CH4 oxidation using CuFe/ZSM-5, where •CH3 radicals were not observed [31].
Recently, further experiments showed that colloidal Au-Pd nanoparticles can oxidize CH4 in aqueous solution at mild temperatures with 92% CH3OH selectivity, in the presence of both H2O2 and O2 [23]. Different components of the catalyst were studied for the H2O2 degradation rates. H2O2 degradation rates of bare TiO2 and unsupported Au-Pd colloidal NPs were low, while the Au-Pd/TiO2 catalyst exhibited a high rate of H2O2 degradation (73%), indicating that either the interfacial sites at the support/metal interface or a change in the morphology of the NP led to the high H2O2 degradation rates [67,68,69,70]. The Au-Pd colloid catalyst decomposes H2O2 at a much lower rate (38%) but makes substantially more products. Moreover, the addition of 5 bar of O2 pressure to the reaction resulted in an increased yield of oxygenate products (e.g., CH3OH, CH3OOH and HCOOH) compared with the H2O2-only reaction. Isotopically labeled oxygen (O2) as oxidant in the presence of H2O2 indicated that the produced CH3OH incorporated a substantial fraction (70%) of gas-phase O2 under optimized conditions, which can react with •CH3 radicals generated via H abstraction by •OH from H2O2 as initiation step of CH4 activation (Figure 9). Additionally, more oxygenated products were formed than the amount of H2O2 consumed, suggesting that the controlled breakdown of H2O2 activates CH4, which subsequently incorporates O2 through a radical process [23]. Titania-supported AuPdCu catalysts are active for the oxidation of CH4 under mild reaction conditions by using H2O2 as oxidant. After depositing Cu together with Au/Pd on the surface of TiO2 the rate of CH4 oxidation with addition of H2O2 is significantly enhanced. In particular, 2.5% Au 2.5% Pd 1.0% Cu/TiO2 showed ca. 83% selectivity to CH3OH [71].
To make CH4 economically competitive as a source of energy, it is crucial to increase the productivity of CH3OH and the efficiency of H2O2 utility [72]. DOMTM by AuPd@ZSM-5 catalyst (Figure 10) using in situ generated H2O2 at mild temperature (70 °C) has been reported, which can prevent H2O2 dilution, and thereby keep a high local concentration of H2O2 around the AuPd nanoparticles [29]. This hydrophobic zeolite was synthesized by AuPd alloy nanoparticles within aluminosilicate zeolite crystals, where the external surface of the zeolite was modified by organosilanes, as depicted in Figure 10. The hydrophobic sheath appears to allow diffusion of H2, O2, and CH4 to the catalyst active sites, while it hinders the diffusion of generated H2O2 from the encapsulated AuPd nanoparticles. As shown in Figure 11A, the AuPd@ZSM-5-C3 catalyst gave a CH4 conversion of 16.4% with CH3OH selectivity of 90%, and higher conversions were realized over AuPd@ZSM-5-C6 and AuPd@ZSM-5-C16 catalysts that contained longer organic chains modified on the zeolite crystals. The AuPd@ZSM-5-C16 catalysts exhibited the best performance with 17.3% CH4 conversion and 92% CH3OH selectivity, corresponding to CH3OH productivity up to 91.6 millimoles per gram of AuPd per hour. Additionally, the formation of H2O2 from H2 and O2 over AuPd@ZSM-5 catalysts was also studied, as shown in Figure 11B. Washing the solid catalysts by cold CH3OH and tetrahydrofuran (~0 °C), which can liberate the H2O2 within the zeolite crystals, 8.1, 68.4, and 78.3 μmol of H2O2 was obtained during this process for AuPd@ZSM-5, AuPd@ZSM-5-C3, and AuPd@ZSM-5-C16, respectively. In contrast, the H2O2 quantity in the water solution reached 97.9 mmol when the AuPd@ZSM-5 catalyst was used, which further suggests that AuPd@ZSM-5-C16 catalyst can enrich the H2O2 within the zeolite crystals. This molecular-fence concept seems interesting and represents a large step toward the application of direct CH4 oxidation to valuable products. However, the CH3OH productivity still needs further improvement, and the batchwise operation restricts the commercial application [29].
In addition to the above-mentioned precious metal catalysts, some cheap metal catalysts have also been studied in DOMTM using H2O2 as oxidant. Xie and co-workers studied the performance of TiO2-supported iron catalysts in DOMTM at ambient conditions [73]. It was observed that the H2O2 amount was vital for controlling the CH3OH selectivity. When no H2O2 was present, no CH4 was converted. Increasing the H2O2 amount results in a higher CH4 conversion, but the CH3OH selectivity decreased. The optimal ratio of H2O2 to CH4 was 0.11, showing a 15% CH4 conversion and an alcohol selectivity over 97% (CH3OH selectivity over 90%) [73]. Xu and coworkers studied DOMTM over Cu- and Fe-modified ZSM-5 catalysts in a flow reactor using H2O2 as the oxidant under continuous flow operation. Co-impregnation of ZSM-5 with Fe and Cu by chemical vapour impregnation yielded catalysts that showed high CH3OH selectivity (>92% selectivity, 0.5% conversion) [74]. In another study, Kim et al. reported partial oxidation of CH4 over Fe/ZSM-5 catalyst using H2O2 as oxidant, and found that the total product yield and the amount of H2O2 consumed increased with increasing Fe content in the Fe/ZSM-5 catalyst prepared using an ion-exchange method [75]. The effect of Si:Al molar ratio of Cu- and Fe-exchanged zeolites on DOMTM was also studied and the results demonstrated that high CH3OH production can be realized when catalysts with low Si:Al ratio were used [76]. The Fe-only ZSM-5 catalysts exhibited the highest catalytic activity (total oxygenated products) with HCOOH being the major product, which can be explained by the increased amount of Brønsted acid sites. The presence of Cu aims to maintain high MeOH selectivity by suppressing the production of the deeper oxidation product like CH3COOH and HCOOH [76].
A graphene-confined single Fe atom catalyst (FeN4/GN) was screened from a series of 3d metal–N4 embedded in the lattice structure of graphene nano-sheets, as shown in Figure 12A,B. The unique O–FeN4–O structure formed in graphene can directly convert CH4 to C1 oxygenated products (e.g., CH3OH, CH3OOH, HCOOH and HOCH2OOH), with total selectivity around 94% [10]. The CH4 oxidation reaction was carried out in a specifically designed high-pressure reactor connected with an operando time-of-flight mass spectrometer (TOF-MS) (Figure 12C), which can successfully accomplish the qualitative and quantitative identification of products during reaction and can detect how CH4 is exactly oxidized stepwise at a more real-time analysis. The CH3 radical is first converted into CH3OH and CH3OOH, and CH3OH can be further converted to HOCH2OOH and HCOOH on the O–FeN4–O site (Figure 12D,E), as illustrated by TOF-MS, 13C NMR, and DFT calculations. The intermediate HOCH2OOH was the first time to be identified as a product in CH4 oxidation. Comparison studies between the formation energies of O–MN4–O active sites illustrated that among all O–MN4–O (M=Cr, Mn, Fe, and Co) structures, O–FeN4–O has a moderate formation energy and the highest CH4 activation rate (Figure 12F), corroborating well with the best activity of the FeN4 site in the experimental studies. Recently, Meysam et al. studied the influence of zeolite acidity on partial oxidation of CH4 over M-Fe-MFI (M: Ga, Al, B) zeolites [77]. The results indicated that the HCOOH production rate and total formation rate of oxygenated compounds correlated with total acidity. The samples with weaker acidity showed much lower oxygenate productivity and selectivity. Partial oxidation of C2H6 to oxygenates using Fe- and Cu-containing ZSM-5 catalysts revealed that the Fe/ZSM-5 catalyst is highly selective for the conversion of C2H6 to a range of oxygenates at appreciable levels of conversion under mild conditions [78]. The reaction pathway is more complicated than that previously proposed for the CH4 oxidation reaction using similar zeolite catalysts.
2.3. Oxygen as Oxidants
As one of the main components in the air, oxygen (O2) is an inexpensive oxidant, which is conducive to large-scale application from an economic point of view. However, as an oxidant, oxygen has a high reaction energy barrier and requires a high temperature to activate CH4 to CH3OH formation (1). In order to avoid excessive oxidation of CH3OH or its derivatives, researchers proposed a stepwise process (Figure 13) (i.e., stoichiometric chemical looping), which involves three separate steps: (1) Activation of the metal-zeolite catalyst by an oxidant at a relative high temperature (250–500 °C), (2) CH4 reaction at a relatively low temperature (25–200 °C), and (3) CH3OH extraction using a solvent or steam at a relatively low temperature (25–200 °C) [3,32,78]. Both O2 and N2O can be used as oxidant for the metal-zeolite catalyst activation (Step 1), N2O could generally operate under lower temperature than O2, but in this section we mainly focus on the selected catalysts based on O2 as oxidant. Currently, Cu and Fe exchanged zeolites have been extensively studied, and significant attention was given to elucidating the nature of the active sites. The identity of the catalytic sites (Figure 14) in the active Cu-zeolites was subject of many spectroscopic studies, which mainly include copper monomers, dimers in the form of mono-μ-oxo and bis-μ-oxo dicopper cores, trimers and larger clusters on the basis of powder XRD, EXAFS, Raman, and UV–Vis data [8,79].
Cu/ZSM-5 have been intensively studied in CH4 partial oxidation, and the bis(μ-oxo)dicopper site, [Cu2(μ-O)2]2+, was firstly identified as the active site, evidenced from the absorption band at 22,700 cm−1 in UV–Visible spectra [32,80]. Later, the active species in Cu/ZSM-5 catalyst was further ascribed to a bent mono-(μ-oxo) dicupric site, [Cu2(μ-O)]2+. DFT calculation and normal coordinate analysis of symmetric and anti-symmetric vibrations on resonance enhanced Raman spectroscopy provides insight into how the constrained lattice of ZSM-5 restricts the coordination environment of the bound Cu atoms and their spatial orientation in the formation of active sites for DOMTM [81,82]. This mono(μ-oxo)dicopper core can be formed by activation in both N2O and O2 atmosphere as evidenced by observation of the UV–Vis NIR band at 22,700 cm−1 [83]. Additionally, the existence of trinuclear active species, [Cu3(μ-O)3]2+, has been demonstrated and this trinuclear species in the non-frame-work of the zeolite structure is indeed more stable than the binuclear in the MFI zeolite framework indicated by DFT [84]. In general, the composition and varying topology of copper-modified zeolites are thought to have a large effect on the nature of active sites and on the performance in terms of CH3OH yield. Higher Si:Al ratios with highly dispersed Al atoms on copper-modified zeolites are more likely to support monomeric active sites, whereas lower Si:Al ratios are likely to stabilize multinuclear copper clusters due to the close proximity of Al atoms on zeolites [85].
Cu-MOR is typically observed to produce a higher CH3OH yield than Cu/ZSM-5, but more than one site is responsible for CH3OH formation. Cu/MOR has been suggested to possess both binuclear and trinuclear clusters capable of performing CH4 partial oxidation [84,86,87]. Whereas a binuclear active site in Cu/ZSM-5 is located in the intersection of the two ten-member-rings, Cu-MOR provides two distinct local structures, situated in the eight-member-ring windows of the side pockets, suggested by spectroscopic observations and DFT simulations [88]. The improved performance in reactivity can be ascribed to subtle difference in the ground states of the Cu-O-Cu sites, indicating the participation of the zeolite lattice in the reaction coordinate. In addition, operando X-ray absorption spectroscopy (XAS) and high-energy-resolution fluorescence-detected (HERFD) and XANES spectroscopy provide a novel perspective on the complex nature and dynamics of Cu-species present in the MOR framework, which enabled an accurate quantification of Cu-speciation on zeolites [33]. The results demonstrated that the productivity increases with a slope of exactly 0.5 when the spectroscopically-determined concentration of active Cu is increased, across a series of materials and activation protocols, suggesting the active site on Cu-mordenite for selective CH4 oxidation is a dicopper site (Figure 15). The proportion of active Cu in Cu-MOR has been quantified by a spectroscopic method and correlated with reaction performance, and the highest CH3OH yield (170 μmolMeOH gcat−1) has been achieved over Cu-MOR with Si:Al = 7 and Cu:Al = 0.18 [33].
Different framework topologies of Cu-zeolites have been studied in stepwise partial oxidation of CH4 to CH3OH, and Cu2+ ion-exchanged zeolite omega with MAZ type showed the highest yield (86 μmol·gcat.−1) among these zeolite materials. Further experimental results revealed the highest CH3OH yield ever reported (i.e., 150 μmol CH3OH per gram zeolite under 1 bar CH4, and as high as 200 μmol CH3OH per gram zeolite under 30 bar CH4 were achieved on copper-exchanged omega (MAZ) zeolite) [35,86]. The remarkable CH3OH yield of Cu-omega is attributed to the relatively high density of copper-oxo active species formed on three-dimensional eight-member-ring channels. Additionally, highly dispersed Cu2+ ions in the zeolite pores are essential, which depends on the Si:Al ratio and the preparation method. Higher activation temperature leads to the formation of more copper oxo species, whereas too much copper is not favorable for CH3OH synthesis [86]. Copper-modified small pore zeolites, including SSZ-13, Cu/SSZ-16, Cu/SSZ-39, and Cu/SAPO-34, have been reported for DOMTM, and the improved CH3OH yield is attributed in part to the high-temperature water vapor extraction protocol [89]. In particularly, Cu/SSZ-13 could produce competitive quantities of CH3OH per copper atom, with the highest CH3OH yield of 125 μmolMeOH gcat−1 (0.2 mol CH3OH/mol Cu) [34]. Optimal CH3OH productivity is correlated with optimal reducibility of Cu/SSZ-13, which is highly dependent on the composition, in terms of Si:Al and Cu:Al ratios (Figure 16). Intermediate Si:Al ratios (∼12−15) and high Cu loading (∼0.5) are prone to produce a high population of Z[CuIIOH] precursor sites stabilized within an 8MR CHA zeolite, ultimately resulting in high CH3OH yields. The combination of testing and XAS measurements clearly evidence a positive linear correlation between the CH3OH productivity and the reducibility of the Cu centers.
The isothermal conditions were proposed to avoid the time-consuming heating and cooling step during the stepwise cycles. Several studies have shown the ability to run this reaction in an isothermal regime using O2 or NO as oxidant at 473 and 423 K, respectively [90,91,92]. By operated in an isothermal regime, Cu-FAU realized a stable CH3OH yield of 90 μmol/g and 92% selectivity at 633 K and ambient pressure without suffering long cycling time and temperature swing [92]. Furthermore, increasing the CH4 pressure to 15 bar results in a CH3OH yield of 360 μmol/g, which is the highest value achieved over copper-exchanged zeolite in one cycle. The redox properties of CuII-oxo species vary with the structure of the sites, exhibiting different reduction temperature for copper-oxo aggregates and isolated sites, which is an important factor for CH3OH yield and selectivity. High reduction temperature leads to low activity and CH3OH yield, while a low copper oxo reduction temperature may tend to high activity toward CH4 conversion, but with low CH3OH selectivity. This connection between structure and reducibility of the copper-oxo species can be considered for further designing of high-performance catalysts on DOMTM at desired temperature.
Except Cu-exchanged zeolites, some other catalysts (e.g., Fe-, Co- and Ni-exchanged zeolites) have been reported in both spectroscopy and computational studies based on O2 or N2O as oxidants. Co-based catalysts for DOMTM were determined in chemical looping mode analogue to the system with Cu-based catalysts [36,93]. Co/ZSM-5 can realize CH4 oxidation to oxygenates at low temperature by air as oxidant. Two catalyst preparation methods were adopted to tune the catalytic activity and selectivity of the catalytic system. The samples by ion-exchange are selective towards CH2O, while impregnated samples with more oxidic cobalt species (CoO and Co3O4) are favorable for CH3OH synthesis [94]. Zn-promoted Cu-containing mordenite was investigated using O2 as oxidant for DOMTM, and it was observed that Zn addition decreased the CH3OH yield first, followed by a gradual increase upon higher Zn concentration. The promotional effects of Zn addition could be attributed to the fact that is catalytically active by itself, and to promoting O2 activation by small ZnO particles [95].
An inverse CeO2/Cu2O/Cu(111) catalyst was reported that can activate CH4 to produce C, CHX fragments, and COX species at room temperature. The addition of water had a strong effect on CH3OH synthesis by DOMTM (Figure 17), mainly due to OH groups formed by the dissociation of water on the catalyst surface, which could adsorb easily on the active sites with special electronic properties, and thus inhibit the CHX full decomposition [96]. A further study showed that the site blocking effect of water on Ni/CeO2 catalyst can improve the CH3OH selectivity, in which the production of CO and CO2 are inhibited [39]. Another factor that needs to be considered is the effect of metal−support interaction to bind and activate CH4 and H2O, which provides a new insight for designing metal/oxide catalysts for DOMTM.
O2 as oxidant has been intensively studied during the past decades, and many catalysts has been explored in DOMTM under low temperature and atmosphere pressure. Recently, the economic potential and guidance have been reported on DOMTM via a chemical looping (or redox) process by comparison with the industrial performance criteria [97]. Low CH3OH productivity and long cycle time lead to an overall production rate that is a factor of ∼50 below the industrial threshold; therefore, enhancing productivity and reducing the cycle time are highly recommended for future studies.
2.4. Water as Oxidant or Co-Oxidant
The oxidizing character of water (H2O) is weak and; therefore, its use has been rarely reported for DOMTM (4), since it is a strong endothermic reaction.
CH4 + H2O → CH3OH + H2 ΔH (298K) = 262.2 kJ·mol−1
Van Bokhoven and co-workers proposed a direct stepwise method for DOMTM over a copper-containing zeolite, based on H2O as an oxidant [98,99]. Firstly, Cu-MOR was activated at 673 K with helium (Figure 18, Top), and then exposed to 7 bars of CH4, and finally reacting with H2O at 473 K. Noticeably, both CH3OH selectivity and yield remained constant over three additional cycles; the CH3OH productivity stabilized at 0.202 molCH3OH/molCu, and the selectivity reached 97%. In this anaerobic process, H2O as the source of oxygen can regenerate the zeolite active centers and ensures energetically favorable desorption of CH3OH. An excess of H2O can stabilize the reaction intermediates, indicated by isotopic labeling and DFT calculation. The reaction pathway predicted by DFT is illustrated in Figure 18, and the proposed mechanism shows that CH4 oxidation occurs at CuII oxide active centers, followed by CuI reoxidation by H2O with the formation of H2 [98]. Further insight of the active sites structure has been studied on Cu-MOR [86,100]. Monomeric and oligomeric Cu active sites were synthesized by mordenite with varied Si:Al ratio. Copper oligomer species shows a high activity under both aerobic (O2 as oxidant) and anaerobic (H2O as oxidant) activation conditions, whereas copper monomer sites produce CH3OH only in the aerobic process. This difference is most possibly associated with the stabilization effect of the H2O molecules on active copper sites [86]. Furthermore, Cu(II)-exchanged mordenites (i.e., Cu/H-MOR and Cu/Na-MOR) were compared with varied Si:Al ratio and Cu:Al ratio, and the results exhibited Cu/H-MOR had a higher methanol yield, which contributed to suppression of CO2 formation from the methoxy species at the final oxidation step indicated by DFT calculation [100]. Additionally, the activity of Cu-MOR is in line with the amount of Brønsted acid sites in the catalysts on DOMTM oxidized by H2O. Inspired by the study of anaerobic process on methane oxidation on Cu/MOR, other catalyst, e.g., Cu/SSZ-13, was also studied in a continuous flow reactor at ambient pressure and low temperature (≤250 °C) [101]. Undoubtedly, the DOMTM process with H2O as oxidant demonstrated here is promising from the perspective of cost and local on-site applications, which may contribute to the development of an industrial process for DOMTM. However, this work has also given rise to several discussions among researchers, mainly on several technical comments and replies questioning and defending the thermodynamic feasibility of the proposed mechanisms [98,102,103,104].
Except for H2O as a sole oxidant, the H2O-promoted DOMTM process exhibits a big potential to improve the CH3OH productivity and selectivity [38,39,87,95,105,106,107]. Recently, a CeO2/Cu2O/Cu(111) catalyst was reported for which the reaction can be operated using a mixture of CH4, O2 and H2O as feedstock. H2O participated in the reaction as the actual oxygen provider and enabled direct CH4 to CH3OH conversion. Additionally, H2O can act as a site blocker, where the facile dissociation at the interfacial Ce sites produced the active *OH, which promotes the DOMTM process [38]. Furthermore, H2O also functions as an extractor in the hydrogenation of *CH3O or blocks surface sites to preventing its decomposition, thus facilitating the extraction of CH3OH [39,95].
In this section, we showed the latest progress of different oxidants (N2O, H2O2, O2, and H2O) for DOMTM in heterogeneous catalysis. Attempts on how to reduce the kinetic energy barrier (Ea) of DOMTM are key to improve the CH4 conversion and CH3OH productivity, by using suitable oxidants. Generally, strong oxidants result in a lower Ea and lower reaction temperature. However, this is not always true, because different catalysts reduce the Ea to different degrees. Furthermore, most Ea values for DOMTM have been obtained by DFT calculations, but they are subject to uncertainties (e.g., due to the used functionals and dispersion corrections). Therefore, unified methods and standards need to be established to compare the role of catalysts in reducing the Ea of DOMTM. Recently, RuCu nano-sheets were reported on which CH4 can be efficiently converted to CH3OH and CH3OOH (methylhydroperoxide) under mild conditions in the presence of O2 and trace amount of H2O2 [40]. The combination of two or more oxidants (e.g., O2 + H2O, O2 + H2O2) may be effective in lowering the energy barrier of CH4 oxidation in DOMTM, avoiding the over-oxidation of CH3OH. Additionally, the exploration of efficient catalytic system remains a long-pursued target for DOMTM in heterogeneous catalysis.
3. Plasma Catalysis
Plasma catalysis provides an alternative to heterogeneous catalysis, where the catalytic process is complemented by plasma technology to activate the source gas [108,109,110,111]. This combination is often observed to result in a synergy between plasma and catalyst, which is increasingly gaining attention in many fields, such as CO2 and CH4 conversion, NOx decomposition, NH3 synthesis, H2O2 synthesis, Fischer–Tropsch synthesis, volatile organic compounds removal, wastewater treatment and degradation of pesticide residues [110,111,112]. Typically, the function of a catalyst in a given process is to reduce the activation energy barrier for the rate-limiting reaction and regulate the product distribution. This clear-cut function could also apply to plasma catalysis [113]. However, the effects of a catalyst in plasma catalysis are somewhat different than in traditional heterogeneous catalysis. In general, plasma as the “fourth state of matter” consists of many reactive species (i.e., electrons, molecules, radicals, excited species and ions), which could be adsorbed on the catalyst surface. The surface reactions happening in plasma catalysis are more complex than in conventional thermal catalysis and the possible interaction mechanisms between plasma and catalyst are illustrated in Figure 19 [113]. In this section, we mainly focus on the insights obtained by heterogeneous catalysis based on different oxidants for the further development of the emerging field of plasma catalysis for DOMTM.
The kind of plasma used in plasma catalysis is so-called non-thermal plasma (NTP), where the gas temperature remains near room temperature, while the temperature of the electrons is extremely high (usually in the range of 1–10 eV). This electron temperature is sufficient to activate stable gas molecules (e.g., CH4, CO2 and O2). Hence, NTP offers a distinct approach to enable thermodynamically unfavorable chemical reactions to proceed at low temperature by breaking thermodynamic limits [113,114,115,116,117]. The direct oxidation of CH4 in NTP seems a promising way of forming oxygenates (e.g., CH3OH or CH2O). Activated by plasma, the neutral CH4 and O2 molecules can form reactive species, and the major dissociation intermediates are the CH3 radicals and O atoms [118]. As mentioned above, CH3OH is more reactive than CH4 and more likely to undergo oxidation; therefore, moderate conditions (i.e., low temperature) are suitable, which can be provided by NTP. There exist various types of plasmas, such as microwave discharges, glow discharges, gliding arc discharges and dielectric barrier discharges (DBD). The latter are particularly suitable for plasma catalysis, because of their simple operation (atmospheric pressure and near room temperature) and simple design, allowing the easy integration of catalysts in the plasma reactor [108,112], Thus plasma-driven DOMTM is mostly based on DBD plasmas, and recent reports have shown the prospect in CH4 conversion assisted by plasma technology. An overview of the representative results is summarized in Table 2. Noticeably, DOMTM by plasma catalysis mostly can be operated near room temperature. Hence, H2O2 is rarely reported as oxidant, due to its high boiling point (150.2 °C). In addition, CO2 can be used as a soft oxidant for CH4 conversion in plasma catalysis (i.e., dry reforming of methane (DRM). This is not the focus of our review, as it has been elaborated in recent reviews [119,120].
3.1. Nitrous Oxide as an Oxidant
Co-processing of CH4 and nitrous oxide into high-value products, like CH3OH, CH2O, etc., was attempted in plasma under ambient conditions [121,122,123,136]. In a DBD reactor, it was observed that the mole ratio of CH4/N2O significantly affected the conversion and the product selectivity, and the highest selectivity (28%) of CH3OH could be realized at 5:1 CH4/N2O mole ratio, decreasing to 23% and 13%, respectively, for 1: 1 and 1: 5 ratios. A low ratio of CH4/N2O (i.e., 1:5) contains more N2O in the feed, which results in deep oxidation of the formed oxygenates to CO and CO2 [123]. Additionally, argon carrier gas played an important role in DOMTM for CH4/N2O plasma, by energy transfer from excited Ar atoms to the reactant molecules in this DBD system [121]. The formation of small amounts of C2H6 and carbonaceous materials was observed at higher input voltages, next to the main products * i.e., CH3OH, HCHO H2, CO and CO2). Under the conditions of 5% CH4 and 5% N2O in Ar, 50 cm3/min total flow rate and 7 kV input voltage, the combined yield of CH3OH and HCHO reached 10 mol%, but a further rise in residence time led to a lower selectivity of CH3OH and HCHO [121].
In order to improve the selectivity to CH3OH, CuO(10%)/CeO2, NiO(10%)/CeO2 and Cu-Ni(5-5%)/CeO2 catalysts were packed in a DBD reactor [122]. The catalytic activity followed the order of CuO-NiO/CeO2 > NiO/CeO2 > CuO/CeO2, with the best CH3OH selectivity of ∼36% on NiO/CeO2 catalyst. The best performance is highly attributed to the oxygen vacancies (proven by Raman results), and the increasing amount of oxygen vacancies may facilitate oxygen transfer and improve the catalytic activity [122]. Generally, the generation of energetic electrons is considered as initial step in plasma-initiated reactions [110,136]. In the plasma zone, the presence of abundant electrons could active NO2 and CH4 into various radicals. Based on molecular modelling results, the energy barrier for the formation of formyl radical (HCO) is much lower than that of other species [136]. Therefore, the authors suggested that HCO as an intermediate could be the major reason for CH3OH production [136]. Subsequently, further H addition to form CH3OH was found to have a very low energy barrier. This investigation confirms that DOMTM with NO2 as oxidant in non-thermal plasma is a simple and effective way for CH3OH production. However, the competing reaction (Figure 20) producing toxic gas (HCN and nitric acids) is a main challenge, although higher CH3OH selectivity can be obtained, compared with other oxidants (e.g., O2 [136]).
3.2. Oxygen or Air as an Oxidant
Molecular oxygen (O2) could be a perfect oxidant for plasma catalysis, due to its low cost and lack of harmful by-products, so it can meet the industry’s main goals in “green chemistry”. In CH4/O2 or CH4/air plasmas without catalyst, the effects of various parameters (e.g., reactor configuration, feed ratio, applied voltage, input power, residence time, pressure, specified energy input (SEI) and reactor wall temperature) have been extensively studied [124,126,127,137,138,139,140,141,142]. Oxidative products in these studies mainly included CH3OH, HCHO, HCOOH, CO and CO2, and a small quantity of C2+ oxygenates (e.g., ethanol, acetic acid and acetone). Noticeably, avoiding the decomposition of the produced CH3OH or other oxygenates by the plasma is an important issue [119].
Nozaki et al. reported a single step CH4 conversion into organic oxygenates by a microplasma reactor, and the organic oxygenates reached 5–20% one-pass yield with 70–30% selectivity. Additionally, syngas was found to be the main product with selectivity of 40% [140]. A further study showed that H2O2 with concentration of 0.15 wt.% was also detected in the liquid products [141]. Methyl hydroperoxide (CH3OOH) was also detected by 1H-NMR, and it was considered to be an important intermediate for plasma-assisted partial oxidation of CH4 [141]. The decomposition and further oxidation of liquid organic oxygenates cannot be ignored in plasma-assisted DOMTM. Larkin et al. adopted in situ condensation (a cold trap), as well as reduced residence time, to avoid further reaction of the oxygenates [127,138]. This approach can also be found in subsequent studies [140,142,143]. It can be concluded that a longer residence time may drive total combustion of CH4 to COX and H2O, and there was an optimal residence time for the production of CH3OH or other oxygenates [139]. Additionally, the specified energy input (SEI) was compared to consider the effects of power density on the DOMTM process, in which a lower SEI value favors the selectivity toward methanol and suppresses the formation of carbon oxides [127], but an opposite experimental result can be found that the selectivity to oxygenated products (e.g., CH3OH and HCHO) increased on high SEI value, mainly due to numbers of energetic electrons increased in CH4/O2 or CH4/Air plasma [129,130,133].
Coupling of plasma with catalysts is capable of improving CH3OH selectivity. Indarto et al. found that ZnO/Al2O3 catalyst in a DBD reactor doubled CH3OH selectivity to around 20%, compared with plasma-only conditions [143]. A similar experiment was found on Ni-doped Y-stabilized Zr catalyst, with optimum CH3OH selectivity of 23% [132]. Chawdhury et al. found that a CH4/O2 plasma packed with glass beads provided an optimal CH3OH selectivity of 35.4% [129]. Further work showed an improved CH3OH selectivity of 37% when using CuO/γ-Al2O3 catalyst (5 wt.% copper loading). Noticeably, a high Cu loading favored HCHO formation, with 20 wt.% Cu loading yielding the highest HCHO selectivity of 34%. Clearly, plasma packed with catalysts can produce significant amounts of oxygenates [130]. Compared with thermal reduction, a Pd/SBA-15 catalyst reduced by NTP showed much better catalytic performance (i.e., 70% oxygenates selectivity and CH3OH selectivity of 32%) [125]. The authors suggested the Pd-based catalysts reduced by NTP possess better surface characteristics, which can prevent the recombination and favor surface reactions, such as in-situ coupling, cleavage, H-transfer and CO/CO2 hydrogenation.
The position of the catalyst, embedded in the plasma discharge region, can largely influence the extent of various synergistic effects and thus the conversion rate. Ceramic-supported Pt, Fe2O3 and CeO2 catalysts were located downstream of the discharge zone to promote CH4/air plasma reaction, and the Fe2O3-based catalyst showed the best catalytic activity and the highest stability [128]. A decrease in the molar ratio of CH4 to air resulted in more O2 content available to react with the CH4 molecules, which led to a higher CH4 conversion. Packing of the catalysts had no significant effect on the CH4 conversion, but the CH3OH selectivity was enhanced for all three catalysts. Chen et al., compared in-plasma catalysis (IPC) and post-plasma catalysis (PPC) for CH4 partial oxidation to CH3OH [135]. The experimental results showed that IPC presented a better reaction performance, while PPC exhibited a higher stability because of lower carbon deposition. The reactive species (e.g., O3, O, CH2 and CH3) were identified by in situ optical emission spectroscopy (OES) and FT-IR spectroscopy, and more active species were verified in IPC.
It is known that the synergy between catalytic process and plasma activation depends on both the nature of the packing material and the discharge characteristics. The high CH3OH selectivity (~23%) on Ni/YSZ catalyst can be attributed to the presence of more surface oxygen vacancies on the YSZ surface [132]. Chawdhury et al. compared the catalytic performance of glass beads, Al2O3, TiO2 and CeO2 catalysts [129]. In contrast to the expectations, glass beads exhibited better performance than the other catalysts, and the authors attributed this better performance to a more uniform distribution of micro discharges and improved field strength on the glass beads [129]. A porous structure of the catalyst surface is beneficial to the formation of micro discharge inside the pores, although modeling predicted that the latter should only occur for pore diameters larger than the so-called Debye length, which is typically several 100 nm [144,145,146]. Furthermore, plasma parameters, e.g., electron temperature and densities of radicals, ions and electrons, show significant enhancement with increasing dielectric constant of the packed material [111,147]. Therefore, it is reasonable to expect that catalysts with high dielectric constant, porous structure and highly efficient active sites may promote CH3OH formation in CH4/O2 plasma catalysis.
Recently, three supported transition metal catalysts (Fe/γ-Al2O3, Ni/γ-Al2O3 and Cu/γ-Al2O3) were compared in plasma-catalytic DOMTM, and the Fe/γ-Al2O3 catalyst exhibited the highest CH3OH selectivity of 36%, while the Cu/γ-Al2O3 catalyst improved the selectivity of C2 oxygenates to 9.4%, mainly due to more acid sites on the surfaces of the Cu catalyst, as revealed by NH3-TPD experiments [118]. It is noticed that the plasma-catalytic DOMTM reaction involves both plasma gas-phase reactions and plasma-assisted surface reactions on the catalysts [110,118]. In the plasma gas phase, the reactions are initiated through a variety of inelastic collisions between the reactants (CH4 and O2) and energetic electrons, generating a lot of reactive species (e. g., CHx and O) and excited species, which are believed to play a crucial role for CH3OH production [118]. Combined with the results of optical emission spectroscopy (OES), the plausible reaction pathways in the plasma gas phase were proposed, as illustrated in Figure 21, and the CH3 radicals, mainly created by electron impact dissociation of CH4, were considered as the dominant and critical species in this reaction.
The addition of an Fe/γ-Al2O3 catalyst remarkably changed the OES intensities, and the main characteristic peaks (CH, H, C2, CO and O peaks) of the CH4/O2 plasma were lowered by the catalyst packing [118]. The authors explained this by the adsorption of reactive species on the catalyst surface. Surface discharges are dominant in the plasma-catalytic reactions, while filamentary microdischarges were limited when packing catalysts in the discharge region [116,148]. For plasma-catalytic surface reactions, the reactions may take place via Langmuir-Hinshelwood (L-H) and Eley-Rideal (E-R) mechanisms, whereas in thermal catalytic reactions, the L-H mechanism dominates [110]. As shown in Figure 22, the radicals (e.g., CHx, O and OH) can be formed in the plasma gas phase, and they can be directly adsorbed onto the catalyst surface. These chemisorption CHx species can be further oxidized to form CHxO by O and OH radicals, via E-R and L-H mechanisms, to speed up the CH3OH production by stepwise hydrogenation on the Fe/γ-Al2O3 catalyst surface [118].
Kinetic modeling of CH4 conversion by plasma can help to determine the suitable parameters (such as feed gas ratio, residence time, and discharge power) for optimal performance [149,150]. De Bie et al., developed a one-dimensional fluid model to study the chemistry in a CH4/O2 and CH4/CO2 DBD plasma [149]. The dominant reaction pathways as predicted by the model for CH4 partial oxidation into CH3OH and other oxygenates are shown in Figure 23. Densities of the various plasma species as a function of residence time and gas mixing ratio were studied, and the simulation results showed that CH4/CO2 plasma favors the formation of H2, CH2O, CH3CHO, and CH2CO, while the densities of H2O2, CH3OH, C2H5OH, CH3OOH, and C2H5OOH were higher in CH4/O2 plasma. CO was formed at high density in both gas mixtures. Basically, the modelling results provide a better understanding of the reaction pathways, which is helpful for future experiments to acquire a maximum selectivity/yield with minimum energy consumption.
Next to the plasma chemistry, the reactions at the catalyst surface can also be studied by kinetic modeling. Loenders et al. investigated how different plasma species affect the partial oxidation of CH4 into CH3OH and other oxygenates on a Pt(111) surface [151]. In particular they focused on the effect of vibrationally excited CH4 and O2, as well as plasma-generated radicals and stable intermediates on the reaction kinetics and mechanisms. The calculation results revealed that vibrational excitation enhances the turnover frequency (TOF) of catalytic CH4 dissociation and has good potential for improving the selectivities towards CH3OH, HCOOH, and C2 hydrocarbons, but the plasma-generated radicals mainly govern the surface chemistry. They enhance the TOFs of COx and oxygenates, increase the selectivity towards oxygenates, and make the formation of HCOOH more significant on Pt(111), compared to thermal catalysis. Such modeling is of great interest for obtaining a better understanding of plasma-catalytic DOMTM [151].
3.3. Water as an Oxidant
In a plasma reactor, energetic electrons are capable of dissociating CH4 together with H2O into reactive species (i.e., electrons, radicals, excited species and ions), which makes the direct reaction of CH4 and H2O to CH3OH possible under mild conditions. Tsuchiya et al. investigated a CH4/H2O plasma under different discharge conditions, such as gas flow rate, gas-mixing ratio, and discharge power, with total gas pressure of 1–10 Torr [152]. Several gaseous organic products, such as C2H6, C2H4, C2H2 and CH3OH, were detected, and the CH3OH selectivity was sensitive to the ratio of CH4/H2O. Under optimized conditions, the highest CH3OH selectivity reported was 20%, and CO was detected to be the major product. The possible reaction pathways for DOMTM process in CH4/H2O plasma are illustrated in Figure 24.
More insight in the mechanism in CH4/H2O plasma could be obtained from 2H, 17O and 18O-labeled isotopologues experiment, and the results indicated that O atoms in the 1D state inserted into CH4 could be considered as a possible mechanism for CH3OH production (i.e., CH4 + O (1D) → CH3OH) [153]. The second most abundant isotopologue of CH3OH was probably formed from a reaction (CH2OH + H - CH2DOH) with CH2OH radical. In addition, mMatrix isolation FTIR experiments revealed that CH4/H2O/Ar plasma at 11 K yielded organic molecules, including CH3OH, CH2O, CH2OH, HOCH and HCO, with CH3OH as the major product.
3.4. Discussion
The previous sections gave an overview of results obtained by plasma-catalytic DOMTM based on different oxidants (i.e., NOx, O2 and H2O). It is clear that the synergistic effects in plasma catalysis are quite complicated. Evidently, the reaction kinetics, product yields and selectivities are influenced by alternative reaction pathways compared to classical heterogeneous catalysis, and there is clear need for a more fundamental understanding of the mutual interactions between plasma and catalyst. As mentioned in the introduction, the selective activation and controlled conversion of CH4 into CH3OH are considered the “holy grail” in catalysis, mainly due to the highly stable C–H bond. This activation could be possibly realized by directing energy into C–H bond vibrations, or by creating CH3 radicals, which is both possible in NTP at room temperature and atmosphere pressure [109,151,154]. These vibrationally or electronically excited species, or plasma radicals, may facilitate certain steps at the catalyst surface (e.g., dissociative adsorption), allowing other catalysts to perform better in the overall catalytic process, as compared to classical thermal catalysis [110,155]. Additionally, the lifetime of plasma-produced reactive species is a key parameter in determining their effectiveness in plasma catalysis. Figure 25 illustrates the various processes occurring in plasma catalysis, in the full range of time scale, from picoseconds to minutes [112]. The lifetime of reactive species in plasma catalysis is largely influenced by the operating conditions. Therefore, various parameters, such as reactor configuration, feed ratio, applied voltage, residence time, pressure and reactor wall temperature, should be fully considered and adjusted for the desired reaction.
The activity of different catalysts inside the plasma region is determined by multiple factors ranging from chemical composition to physical properties. For DOMTM, another important factor is how to avoid deep oxidation of CH3OH to CO or CO2. Based on the mechanism of plasma catalysis, the adsorption probability is essential for all surface-mediated reaction mechanisms, namely the Langmuir–Hinshelwood (L-H), Eley–Rideal (E-R) and Mars–van Krevelen (MvK) mechanisms [110]. Recent work on plasma catalysis for HCN production by CH4/NH3 NTP corroborated well with the barrierless Eley-Rideak (E-R) reactions between radicals generated in plasma and adsorbed species over the Cu surface, indicated by DFT calculation [156]. As for DOMTM, it is believed that E-R and L-H mechanism may occur in the case of plasma-catalysis on catalyst surfaces, whereas the L-H mechanism dominates in thermal catalytic reactions [118]. These findings mean that once CH3 radicals are formed, the subsequent reactions will be rapid. That is, the contribution of both E-R and L-H mechanism will be limited by the dissociation rate of CH4 through electron impact dissociation, implying that advanced plasma systems with fast dissociation rate of CH4 will promote CH4 conversion. On the other hand, pre-adsorption of reactive species is necessary for both E-R and L-H mechanisms to produce CH3OH. This is important in further studies for rational design of catalysts, and the key is to seek advanced catalytic systems which could accelerate the hydrogenation of important intermediates, such as, CH3O, as well as the desorption of CH3OH from the catalyst surface, improving CH3OH selectivity.
4. Outlook and Conclusions
This review paper provides a comprehensive overview about the recent progress in direct oxidation of methane to methanol (DOMTM) based on different oxidants, by both heterogeneous catalysis and plasma catalysis. Compared with the numerous studies in heterogeneous catalysis, much more research should be carried out in the field of plasma catalysis, to improve the CH4 conversion and target product selectivity. For this purpose, a better understanding of the underlying mechanisms is required, as they are much more complicated than in heterogeneous catalysis. This should be obtained by modeling, as well as by dedicated experiments. Fundamental studies on the interaction between plasma and catalysts are often limited by the lack of available instruments, especially for in situ measurements, which can provide the fundamental information about the behavior of the different species created in the gas-phase and on the catalytic surface.
As shown in Table 2, the reported selectivity of CH3OH remains low (<40%) and further work needs to improve the plasma-catalytic performance. Innovations in new catalysts and concepts are needed to seek cost-effective, highly active and stable catalysts. A better catalyst selection strategy can be achieved only by combining advanced level simulation on plasma, catalysis and plasma–surface interactions and validate them with dedicated experiments. To achieve this goal, the advantage of plasma should be fully exploited, and at the same time, insights from heterogeneous catalysis (e.g., catalyst combination, reaction combination, active sites design, etc.) can help to further improve the potential of the promising field of plasma catalysis.
In summary, CH3OH is an important building block for the chemical industry, and DOMTM by several possible oxidants (N2O, H2O2, O2 and H2O) could be interesting for this purpose. However, before DOMTM can be exploited commercially, the CH4 conversion and CH3OH yield in heterogeneous catalysis must be further improved, especially by a combination of various oxidants, which may effectively lower the energy barrier of CH4 oxidation, and avoid the over-oxidation of CH3OH. For plasma-catalytic DOMTM, a much deeper fundamental understanding of the process is required by means of strong interdisciplinary studies. A combination of computer simulations with experiments will be needed for an in-depth understanding of the reaction mechanisms responsible for the synergy between plasma and catalysts. Therefore, future research should focus on a better understanding and rational screening of highly active catalysts.
Author Contributions
Conceptualization: S.L., R.A., Y.Y. and A.B.; validation: S.L., R.A., Y.Y. and A.B.; formal analysis: S.L., R.A., Y.Y. and A.B.; resources: Y.Y. and A.B.; data curation: Y.Y. and A.B.; writing—original draft preparation: S.L., R.A.; writing—review and editing: Y.Y. and A.B.; supervision: Y.Y. and A.B.; funding acquisition: Y.Y. and A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fundamental Research Funds for the Central Universities of China (DUT18JC42), the National Natural Science Foundation of China (21503032) PetroChina Innovation Foundation (2018D-5007-0501) and the TOP-BOF research project of the Research Council of the University of Antwerp (grant ID 32249). This research was supported by the China Scholarship Council (CSC). The authors warmly acknowledge CSC for their support.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IEA. World Energy-Outlook 2019; Part A: World Global Energy Trend, Outlook for Natural Gas; IEA: Paris, France, 2019; pp. 176–187. [Google Scholar]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; Van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Yang, Y.; Xu, Y.; Etim, U.J.; Qiao, K.; Xu, B.; Yan, Z. A review of the direct oxidation of methane to methanol. Chin. J. Catal. 2016, 37, 1206–1215. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent advances in heterogeneous selective oxidation catalysis for sustainable chemistry. Chem. Soc. Rev. 2014, 43, 3480–3524. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Perez, P.J. Methane as a raw material in synthetic chemistry the final frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef]
- Zakaria, Z.; Kamarudin, S.K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Raynes, S.; Shah, M.A.; Taylor, R.A. Direct conversion of methane to methanol with zeolites: Towards understanding the role of extra-framework d-block metal and zeolite framework type. Dalton Trans. 2019, 48, 10364–10384. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-Electro or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-Temperature Methane Conversion by Graphene-Confined Single Iron Atoms. Chem 2018, 4, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Loffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A Mercury Catalyzed High Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Periana, R.A.; Tauba, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum Catalysts for the High Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Muehlhofer, M.; Strassner, T.; Herrmann, W.A. New Catalyst Systems for the Catalytic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2002, 41, 1745–1747. [Google Scholar] [CrossRef]
- Smith, M.R.; Ozkan, U.S. The Partial Oxidation of Methane to Formaldehyde: Role of different Crystal Planes of MoO3. J. Catal. 1993, 141, 124–139. [Google Scholar] [CrossRef]
- Weng, T.; Wolf, E.E. Partial oxidation of methane on Mo/Sn/P silica supported catalysts. Appl. Catal. A Gen. 1993, 96, 383–396. [Google Scholar] [CrossRef]
- Millner, T.; Neugebauer, J. Volatility of the oxides of tungsten and molybdenum in the presence of water vapour. Nature 1949, 163, 601–602. [Google Scholar] [CrossRef]
- Barbero, J.A.; Alvarez, M.C.; Banares, M.A.; Pena, M.A.; Fierro, J.L.G. Breakthrough in the direct conversion of methane into C-1 Oxygenates. Chem. Commun. 2002, 11, 1184–1185. [Google Scholar] [CrossRef]
- Chen, S.Y.; Willcox, D. Effect of vanadium oxide loading on the selective oxidation of methane over vanadium oxide (V2O5)/silica. Ind. Eng. Chem. Res. 1993, 32, 584–587. [Google Scholar] [CrossRef]
- Marturano, P.; Drozdová, L.; Kogelbauer, A.; Prins, R. Fe/ZSM-5 Prepared by Sublimation of FeCl3 The Structure of the Fe Species as Determined by IR, 27Al MAS NMR, and EXAFS Spectroscopy. J. Catal. 2000, 192, 236–247. [Google Scholar] [CrossRef]
- Battiston, A.A.; Bitter, J.H.; De-Groot, F.M.F.; Overweg, A.R.; Stephan, O.; Van Bokhoven, J.A.; Kooyman, P.J.; van-der-Spek, C.; Vanko, G.; Koningsberger, D.C. Evolution of Fe species during the synthesis of over-exchanged Fe/ZSM5 obtained by chemical vapor deposition of FeCl3. J. Catal. 2003, 213, 251–271. [Google Scholar] [CrossRef] [Green Version]
- Groothaert, M.H.; Van Bokhoven, J.A.; Battiston, A.A.; Weckhuysen, B.M.; Schoonheydt, R.A. Bis(mu-oxo)dicopper in Cu-ZSM-5 and its role in the decomposition of NO: A combined in situ XAFS, UV-Vis-Near-IR, and kinetic study. J. Am. Chem. Soc. 2003, 125, 7629–7640. [Google Scholar] [CrossRef]
- Vanelderen, P.; Hadt, R.G.; Smeets, P.J.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F. Cu-ZSM-5: A biomimetic inorganic model for methane oxidation. J. Catal. 2011, 284, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, J.; Li, M.; Allard, L.F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 2017, 551, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Starokon, E.V.; Parfenov, M.V.; Arzumanov, S.S.; Pirutko, L.V.; Stepanov, A.G.; Panov, G.I. Oxidation of methane to methanol on the surface of FeZSM-5 zeolite. J. Catal. 2013, 300, 47–54. [Google Scholar] [CrossRef]
- Bols, M.L.; Hallaert, S.D.; Snyder, B.E.R.; Devos, J.; Plessers, D.; Rhoda, H.M.; Dusselier, M.; Schoonheydt, R.A.; Pierloot, K.; Solomon, E.I.; et al. Spectroscopic Identification of the alpha-Fe/alpha-O Active Site in Fe-CHA Zeolite for the Low-Temperature Activation of the Methane C–H Bond. J. Am. Chem. Soc. 2018, 140, 12021–12032. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, T.Y.; Lee, H.; Yi, J. Distinct activation of Cu-MOR for direct oxidation of methane to methanol. Chem. Commun. 2017, 53, 4116–4119. [Google Scholar] [CrossRef] [PubMed]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold–Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, T.Y.; Kwon, G.; Yi, J.; Lee, H. Selective Activation of Methane on Single-Atom Catalyst of Rhodium Dispersed on Zirconia for Direct Conversion. J. Am. Chem. Soc. 2017, 139, 17694–17699. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(µ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; et al. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef] [PubMed]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure-Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef] [Green Version]
- Knorpp, A.J.; Pinar, A.B.; Newton, M.; Sushkevich, V.L.; Van Bokhoven, J.A. Copper-Exchanged Omega (MAZ) Zeolite: Copper-concentration Dependent Active Sites and its Unprecedented Methane to Methanol Conversion. ChemCatChem 2018, 10, 5593–5596. [Google Scholar] [CrossRef]
- Beznis, N.V.; Weckhuysen, B.M.; Bitter, J.H. Partial Oxidation of Methane Over Co-ZSM-5: Tuning the Oxygenate Selectivity by Altering the Preparation Route. Catal. Lett. 2010, 136, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Huang, W.; Nguyen, L.; Yu, Y.; Zhang, S.; Li, Y.; Frenkel, A.I.; Tao, F.F. Conversion of methane to methanol with a bent mono(mu-oxo)dinickel anchored on the internal surfaces of micropores. Langmuir 2014, 30, 8558–8569. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, E.; Orozco, I.; Liao, W.; Palomino, R.M.; Rui, N.; Duchon, T.; Nemsak, S.; Grinter, D.C.; Mahapatra, M.; et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst. Science 2020, 368, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Lustemberg, P.G.; Palomino, R.M.; Gutiérrez, R.A.; Grinter, D.C.; Vorokhta, M.; Liu, Z.; Ramírez, P.J.; Matolín, V.; Ganduglia-Pirovano, M.V.; Senanayake, S.D.; et al. Direct Conversion of Methane to Methanol on Ni-Ceria Surfaces: Metal–Support Interactions and Water-Enabled Catalytic Conversion by Site Blocking. J. Am. Chem. Soc. 2018, 140, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Yao, Q.; Xu, Y.; Cao, K.; Huang, X. Strong synergy in a lichen-like RuCu nanosheet boosts the direct methane oxidation to methanol. Nano Energy 2020, 71, 104566. [Google Scholar] [CrossRef]
- Liu, R.S.; Iwamoto, M.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over Molybdenum Oxide supported on Silica. J. Chem. Soc. Chem. 1982, 1, 78–79. [Google Scholar] [CrossRef]
- Rosenzweig, A.; Frederick, C.; Lippard, S.; Nordlund, P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 1993, 366, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Panov, G.I.; Soboley, V.I.; Kharitonov, A.S. The role of iron in N2O decomposition on ZSM-5 zeolite and reactivity of the surface oxygen formed. J. Mol. Catal. 1990, 61, 85–97. [Google Scholar] [CrossRef]
- Panov, G.I.; Sheveleva, G.A.; Kharitonov, A.S.; Romannikov, V.N.; Vostrikova, L.A. Oxidation of benzene to phenol by nitrous oxide over Fe-ZSM-5 zeolites. Appl. Catal. A Gen. 1992, 82, 31–36. [Google Scholar] [CrossRef]
- Panov, G.I.; Soboley, V.I.; Dubkov, K.A.; Parmon, V.N.; Ovanesyan, N.S.; Shilov, A.E.; Shteinman, A.A. Iron complexes in zeolites as a new model of methane monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.L.; Hallaert, S.D.; Bottger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Michalkiewicz, B. Partial oxidation of methane to formaldehyde and methanol using molecular oxygen over Fe-ZSM-5. Appl. Catal. A Gen. 2004, 277, 147–153. [Google Scholar] [CrossRef]
- Goltl, F.; Michel, C.; Andrikopoulos, P.C.; Love, A.M.; Hafner, J.; Hermans, I.; Sautet, P. Computationally Exploring Confinement Effects in the Methane-to-Methanol Conversion Over Iron Oxo Centers in Zeolites. ACS. Catal. 2016, 6, 8404–8409. [Google Scholar] [CrossRef]
- Andrikopoulos, P.C.; Sobalik, Z.; Novakova, J.; Sazama, P.; Sklenak, S. Mechanism of framework oxygen exchange in Fe-zeolites: A combined DFT and mass spectrometry study. ChemPhysChem 2013, 14, 520–531. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Yoshizawa, K. Methane selective oxidation to methanol by metal-exchanged zeolites: A review of active sites and their reactivity. Catal. Sci. Techol. 2019, 9, 1744–1768. [Google Scholar] [CrossRef]
- Zhao, G.; Adesina, A.A.; Kennedy, E.M.; Stockenhuber, M. Formation of Surface Oxygen Species and the Conversion of Methane to Value-Added Products with N2O as Oxidant over Fe-Ferrierite Catalysts. ACS Catal. 2020, 10, 1406–1416. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, J.H.; Park, S.H.; Moon, D.J.; Roh, H.-S.; Chung, C.-H.; Um, S.H.; Choi, J.-H.; Bae, J.W. Direct activation of CH4 to oxygenates and unsaturated hydrocarbons using N2O on Fe-modified zeolites. J. Mol. Catal. A Chem. 2017, 426, 130–140. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Pirutko, L.V.; Abornev, S.I.; Panov, G.I. Room Temperature Oxidation of Methane by R Oxygen and Extraction of Products from the FeZSM-5 Surface. J. Phys. Chem. 2011, 115, 2155–2161. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Jenkins, R.; Lopez-Sanchez, T.; Kondrat, S.; Ab-Rahim, M.H.; Forde, M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 3, 689–699. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct Conversion of Methane to Methanol by Metal-Exchanged ZSM-5 Zeolite (Metal=Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Li, S.; Fan, L.; Song, L.; Cheng, D.; Chen, F. Influence of extra-framework Al in Fe-MOR catalysts for the direct conversion of methane to oxygenates by nitrous oxide. Chin. J. Chem. Eng. 2020. [Google Scholar] [CrossRef]
- Parfenov, M.V.; Starokon, E.V.; Pirutko, L.V.; Panov, G.I. Quasicatalytic and catalytic oxidation of methane to methanol by nitrous oxide over FeZSM-5 zeolite. J. Catal. 2014, 318, 14–21. [Google Scholar] [CrossRef]
- Liu, H.F.; Liu, R.S.; Liew, K.Y.; Johnson, R.E.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over molybdenum on silica. J. Am. Chem. Soc. 1984, 106, 4117–4121. [Google Scholar] [CrossRef]
- Ipek, B.; Lobo, R.F. Catalytic conversion of methane to methanol on Cu-SSZ-13 using N2O as oxidant. Chem. Commun. 2016, 52, 13401–13404. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, P.; Neyts, E.C. Direct oxidation of methane to methanol on Co embedded N-doped graphene: Comparing the role of N2O and O2 as oxidants. Appl. Catal. A Gen. 2020, 602, 117716. [Google Scholar] [CrossRef]
- Montzka, S.A.; Dlugokencky, E.J.; Butler, J.H. Non-CO2 greenhouse gases and climate change. Nature 2011, 476, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Goor, G.; Glenneberg, J.; Jacobi, S.; Dadabhoy, J.; Candido, E. Hydrogen Peroxide. Ullmann’s Encycl. Ind. Chem. 2019. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Song, S.; Wang, X.; Zhang, H. CeO2-encapsulated noble metal nanocatalysts: Enhanced activity and stability for catalytic application. NPG Asia Mater. 2015, 7, e179. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhang, S.; Tang, Y.; Li, Y.; Nguyen, L.; Li, Y.; Shan, J.; Xiao, D.; Gagne, R.; Frenkel, A.I.; et al. Low-Temperature Transformation of Methane to Methanol on Pd1 O4 Single Sites Anchored on the Internal Surface of Microporous Silicate. Angew. Chem. Int. Ed. 2016, 55, 13441–13445. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, Y.; Fung, V.; Jiang, D.E.; Huang, W.; Zhang, S.; Iwasawa, Y.; Sakata, T.; Nguyen, L.; Zhang, X.; et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 2018, 9, 1231. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.K.; Solsona, B.E.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au–Pd catalysts. J. Catal. 2005, 236, 69–79. [Google Scholar] [CrossRef]
- Kesavan, L.; Tiruvalam, R.; Ab Rahim, M.H.; Bin Saiman, M.I.; Enache, D.I.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Knight, D.W.; et al. Solvent-free oxidation of primary carbon-hydrogen bonds in toluene using Au-Pd alloy nanoparticles. Science 2011, 331, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Piccinini, M.; Tiruvalam, R.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Edwards, J.K.; Kiely, C.J.; et al. Effect of heat treatment on Au–Pd catalysts synthesized by sol immobilisation for the direct synthesis of hydrogen peroxide and benzyl alcohol oxidation. Catal. Sci. Technol. 2013, 3, 308–317. [Google Scholar] [CrossRef]
- Thetford, A.; Hutchings, G.J.; Taylor, S.H.; Willock, D.J. The decomposition of H2O2 over the components of Au/TiO2 catalysts. Proc. Math. Phys. Eng. Sci. 2011, 467, 1885–1899. [Google Scholar]
- Ab-Rahim, M.H.; Armstrong, R.D.; Hammond, C.; Dimitratos, N.; Freakley, S.J.; Forde, M.M.; Morgan, D.J.; Lalev, G.; Jenkins, R.L.; Lopez-Sanchez, J.A.; et al. Low temperature selective oxidation of methane to methanol using titania supported gold palladium copper catalysts. Catal. Sci. Technol. 2016, 6, 3410–3418. [Google Scholar] [CrossRef] [Green Version]
- Serra-Maia, R.; Michel, F.M.; Kang, Y.; Stach, E.A. Decomposition of Hydrogen Peroxide Catalyzed by AuPd Nanocatalysts during Methane Oxidation to Methanol. ACS Catal. 2020, 10, 5115–5123. [Google Scholar] [CrossRef]
- Xie, J.; Jin, R.; Li, A.; Bi, Y.; Ruan, Q.; Deng, Y.; Zhang, Y.; Yao, S.; Sankar, G.; Ma, D.; et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 2018, 1, 889–896. [Google Scholar] [CrossRef]
- Xu, J.; Armstrong, R.D.; Shaw, G.; Dummer, N.F.; Freakley, S.J.; Taylor, S.H.; Hutchings, G.J. Continuous selective oxidation of methane to methanol over Cu- and Fe-modified ZSM-5 catalysts in a flow reactor. Catal. Today 2016, 270, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Park, K.H.; Cho, S.J.; Park, E.D. Partial oxidation of methane with hydrogen peroxide over Fe-ZSM-5 catalyst. Catal. Today 2020. [Google Scholar] [CrossRef]
- Kalamaras, C.; Palomas, D.; Bos, R.; Horton, A.; Crimmin, M.; Hellgardt, K. Selective Oxidation of Methane to Methanol Over Cu- and Fe-Exchanged Zeolites: The Effect of Si/Al Molar Ratio. Catal. Lett. 2016, 146, 483–492. [Google Scholar] [CrossRef]
- Shahami, M.; Shantz, D.F. Zeolite acidity strongly influences hydrogen peroxide activation and oxygenate selectivity in the partial oxidation of methane over M,Fe-MFI (M: Ga, Al, B) zeolites. Catal. Sci. Technol. 2019, 9, 2945–2951. [Google Scholar] [CrossRef]
- Forde, M.M.; Armstrong, R.D.; Hammond, C.; He, Q.; Jenkins, R.L.; Kondrat, S.A.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Willock, D.; et al. Partial oxidation of ethane to oxygenates using Fe- and Cu-containing ZSM-5. J. Am. Chem. Soc. 2013, 135, 11087–11099. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, P.; Ranocchiari, M.; Van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Markovits, M.A.C.; Jentys, A.; Tromp, M.; Sanchez-Sanchez, M.; Lercher, J.A. Effect of Location and Distribution of Al Sites in ZSM-5 on the Formation of Cu-Oxo Clusters Active for Direct Conversion of Methane to Methanol. Top. Catal. 2016, 59, 1554–1563. [Google Scholar] [CrossRef]
- Smeets, P.J.; Groothaert, M.H.; Schoonheydt, R.A. Cu based zeolites: A UV–vis study of the active site in the selective methane oxidation at low temperatures. Catal. Today 2005, 110, 303–309. [Google Scholar] [CrossRef]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [Green Version]
- Smeets, P.J.; Hadt, R.G.; Woertink, J.S.; Vanelderen, P.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Oxygen Precursor to the Reactive Intermediate in Methanol Synthesis by Cu-ZSM-5. J. Am. Chem. Soc. 2010, 132, 14736–14738. [Google Scholar] [CrossRef] [Green Version]
- Groothaert, M.H.; Lievens, K.; Leeman, H.; Weckhuysen, B.M.; Schoonheydt, R.A. An operando optical fiber UV–vis spectroscopic study of the catalytic decomposition of NO and N2O over Cu-ZSM-5. J. Catal. 2003, 220, 500–512. [Google Scholar] [CrossRef] [Green Version]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Palagin, D.; Van Bokhoven, J.A. The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2018, 57, 8906–8910. [Google Scholar] [CrossRef] [PubMed]
- Park, M.B.; Ahn, S.H.; Ranocchiari, M.; Van Bokhoven, J.A. Comparative Study of Diverse Cu-Zeolites for Conversion of Methane-to-Methanol. ChemCatChem 2017, 9, 3705–3713. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Vanelderen, P.; Snyder, B.E.R.; Tsai, M.L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic definition of the copper active sites in mordenite: Selective methane oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef] [Green Version]
- Wulfers, M.J.; Teketel, S.; Ipek, B.; Lobo, R.F. Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes. Chem. Commun. 2015, 51, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, T.; Hamill, C.D.; Goguet, A.; Rooney, D.W.; Thompson, J.M. A low temperature, isothermal gas-phase system for conversion of methane to methanol over Cu-ZSM-5. Chem. Commun. 2014, 50, 11053–11055. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; Van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 55, 5467–5471. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Van Bokhoven, J.A. Methane-to-Methanol: Activity Descriptors in Copper-Exchanged Zeolites for the Rational Design of Materials. ACS Catal. 2019, 9, 6293–6304. [Google Scholar] [CrossRef]
- Krisnandi, Y.K.; Putra, B.A.P.; Bahtiar, M.; Zahara; Abdullah, I.; Howe, R.F. Partial Oxidation of Methane to Methanol over Heterogeneous Catalyst Co/ZSM-5. Procedia Chem. 2015, 14, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Meesub, C.; Tannititam, P.; Kruemek, P.; Triamnak, N.; Chotigkrai, N. Direct stepwise oxidation of methane to methanol via zinc promoted copper containing mordenite. In Proceedings of the 8th International Thai Institute of Chemical Engineering and Applied Chemistry Conference, Pattaya, Thailand, 8–9 November 2018; pp. 45–50. [Google Scholar]
- Zuo, Z.; Ramirez, P.J.; Senanayake, S.; Liu, P.; Rodriguez, J.A. Low-Temperature Conversion of Methane to Methanol on CeOx/Cu2O Catalysts: Water Controlled Activation of the C–H Bond. J. Am. Chem. Soc. 2016, 138, 13810–13813. [Google Scholar] [CrossRef]
- Lange, J.P.; Sushkevich, V.L.; Knorpp, M.J.; Van Bokhoven, J.A. Methane to Methanol via Chemical Looping Economic Potential and Guidance for Future Research. Ind. Eng. Chem. Res. 2019, 58, 8674–8680. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Palagin, D.; Sushkevich, V.L.; van Bokhoven, J.A. Water Molecules Facilitate Hydrogen Release in Anaerobic Oxidation of Methane to Methanol over Cu/Mordenite. ACS Catal. 2019, 9, 10365–10374. [Google Scholar] [CrossRef]
- Jeong, Y.R.; Jung, H.; Kang, J.; Han, J.W.; Park, E.D. Continuous Synthesis of Methanol from Methane and Steam over Copper-Mordenite. ACS Catal. 2021, 11, 1065–1070. [Google Scholar] [CrossRef]
- Koishybay, A.; Shantz, D.F. Water Is the Oxygen Source for Methanol Produced in Partial Oxidation of Methane in a Flow Reactor over Cu-SSZ-13. J. Am. Chem. Soc. 2020, 142, 11962–11966. [Google Scholar] [CrossRef] [PubMed]
- Periana, R.A. Comment on “Selective anaerobic oxidation of methane enables direct synthesis of methanol”. Science 2017, 358, eaan5970. [Google Scholar] [CrossRef] [Green Version]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Response to Comment on “Selective anaerobic oxidation ofmethane enables direct synthesis ofmethanol”. Science 2017, 358, eaan6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labinger, J.A. Comment on “Selective anaerobic oxidation of methane enables direct synthesis of methanol”. Science 2018, 359, eaar4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dinca, M.; Roman-Leshkov, Y. Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef] [PubMed]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Ahi, H.; Trunschke, A.; Schomacker, R.; Thomas, A. Stepwise Methane-to-Methanol Conversion on CuO/SBA-15. Chemistry 2018, 24, 12592–12599. [Google Scholar] [CrossRef] [PubMed]
- Yumura, T.; Hirose, Y.; Wakasugi, T.; Kuroda, Y.; Kobayashi, H. Roles of Water Molecules in Modulating the Reactivity of Dioxygen-Bound Cu-ZSM-5 toward Methane: A Theoretical Prediction. ACS Catal. 2016, 6, 2487–2495. [Google Scholar] [CrossRef]
- Snoeckx, R.; Bogaerts, A. Plasma technology—A novel solution for CO2 conversion? Chem. Soc. Rev. 2017, 46, 5805–5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, J.C. Plasma catalysis the known knowns, the known unknowns and the unknown unknowns. J. Phys. D Appl. Phys. 2016, 49, 243001. [Google Scholar] [CrossRef]
- Bogaerts, A.; Tu, X.; Whitehead, J.C.; Centi, G.; Lefferts, L.; Guaitella, O.; Azzolina-Jury, F.; Kim, H.-H.; Murphy, A.B.; Schneider, W.F.; et al. The 2020 plasma catalysis roadmap. J. Phys. D Appl. Phys. 2020, 53, 443001. [Google Scholar] [CrossRef]
- Neyts, E.C.; Ostrikov, K.K.; Sunkara, M.K.; Bogaerts, A. Plasma Catalysis: Synergistic Effects at the Nanoscale. Chem. Rev. 2015, 115, 13408–13446. [Google Scholar] [CrossRef]
- Kim, H.H.; Teramoto, Y.; Ogata, A.; Takagi, H.; Nanba, T. Plasma Catalysis for Environmental Treatment and Energy Applications. Plasma Chem. Plasma Process. 2015, 36, 45–72. [Google Scholar] [CrossRef]
- Liu, M.; Yi, Y.; Wang, L.; Guo, H.; Bogaerts, A. Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2019, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yi, Y.; Wang, L.; Yan, J.; Guo, H. Pt/TS-1 Catalyst Promoted C–N Coupling Reaction in CH4–NH3 Plasma for HCN Synthesis at Low Temperature. ACS Catal. 2018, 8, 10219–10224. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Wu, C.; Guo, H.; Tu, X. One-Step Reforming of CO2 and CH4 into High-Value Liquid Chemicals and Fuels at Room Temperature by Plasma-Driven Catalysis. Angew. Chem. Int. Ed. 2017, 56, 13679–13683. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Whitehead, J.C. Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: Understanding the synergistic effect at low temperature. Appl. Catal. B Environ. 2012, 125, 439–448. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Guo, H.; Tu, X. Atmospheric Pressure and Room Temperature Synthesis of Methanol through Plasma-Catalytic Hydrogenation of CO2. ACS Catal. 2017, 8, 90–100. [Google Scholar] [CrossRef]
- Chawdhury, P.; Wang, Y.; Ray, D.; Mathieu, S.; Wang, N.; Harding, J.; Bin, F.; Tu, X.; Subrahmanyam, C. A promising plasma-catalytic approach towards single-step methane conversion to oxygenates at room temperature. Appl. Catal. B Environ. 2021, 284, 119735. [Google Scholar] [CrossRef]
- Puliyalil, H.; Jurkovic, D.L.; Dasireddy, V.D.B.C.; Likozar, B. A review of plasma-assisted catalytic conversion of gaseous carbon dioxide and methane into value-added platform chemicals and fuels. RSC Adv. 2018, 8, 27481–27508. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Winter, L.R.; Chen, J.G. Review of plasma-assisted catalysis for selective generation of oxygenates from CO2 and CH4. ACS Catal. 2020, 10, 2855–2871. [Google Scholar] [CrossRef]
- Matsumoto, H.; Tanabe, S.; Okitsu, K.; Hayashi, Y.; Suib, S.L. Selective Oxidation of Methane to Methanol and Formaldehyde with Nitrous Oxide in a Dielectric-Barrier Discharge-Plasma Reactor. J. Phys. Chem. A 2001, 105, 5304–5308. [Google Scholar] [CrossRef]
- Mahammadunnisa, S.; Krushnamurty, K.; Subrahmanyam, C. Catalytic nonthermal plasma assisted co-processing of methane and nitrous oxide for methanol production. Catal. Today 2015, 256, 102–107. [Google Scholar] [CrossRef]
- Mahammadunnisa, S.; Reddy, P.M.K.; Subrahmanyam, C. Nonthermal plasma assisted co-processing of CH4 and N2O for methanol production. RSC Adv. 2014, 4, 4034–4036. [Google Scholar] [CrossRef]
- Yao, S.L.; Takemoto, T.; Ouyang, F.; Nakayama, A.; Suzuki, E.; Mizuno, A.; Okumoto, M. Selective Oxidation of Methane Using a Non-Thermal Pulsed Plasma. Energy Fuels 2000, 14, 459–463. [Google Scholar] [CrossRef]
- Chawdhury, P.; Bhargavi, K.V.S.S.; Subrahmanyam, C. Enhanced synergy by plasma reduced Pd nanoparticles on in-plasma catalytic methane conversion to liquid oxygenates. Catal. Commun. 2020, 147, 106139. [Google Scholar] [CrossRef]
- Larkin, D.W.; Lobban, L.L.; Mallinson, R.G. The direct partial oxidation of methane to organic oxygenates using a dielectric barrier discharge reactor as a catalytic reactor analog. Catal. Today 2001, 71, 199–210. [Google Scholar] [CrossRef]
- Zhou, L.M.; Xue, B.; Kogelschatz, U.; Eliasson, B. Partial Oxidation of Methane to Methanol with Oxygen or Air in a Nonequilibrium Discharge Plasma. Plasma Chem. Plasma Process. 1998, 18, 375–393. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.-W.; Huang, L.; Lei, L.-C. Partial oxidation of methane with air for methanol production in a post-plasma catalytic system. Chem. Eng. Process. 2009, 48, 1333–1340. [Google Scholar] [CrossRef]
- Chawdhury, P.; Ray, D.; Subrahmanyam, C. Single step conversion of methane to methanol assisted by nonthermal plasma. Fuel Process. Technol. 2018, 179, 32–41. [Google Scholar] [CrossRef]
- Chawdhury, P.; Ray, D.; Vinodkumar, T.; Subrahmanyam, C. Catalytic DBD plasma approach for methane partial oxidation to methanol under ambient conditions. Catal. Today 2019, 337, 117–125. [Google Scholar] [CrossRef]
- Indarto, A. Methanol synthesis from methane and oxygen with [Ga Cr]/Cu–Zn–Al catalyst in a dielectric barrier discharge. Ionics 2014, 20, 445–449. [Google Scholar] [CrossRef]
- Indarto, A.; Lee, H.; Choi, J.W.; Song, H.K. Partial Oxidation of Methane with Yttria-stabilized Zirconia Catalyst in a Dielectric Barrier Discharge. Energy Sources Part A 2008, 30, 1628–1636. [Google Scholar] [CrossRef]
- Chawdhury, P.; Kumar, D.; Subrahmanyam, C. NTP reactor for a single stage methane conversion to methanol: Influence of catalyst addition and effect of promoters. Chem. Eng. J. 2019, 372, 638–647. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.; Huang, L.; Lei, L. Post-Plasma Catalysis for Methane Partial Oxidation to Methanol: Role of the Copper-Promoted Iron Oxide Catalyst. Chem. Eng. Technol. 2010, 33, 2073–2081. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.; Huang, L.; Lei, L. Application of in-plasma catalysis and post-plasma catalysis for methane partial oxidation to methanol over a Fe2O3-CuO/γ-Al2O3 catalyst. J. Nat. Gas Chem. 2010, 19, 628–637. [Google Scholar] [CrossRef]
- Indarto, A. Partial oxidation of methane to methanol with nitrogen dioxide in dielectric barrier discharge plasma: Experimental and molecular modeling. Plasma Sources Sci. Technol. 2016, 25, 025002. [Google Scholar] [CrossRef]
- Aghamir, F.M.; Matin, N.S.; Jalili, A.H.; Esfarayeni, M.H.; Khodagholi, M.A.; Ahmadi, R. Conversion of methane to methanol in an ac dielectric barrier discharge. Plasma Sources Sci. Technol. 2004, 13, 707–711. [Google Scholar] [CrossRef]
- Larkin, D.W.; Lobban, L.L.; Mallinson, R.G. Production of Organic Oxygenates in the Partial Oxidation of Methane in a Silent Electric Discharge Reactor. Ind. Eng. Chem. Res. 2001, 40, 1594–1601. [Google Scholar] [CrossRef]
- Indarto, A. A review of direct methane conversion to methanol by dielectric barrier discharge. IEEE Trans. Dielectr. Electr. Insul. 2008, 15, 1038–1043. [Google Scholar] [CrossRef]
- Nozaki, T.; Ağıral, A.; Yuzawa, S.; Han Gardeniers, J.G.E.; Okazaki, K. A single step methane conversion into synthetic fuels using microplasma reactor. Chem. Eng. J. 2011, 166, 288–293. [Google Scholar] [CrossRef]
- Nozaki, T.; Goujard, V.; Yuzawa, S.; Moriyama, S.; Agıral, A.; Okazaki, K. Selective conversion of methane to synthetic fuels using dielectric barrier discharge contacting liquid film. J. Phys. D Appl. Phys. 2011, 44, 274010. [Google Scholar] [CrossRef]
- Nozaki, T.; Hattori, A.; Okazaki, K. Partial oxidation of methane using a microscale non-equilibrium plasma reactor. Catal. Today 2004, 98, 607–616. [Google Scholar] [CrossRef]
- Indarto, A.; Jae Wook, C.; Hwaung, L.; Hyung Keun, S. Methanol Synthesis over Cu and Cu-Oxide-Containing ZnO/Al2O3 Using Dielectric Barrier Discharge. IEEE Trans. Plasma Sci. 2008, 36, 516–518. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Bogaerts, A. Propagation of a plasma streamer in catalyst pores. Plasma Sources Sci. Technol. 2018, 27, 035009. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Wang, W.; Bogaerts, A. Importance of surface charging during plasma streamer propagation in catalyst pores. Plasma Sources Sci. Technol. 2018, 27, 065009. [Google Scholar] [CrossRef] [Green Version]
- Bogaerts, A.; Zhang, Q.Z.; Zhang, Y.-R.; Van Laer, K.; Wang, W. Burning questions of plasma catalysis: Answers by modeling. Catal. Today 2019, 337, 3–14. [Google Scholar] [CrossRef]
- Van Laer, K.; Bogaerts, A. How bead size and dielectric constant affect the plasma behaviour in a packed bed plasma reactor: A modelling study. Plasma Sources Sci. Technol. 2017, 26, 085007. [Google Scholar] [CrossRef]
- Wang, Y.; Craven, M.; Yu, X.; Ding, J.; Bryant, P.; Huang, J.; Tu, X. Plasma-Enhanced Catalytic Synthesis of Ammonia over a Ni/Al2O3 Catalyst at Near-Room Temperature: Insights into the Importance of the Catalyst Surface on the Reaction Mechanism. ACS Catal. 2019, 9, 10780–10793. [Google Scholar]
- De Bie, C.; Verheyde, B.; Martens, T.; Dijk, J.V.; Paulussen, S.; Bogaerts, A. Fluid Modeling of the Conversion of Methane into Higher Hydrocarbons in an Atmospheric Pressure Dielectric Barrier Discharge. Plasma Process. Polym. 2011, 8, 1033–1058. [Google Scholar] [CrossRef]
- De-Bie, C.; Dijk, J.V.; Bogaerts, A. The Dominant Pathways for the Conversion of Methane into Oxygenates and Syngas in an Atmospheric Pressure Dielectric Barrier Discharge. J. Phys. Chem. C 2015, 119, 22331–22350. [Google Scholar] [CrossRef]
- Loenders, B.; Engelmann, Y.; Bogaerts, A. Plasma-catalytic partial oxidation of methane on Pt(111): A microkinetic study of the role of different plasma species. J. Phys. Chem. C 2021, 125, 2966–2983. [Google Scholar] [CrossRef]
- Takayuki Tsuchiya, S.I. Conversion of Methane to Methanol by a Low-Pressure Steam Plasma. J. Environ. Eng. Technol. 2013, 2, 35–39. [Google Scholar]
- Pearce, M.P.; Bussemaker, M.J.; Cooper, P.D.; Lapere, K.M.; Wild, D.A.; McKinley, A.J. Formation of methanol from methane and water in an electrical discharge. Phys. Chem. Chem. Phys. 2012, 14, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, Y.; Mehta, P.; Neyts, E.C.; Schneider, W.F.; Bogaerts, A. Predicted Influence of Plasma Activation on Non-Oxidative Coupling of Methane on Transition Metal Catalysts. ACS Sustain. Chem. Eng. 2020, 8, 6043–6054. [Google Scholar] [CrossRef]
- Nozaki, T.; Okazaki, K. Non-thermal plasma catalysis of methane: Principles, energy efficiency, and applications. Catal. Today 2013, 211, 29–38. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, X.; Jafarzadeh, A.; Wang, L.; Liu, P.; He, B.; Yan, J.; Zhang, R.; Zhang, H.; Liu, X.; et al. Plasma-Catalytic Ammonia Reforming of Methane over Cu-Based Catalysts for the Production of HCN and H2 at Reduced Temperature. ACS Catal. 2021, 11, 1765–1773. [Google Scholar] [CrossRef]
Figure 1.
Significant pathways for value-added utilization of CH4. Commercial (indirect) pathways through syngas are indicated in red. Direct routes to olefins, gasoline or aromatics, by various processes, are in blue, while the direct oxidation to CH3OH is in green.
Figure 1.
Significant pathways for value-added utilization of CH4. Commercial (indirect) pathways through syngas are indicated in red. Direct routes to olefins, gasoline or aromatics, by various processes, are in blue, while the direct oxidation to CH3OH is in green.

Figure 2.
(A) Thermodynamic equilibrium of CH4 conversion upon oxidation by O2 (left axis), and product selectivity (right axis), as a function of reaction temperature at atmospheric pressure (mole ratio: CH4/O2 = 2/1). (B) Product selectivity in logarithmic scale, illustrating the products with lower thermodynamic equilibrium selectivity, including CH3OH.
Figure 2.
(A) Thermodynamic equilibrium of CH4 conversion upon oxidation by O2 (left axis), and product selectivity (right axis), as a function of reaction temperature at atmospheric pressure (mole ratio: CH4/O2 = 2/1). (B) Product selectivity in logarithmic scale, illustrating the products with lower thermodynamic equilibrium selectivity, including CH3OH.

Figure 3.
The mechanism of the C–H bond cleavage of CH4 at low temperature can be classified into two categories. Reproduced from [9] with permission. Copyright 2019 Elsevier.
Figure 3.
The mechanism of the C–H bond cleavage of CH4 at low temperature can be classified into two categories. Reproduced from [9] with permission. Copyright 2019 Elsevier.

Figure 4.
DR-UV–Vis spectra of Fe/BEA: (a) Before activation, (b) after N2O activation at 250 °C, and (c) after reaction with CH4 at room temperature. Adapted with permission from ref [46]. Copyright 2016 Nature.
Figure 4.
DR-UV–Vis spectra of Fe/BEA: (a) Before activation, (b) after N2O activation at 250 °C, and (c) after reaction with CH4 at room temperature. Adapted with permission from ref [46]. Copyright 2016 Nature.

Figure 5.
DFT-optimized structure of α-Fe (iv) = O sites in the S = 2 ground state. Adapted with permission from [46]. Copyright 2016 Nature.
Figure 5.
DFT-optimized structure of α-Fe (iv) = O sites in the S = 2 ground state. Adapted with permission from [46]. Copyright 2016 Nature.

Figure 6.
Scheme of direct activation of CH4 to oxygenates using N2O on Fe-modified zeolites. Adapted with permission from [52]. Copyright 2017 Elsevier.
Figure 6.
Scheme of direct activation of CH4 to oxygenates using N2O on Fe-modified zeolites. Adapted with permission from [52]. Copyright 2017 Elsevier.

Figure 7.
Mechanistic scheme of quasi-catalytic and catalytic oxidation of CH4 by N2O. Reprinted with permission from [57]. Copyright 2014 Elsevier.
Figure 7.
Mechanistic scheme of quasi-catalytic and catalytic oxidation of CH4 by N2O. Reprinted with permission from [57]. Copyright 2014 Elsevier.

Figure 8.
(A) Direct CH4 oxidation results using H2O2 as oxidant in aqueous solution on 0.3 wt% Rh/ZrO2, 2 wt% Rh/ZrO2, and 5 wt% Rh/SiO2 catalysts. (B) Recyclability test results performed with the 0.3 wt% Rh/ZrO2 catalyst. Reaction condition—30 bar of 95%CH4/He, 70 °C, 1 h, 0.5 M H2O2 and catalyst 30 mg. (C) Direct CH4 conversion to C2H6 using O2 as oxidant in gas-phase on 0.3 wt% Rh/ZrO2, 2 wt% Rh/ZrO2 and 5 wt% Rh/SiO2 catalysts. Reaction condition: 9.85 sccm of 91%/0.3%/8.7% CH4/O2/N2, 260 °C atmospheric pressure, and catalyst 0.15 g. Reproduced with permission from [30]. Copyright 2017 American Chemical Society.
Figure 8.
(A) Direct CH4 oxidation results using H2O2 as oxidant in aqueous solution on 0.3 wt% Rh/ZrO2, 2 wt% Rh/ZrO2, and 5 wt% Rh/SiO2 catalysts. (B) Recyclability test results performed with the 0.3 wt% Rh/ZrO2 catalyst. Reaction condition—30 bar of 95%CH4/He, 70 °C, 1 h, 0.5 M H2O2 and catalyst 30 mg. (C) Direct CH4 conversion to C2H6 using O2 as oxidant in gas-phase on 0.3 wt% Rh/ZrO2, 2 wt% Rh/ZrO2 and 5 wt% Rh/SiO2 catalysts. Reaction condition: 9.85 sccm of 91%/0.3%/8.7% CH4/O2/N2, 260 °C atmospheric pressure, and catalyst 0.15 g. Reproduced with permission from [30]. Copyright 2017 American Chemical Society.

Figure 9.
Proposed reaction scheme for CH4 oxidation in the presence of H2O2 and molecular O2. Reproduced with permission from [23]. Copyright 2017 American Association for the advancement of science.
Figure 9.
Proposed reaction scheme for CH4 oxidation in the presence of H2O2 and molecular O2. Reproduced with permission from [23]. Copyright 2017 American Association for the advancement of science.

Figure 10.
Models of the AuPd@ZSM-5-R (left) and AuPd/ZSM-5 (right) catalysts AuPd@Zeolite-R, where the @ symbol denotes encapsulation of the nanoparticles within the zeolite and R indicates the organic substituent of the silane sheath; A conventional ZSM-5 zeolite–supported AuPd nanoparticle catalyst was prepared by impregnating with the same loading, denoted as AuPd/ZSM-5. Reproduced with permission from [29]. Copyright 2020 American Association for the advancement of science.
Figure 10.
Models of the AuPd@ZSM-5-R (left) and AuPd/ZSM-5 (right) catalysts AuPd@Zeolite-R, where the @ symbol denotes encapsulation of the nanoparticles within the zeolite and R indicates the organic substituent of the silane sheath; A conventional ZSM-5 zeolite–supported AuPd nanoparticle catalyst was prepared by impregnating with the same loading, denoted as AuPd/ZSM-5. Reproduced with permission from [29]. Copyright 2020 American Association for the advancement of science.

Figure 11.
(A) Data characterizing the oxidation of CH4 with H2 and O2 over various catalysts. (B) Data showing the amount of H2O2 in the (left) H2O2 synthesis and (right) CH4 oxidation process. The enrichment efficacy is the percentage of H2O2 in zeolite crystals to the total amount of H2O2 in the reactor. Reaction conditions for CH4 oxidation—10 mL of water, 30 min, 70 °C, 27 mg of catalyst, 3.0 MPa of feed gas with 3.3% H2/6.6% O2/1.6% CH4/61.7% Ar/26.8% He, 15 min and 1200 rpm. Each reaction was tested eight times to obtain the error bars. Reproduced with permission from [29]. Copyright 2020 American Association for the advancement of science.
Figure 11.
(A) Data characterizing the oxidation of CH4 with H2 and O2 over various catalysts. (B) Data showing the amount of H2O2 in the (left) H2O2 synthesis and (right) CH4 oxidation process. The enrichment efficacy is the percentage of H2O2 in zeolite crystals to the total amount of H2O2 in the reactor. Reaction conditions for CH4 oxidation—10 mL of water, 30 min, 70 °C, 27 mg of catalyst, 3.0 MPa of feed gas with 3.3% H2/6.6% O2/1.6% CH4/61.7% Ar/26.8% He, 15 min and 1200 rpm. Each reaction was tested eight times to obtain the error bars. Reproduced with permission from [29]. Copyright 2020 American Association for the advancement of science.

Figure 12.
(A) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of FeN4/GN with the model of FeN4/GN in the upper left. (B) Different catalysts for CH4 oxidation at 25 °C. (C) Scheme of operando TOF-MS online connected with high-pressure reactor. (D) Possible reaction path for CH4 oxidation over FeN4/GN catalyst. (E) Reaction pathway of CH4 conversion to CH3OH, CH3OOH, HOCH2OOH and HCOOH (the energy of reaction initiation was set as 0 eV) as well as reactants (black), intermediates (blue), products (green and red) and the activation energy of each step (unit, eV). (F) The relationship between CH4 activation rate (log[rate]) and formation energy of the O–MN4–O active site (Gf) presents a volcano curve. The Gf and activation rate of the calculated O–CrN4–O, O–MnN4–O, O–FeN4–O, and O–CoN4–O structures are shown on the volcano marked by black stars. Reproduced with permission from [10]. Copyright 2018 Cellpress.
Figure 12.
(A) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of FeN4/GN with the model of FeN4/GN in the upper left. (B) Different catalysts for CH4 oxidation at 25 °C. (C) Scheme of operando TOF-MS online connected with high-pressure reactor. (D) Possible reaction path for CH4 oxidation over FeN4/GN catalyst. (E) Reaction pathway of CH4 conversion to CH3OH, CH3OOH, HOCH2OOH and HCOOH (the energy of reaction initiation was set as 0 eV) as well as reactants (black), intermediates (blue), products (green and red) and the activation energy of each step (unit, eV). (F) The relationship between CH4 activation rate (log[rate]) and formation energy of the O–MN4–O active site (Gf) presents a volcano curve. The Gf and activation rate of the calculated O–CrN4–O, O–MnN4–O, O–FeN4–O, and O–CoN4–O structures are shown on the volcano marked by black stars. Reproduced with permission from [10]. Copyright 2018 Cellpress.

Figure 13.
Scheme of stoichiometric chemical looping of CH4 to CH3OH. Reproduced with permission from [79]. Copyright 2017 American Chemical Society.
Figure 13.
Scheme of stoichiometric chemical looping of CH4 to CH3OH. Reproduced with permission from [79]. Copyright 2017 American Chemical Society.

Figure 14.
Cu-oxo complexes proposed as the active sites for CH4 activation in Cu-containing zeolites. Reproduced with permission from [80]. Copyright 2016 Elsevier.
Figure 14.
Cu-oxo complexes proposed as the active sites for CH4 activation in Cu-containing zeolites. Reproduced with permission from [80]. Copyright 2016 Elsevier.

Figure 15.
(A) Normalized productivity (mol CH3OH/mol Cu) as a function of the Cu:Al ratio, comparing the Cu/MOR(7) and Cu/MOR(11) series. (B,C) CH3OH yield and selectivity as a function of Cu concentration (μmol Cu/gcat.) for Cu-MOR materials with Si:Al = 7 (B, Cu/MOR(7) series) and Si:Al = 11 (C, Cu/MOR(11) series). (D) Quantitative correlation between the normalized productivity evaluated at the HERFD XANES testing conditions and the fraction of LT redox-inert fw-CuII from linear combination fit analysis (O2-activation: full colored symbols; He activation+O2: empty colored symbols). All the experimentally determined values match the ideal trend line for stoichiometric conversion over a dicopper active site, reported as a full dark red line. Reprinted with permission from [33]. Copyright 2018 American Chemical Society.
Figure 15.
(A) Normalized productivity (mol CH3OH/mol Cu) as a function of the Cu:Al ratio, comparing the Cu/MOR(7) and Cu/MOR(11) series. (B,C) CH3OH yield and selectivity as a function of Cu concentration (μmol Cu/gcat.) for Cu-MOR materials with Si:Al = 7 (B, Cu/MOR(7) series) and Si:Al = 11 (C, Cu/MOR(11) series). (D) Quantitative correlation between the normalized productivity evaluated at the HERFD XANES testing conditions and the fraction of LT redox-inert fw-CuII from linear combination fit analysis (O2-activation: full colored symbols; He activation+O2: empty colored symbols). All the experimentally determined values match the ideal trend line for stoichiometric conversion over a dicopper active site, reported as a full dark red line. Reprinted with permission from [33]. Copyright 2018 American Chemical Society.

Figure 16.
Rationalization of the effect of composition (Cu:Al and Si:Al ratios) on the productivity for DOMTM over Cu/SSZ-13, reported as a full dark red line. Reprinted with permission from [34]. Copyright 2017 American Chemical Society.
Figure 16.
Rationalization of the effect of composition (Cu:Al and Si:Al ratios) on the productivity for DOMTM over Cu/SSZ-13, reported as a full dark red line. Reprinted with permission from [34]. Copyright 2017 American Chemical Society.

Figure 17.
Production of CH3OH and CO/CO2 as a function of water pressure on a CeO2/Cu2O/Cu(111) catalyst in which ∼40% of the Cu2O was covered by ceria. The samples were exposed to 1 Torr of CH4, 0.5 Torr of O2 and 0, 1, or 4 Torr of H2O at 450 K in a batch reactor. Reprinted with permission from [96]. Copyright 2016 American Chemical Society.
Figure 17.
Production of CH3OH and CO/CO2 as a function of water pressure on a CeO2/Cu2O/Cu(111) catalyst in which ∼40% of the Cu2O was covered by ceria. The samples were exposed to 1 Torr of CH4, 0.5 Torr of O2 and 0, 1, or 4 Torr of H2O at 450 K in a batch reactor. Reprinted with permission from [96]. Copyright 2016 American Chemical Society.

Figure 18.
Mechanism of partial oxidation of CH4 using H2O as oxidant. (Top) Schematic representation of the reaction conditions of the partial oxidation of CH4 by H2O, involving the reduction of the dicopper site of mordenite and providing two electrons to stoichiometrically oxidize CH4 into CH3OH. Subsequent reduction of H2O into H2 returns two electrons for the rejuvenation of the mono(m-oxo) dicopper active core. (Bottom) Reaction pathway predicted by DFT, illustrating the thermodynamic (ΔG, change in Gibbs free energy) and kinetic feasibility of the proposed mechanism. Reprinted with permission from [98]. Copyright 2017 American Association for the advancement of science.
Figure 18.
Mechanism of partial oxidation of CH4 using H2O as oxidant. (Top) Schematic representation of the reaction conditions of the partial oxidation of CH4 by H2O, involving the reduction of the dicopper site of mordenite and providing two electrons to stoichiometrically oxidize CH4 into CH3OH. Subsequent reduction of H2O into H2 returns two electrons for the rejuvenation of the mono(m-oxo) dicopper active core. (Bottom) Reaction pathway predicted by DFT, illustrating the thermodynamic (ΔG, change in Gibbs free energy) and kinetic feasibility of the proposed mechanism. Reprinted with permission from [98]. Copyright 2017 American Association for the advancement of science.

Figure 19.
Overview of the various effects of catalyst on the plasma and of the plasma on the catalyst, which may give rise to a synergistic plasma-catalytic operation. Reproduced with permission from [113]. Copyright 2019 MDPI.
Figure 19.
Overview of the various effects of catalyst on the plasma and of the plasma on the catalyst, which may give rise to a synergistic plasma-catalytic operation. Reproduced with permission from [113]. Copyright 2019 MDPI.

Figure 20.
Schematic diagram of the products of CH4/N2O reaction using non-thermal plasma. Reproduced with permission from [136]. Copyright 2015 Elsevier.
Figure 20.
Schematic diagram of the products of CH4/N2O reaction using non-thermal plasma. Reproduced with permission from [136]. Copyright 2015 Elsevier.

Figure 21.
Proposed reaction pathways in the plasma-only reaction of CH4/O2 towards CH3OH and other oxygenates or hydrocarbons. (Red: Main products, Orange: C2+ oxygenates). Reproduced with permission from [118]. Copyright 2021 Elsevier.
Figure 21.
Proposed reaction pathways in the plasma-only reaction of CH4/O2 towards CH3OH and other oxygenates or hydrocarbons. (Red: Main products, Orange: C2+ oxygenates). Reproduced with permission from [118]. Copyright 2021 Elsevier.

Figure 22.
Proposed mechanisms in plasma-catalytic DOMTM on a Fe/γ-Al2O3 surface [118]. Copyright 2021 Elsevier.
Figure 22.
Proposed mechanisms in plasma-catalytic DOMTM on a Fe/γ-Al2O3 surface [118]. Copyright 2021 Elsevier.

Figure 23.
Schematic overview of the dominant reaction pathways for the conversion of CH4 and O2 into CH3OH and other oxygenates in a 70/30 CH4/O2 gas mixture, as predicted by plasma chemical kinetics modeling. The thickness of the arrows is linearly proportional to the rate of the net reaction. Reproduced with permission from [149]. Copyright 2011 Wiley.
Figure 23.
Schematic overview of the dominant reaction pathways for the conversion of CH4 and O2 into CH3OH and other oxygenates in a 70/30 CH4/O2 gas mixture, as predicted by plasma chemical kinetics modeling. The thickness of the arrows is linearly proportional to the rate of the net reaction. Reproduced with permission from [149]. Copyright 2011 Wiley.

Figure 24.
Major reaction pathways for the conversion of CH4 to CH3OH in CH4/H2O plasma. The upper part shows a schematic of the electron energy variation along the gas flow. Reproduced with permission from [152]. Copyright 2013 Springer.
Figure 24.
Major reaction pathways for the conversion of CH4 to CH3OH in CH4/H2O plasma. The upper part shows a schematic of the electron energy variation along the gas flow. Reproduced with permission from [152]. Copyright 2013 Springer.

Figure 25.
Characteristic time-scales in plasma catalysis. Reproduced with permission from [112]. Copyright, 2015 Springer.
Figure 25.
Characteristic time-scales in plasma catalysis. Reproduced with permission from [112]. Copyright, 2015 Springer.


{kind=link}
{kind=link}
{kind=link}
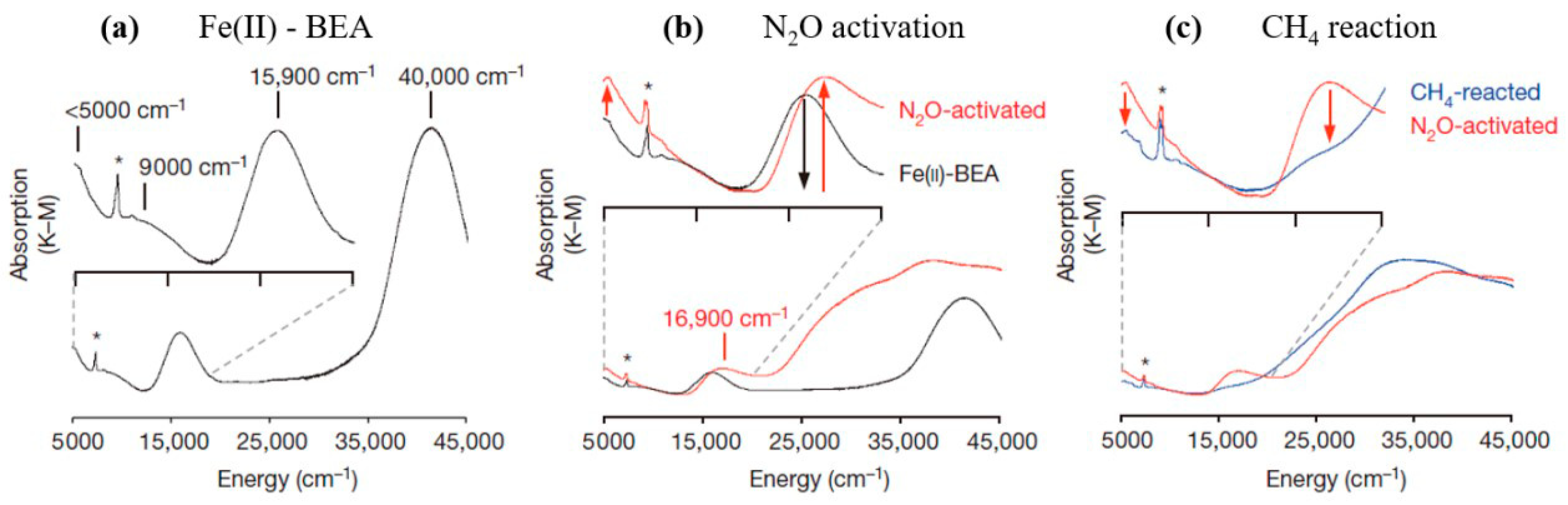{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
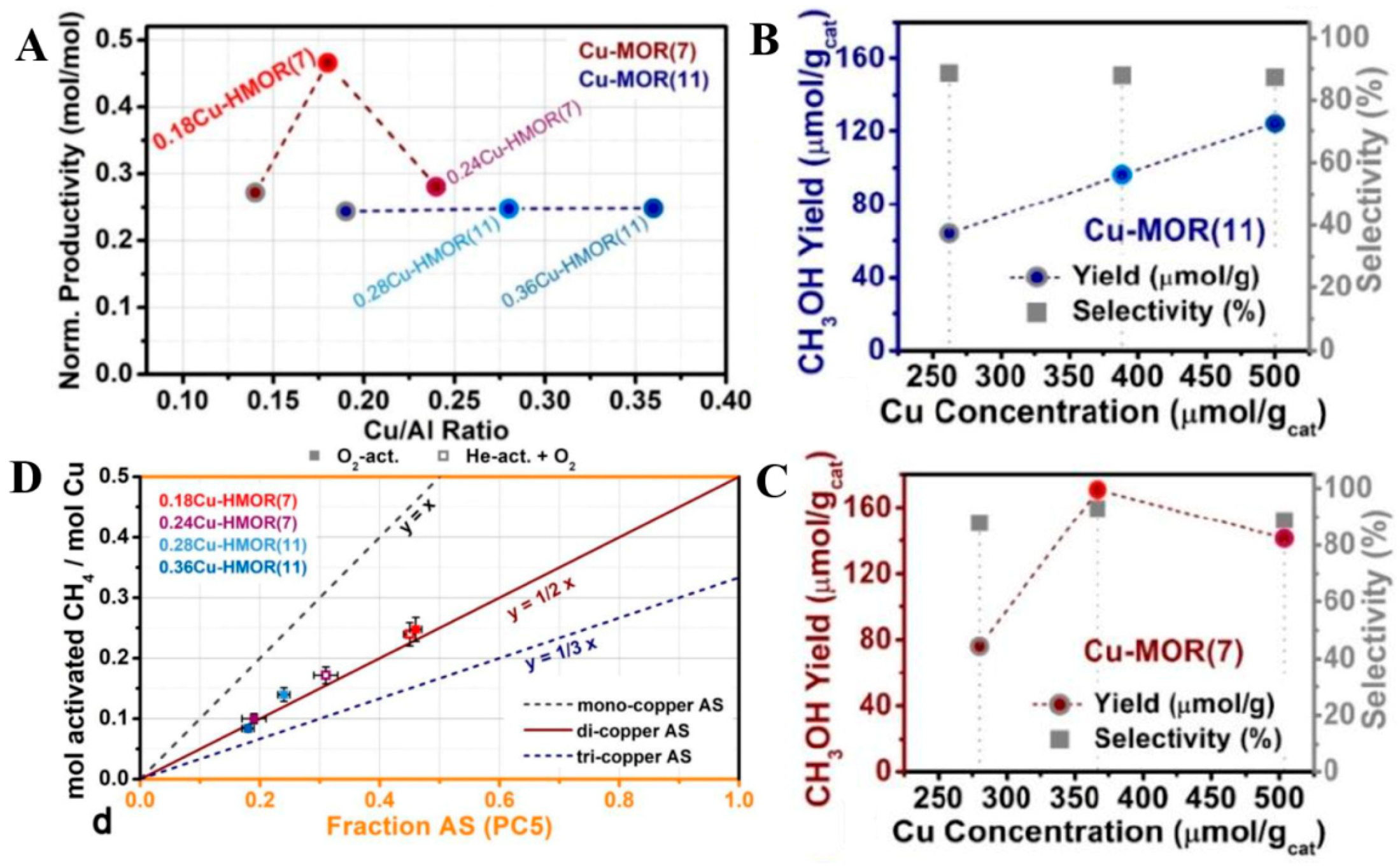{kind=link}
{kind=link}
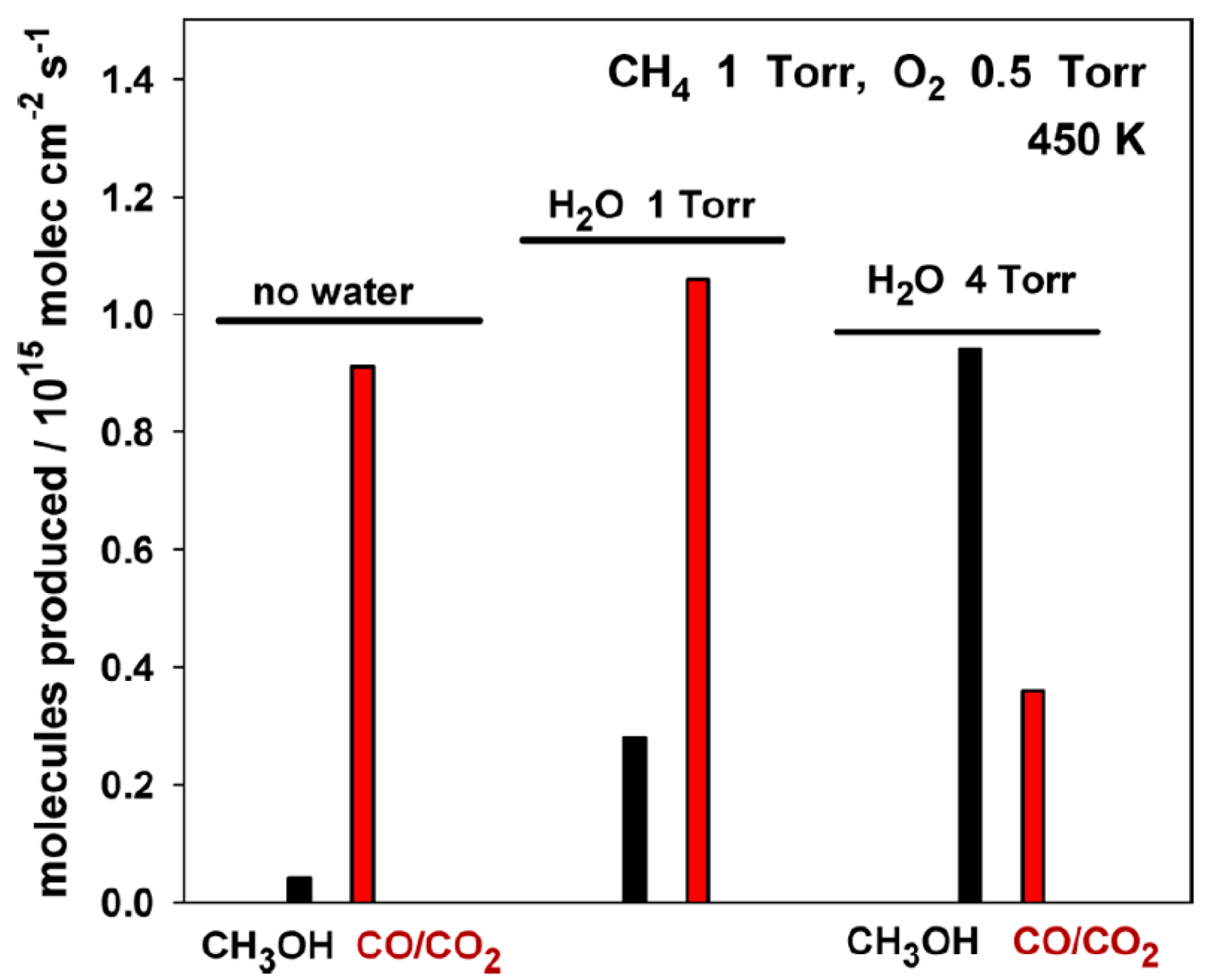{kind=link}
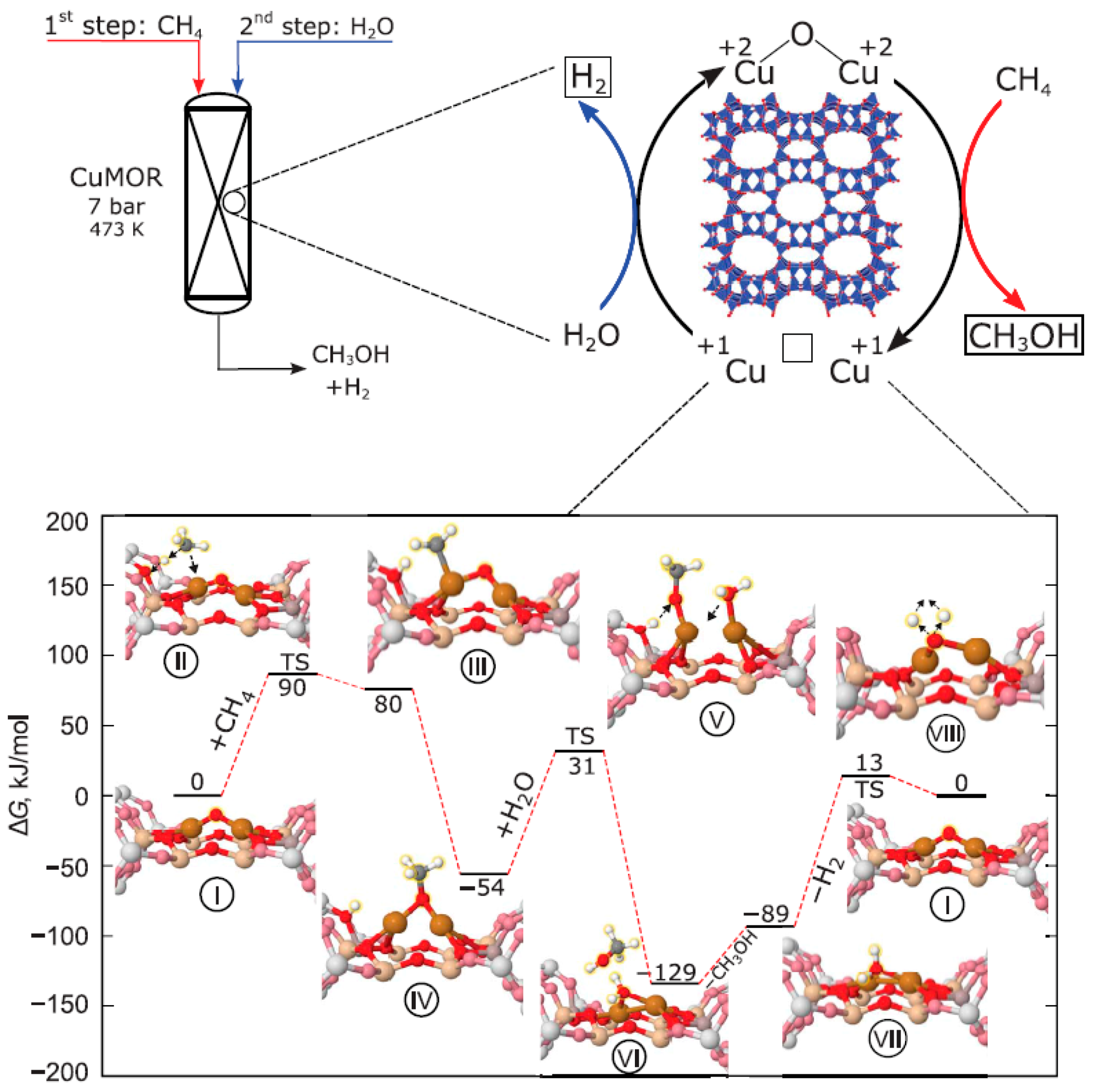{kind=link}
{kind=link}
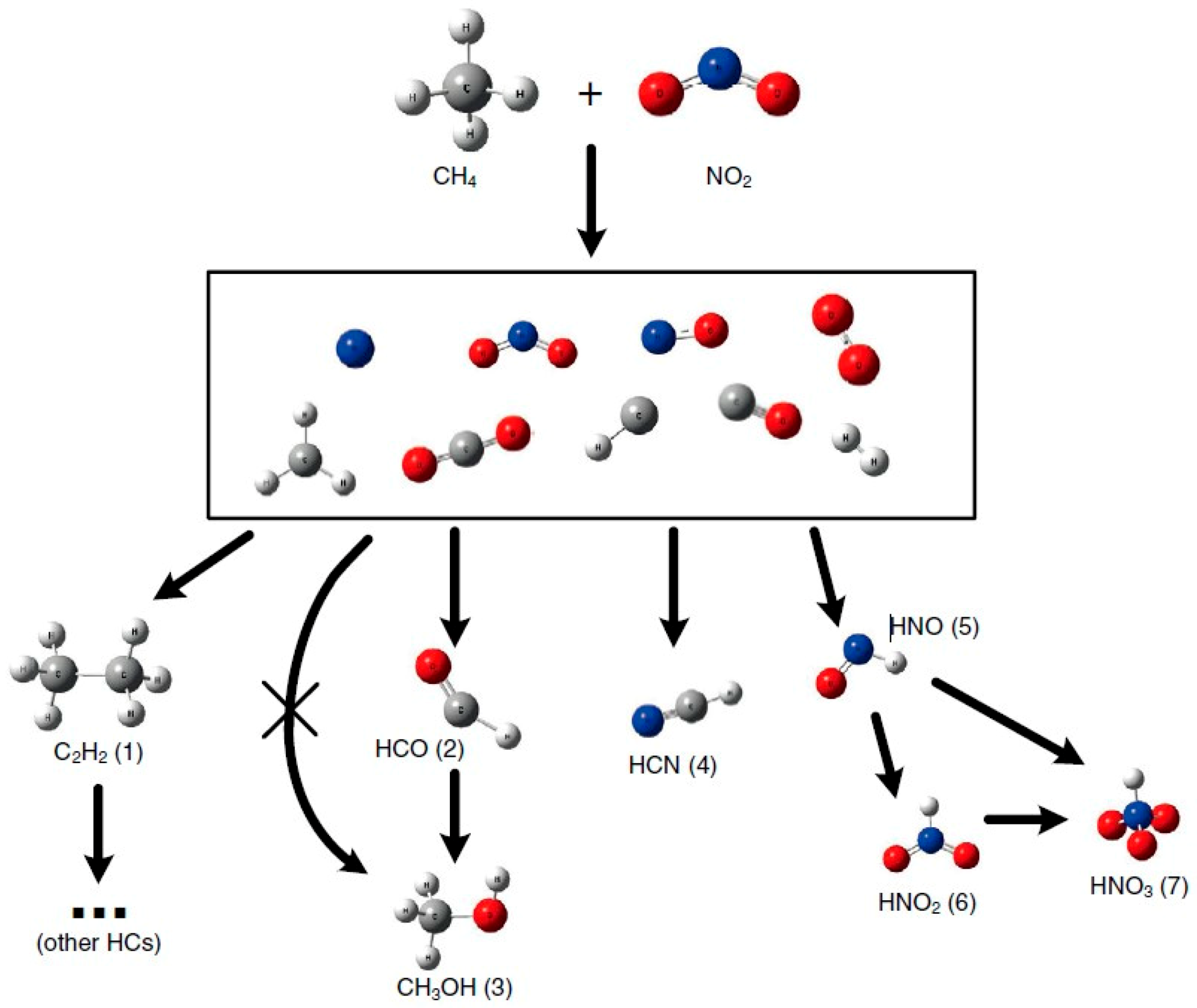{kind=link}
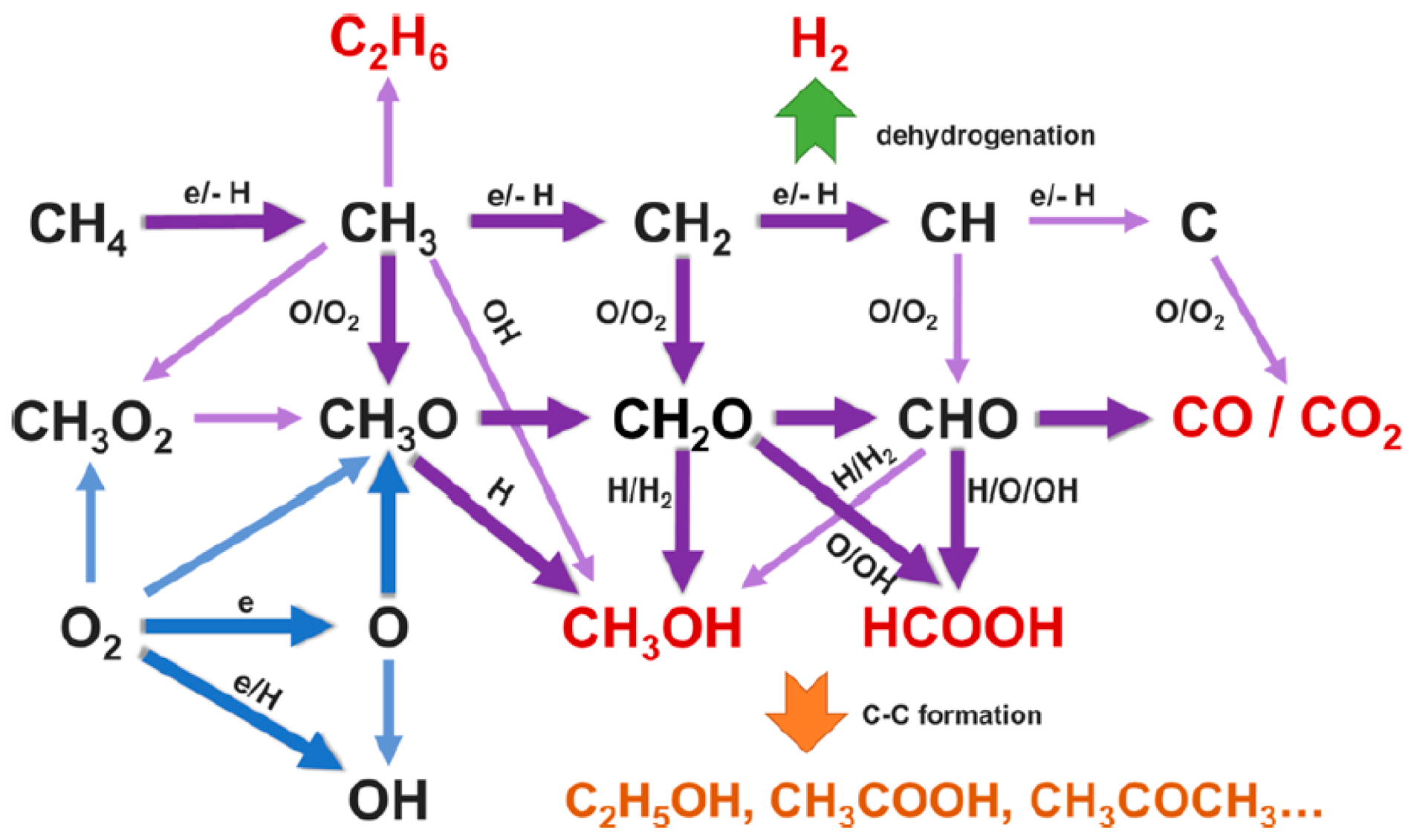{kind=link}
{kind=link}
{kind=link}
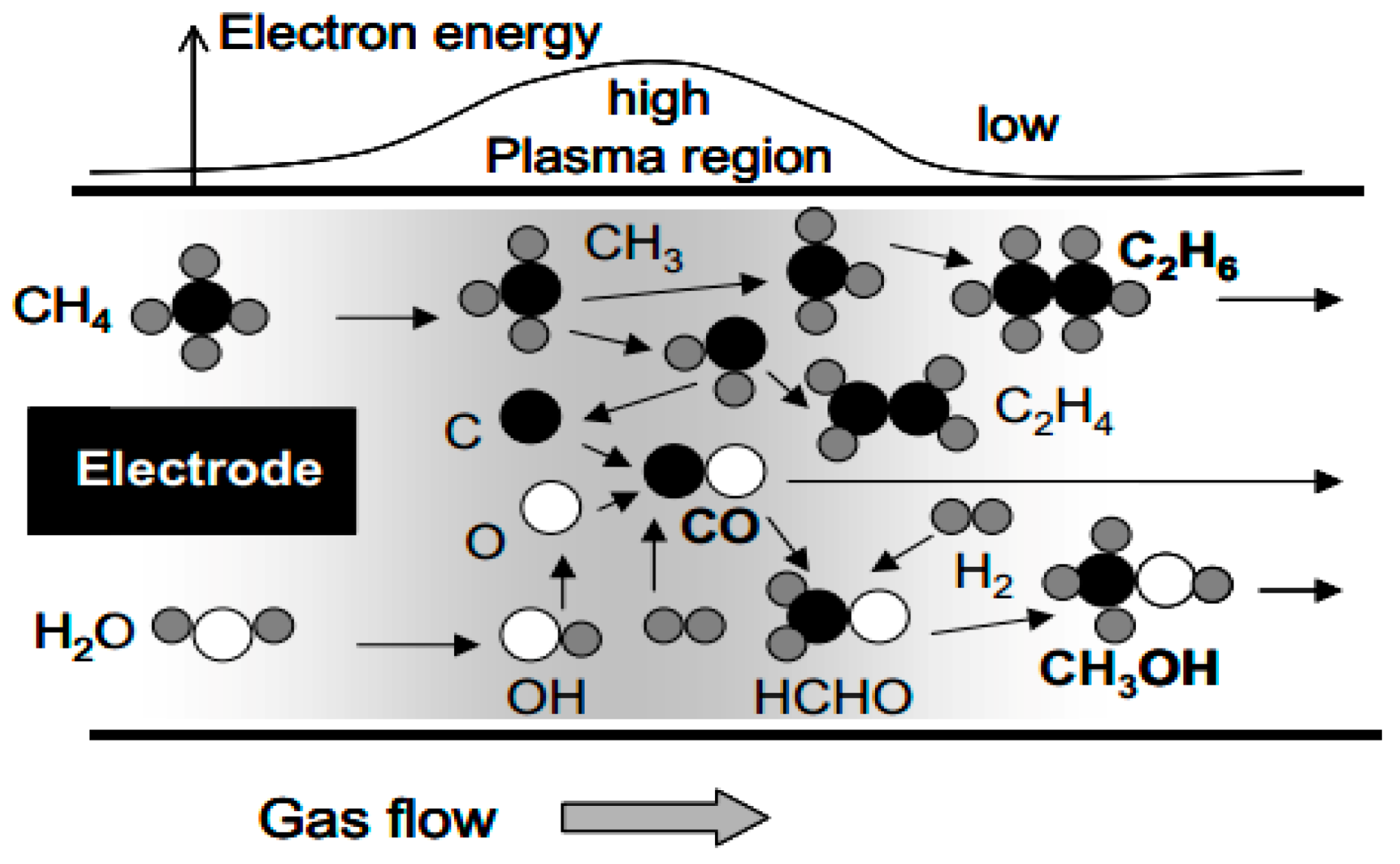{kind=link}
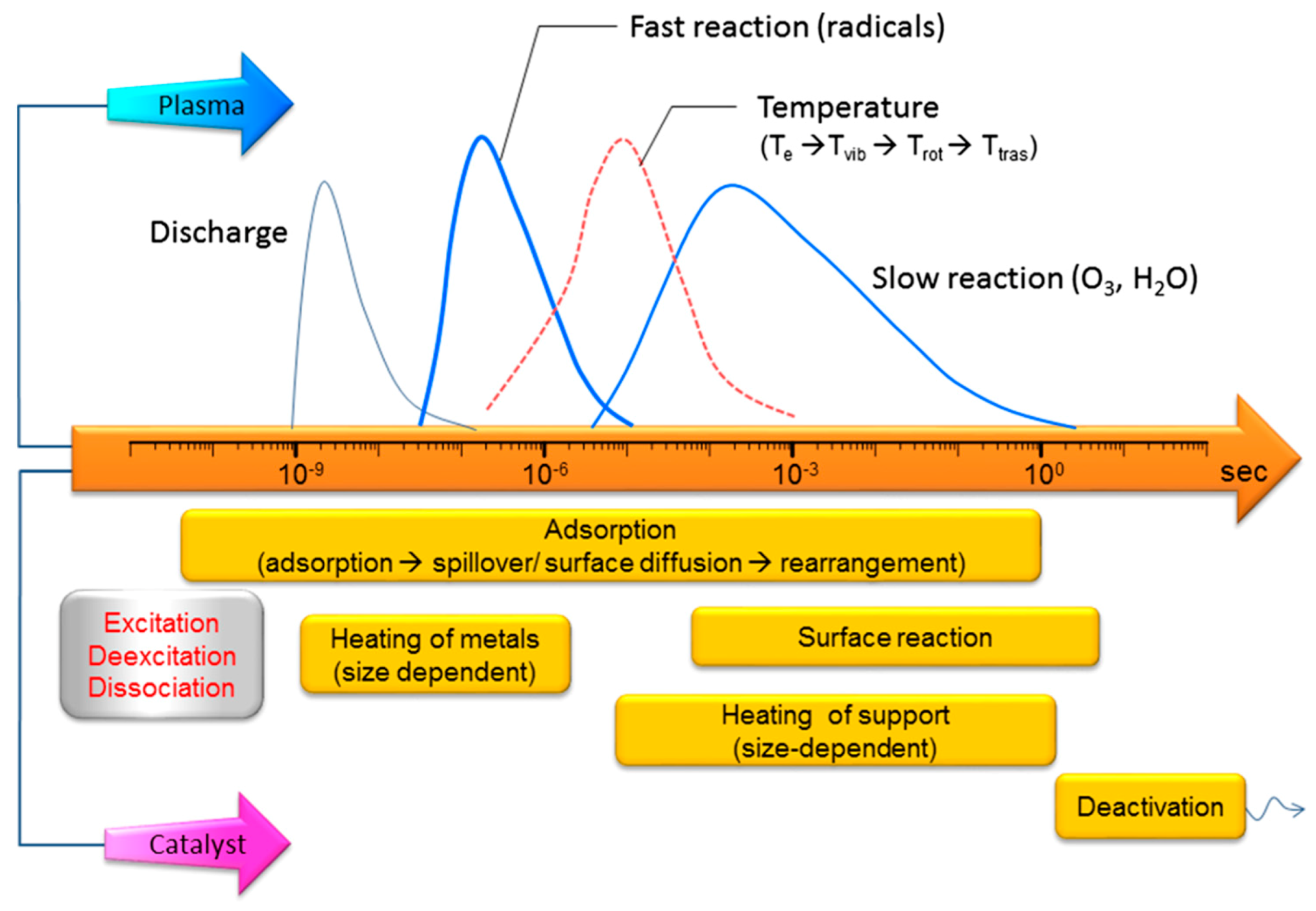{kind=link}
Table 1.
Selected experimental results of direct oxidation of methane to methanol (DOMTM) based on different oxidants.
Table 1.
Selected experimental results of direct oxidation of methane to methanol (DOMTM) based on different oxidants.
| Oxidant | Catalysts | PCH4 (bar) | Temp (°C) a | CH3OH Yield (μmol·gcat−1) | Productivity (mmol /molmetal) | CH3OH Sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| N2O | Fe/ZSM-5 | 6.6 × 10−4 | 160 | 160 | / | 76 | [25] |
| Fe/CHA | / | RT | 26.8 | 681 | / | [26] | |
| Cu/MOR | / | 150 | 97 | >300 | / | [27] | |
| Cu/SSZ-13 | 0.3 | 260 | 19 | / | 27 | [27] | |
| H2O2 | Au-Pd/TiO2 | 30.5 | 2 | 54.6 | 13.1 | 45.2 | [28] |
| Au-Pd colloids | 30 | ≤50 | 2.8–7.6 | / | 14–28.3 | [23] | |
| AuPd@ZSM-5 | 0.48 | 70 | / | 91.6 | 92 | [29] | |
| FeN4/Graphene | 18 | 25 | / | / | / | [10] | |
| Rh/ZrO2 | 28.5 | 70 | / | 1.25 | / | [30] | |
| Cu-Fe/ZSM-5 | 30.8 | 50 | 5.2 × 103 | / | 88 | [31] | |
| O2 | Cu/ZSM-5 | / | 175 | 8.2 | / | 98 | [32] |
| Cu/MOR | / | 200 | 170 | 0.47 | / | [33] | |
| Cu/SSZ-13 | 0.1 | 200 | 125 | 0.2 | >90 | [34] | |
| Cu/Omega | 30 | 200 | 200 | 265 | / | [35] | |
| Co/ZSM-5 | 1 | 150 | 0.3–0.4 | / | 40–100 | [36] | |
| Ni/ZSM-5 | 1 | 175 | 14.9 | / | / | [37] | |
| O2+H2O | CeOx/Cu2O/Cu(111) | 2.7 × 10−5 | 25 | / | / | 70 | [38] |
| Ni/CeO2 | 1.3 × 10−3 | 177 | / | / | <40 | [39] | |
| O2+CO | Rh/ZSM-5 | 20 | 150 | 1224 | / | 6.2 | [24] |
| O2+H2O2 | RuCu/NL b | 25 | 50 | ~1.5 × 103 | / | / | [40] |
| N2O+H2O | Mo/SiO2 | / | 300 | / | 16 | 60 | [41] |
a The temperature of the DOMTM process by chemical looping (more information on Section 2.3) is based on the step of the CH4 reaction for better comparison under the same criteria; b NL: Nanolichens.
Table 2.
Comparison of CH4 conversion and CH3OH selectivity in various plasma and plasma catalysis systems.
Table 2.
Comparison of CH4 conversion and CH3OH selectivity in various plasma and plasma catalysis systems.
| System | Oxidant | Packed Material | Power (W) | SEI (kJ/L) a | Temp. (K) | CH4 Conv. (%) | CH3OH Sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Plasma only | N2O | / | 0.27–7.7 | - | - | 5 | 43 | [121] |
| N2O | / | 6 | 6 | - | 15 | 31 | [122] | |
| N2O | / | 6 | 6 | - | <15 | 28 | [123] | |
| O2 | / | 5.7 | 3.4 | RT b | 1.9 | 47 | [124] | |
| O2 | / | 1.7 | 3.4 | RT | 7 | 20 | [125] | |
| O2 | / | 118 | 35.4 | 288 | 6 | 19 | [126] | |
| O2 | / | 200 | 4.0 | 353 | 3 | 30 | [127] | |
| Air | / | 400 | 11.9 | 353 | 15 | 13.3 | [127] | |
| Air | / | 140 | 28 | 523 | ~25 | ~8 | [128] | |
| Plasma catalysis | N2O | Cu-Ni/CeO2 | 6 | 6 | - | 23 | 36 | [122] |
| O2 | Fe/γ-Al2O3 | 1.8 | 3.6 | RT | 13 | 36 | [118] | |
| O2 | Glass Beads | 1.7 | 5.1 | - | 15.4 | 35.4 | [129] | |
| O2 | Cu/γ-Al2O3 | 1.9 | 3.6 | - | 9 | 37 | [130] | |
| O2 | Ga/CZA | 50 | / | - | 54.5 | 22.2 | [131] | |
| O2 | Ni/YSZ | 80 | 160 | RT | 35.3 | 23.5 | [132] | |
| Air | CuZn/Al2O3 | 1.7 | 3.4 | - | 11 | 28 | [133] | |
| Air | Fe2O3-CuO /CP | 140 | 28 | 473 | ~26 | 11 | [134] | |
| Air | Fe2O3/CuO/Al2O3 | 120 | 24 | 473 | 43 | 3.7 | [135] |
a SEI: Specific energy input; b RT: Room temperature.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, S.; Ahmed, R.; Yi, Y.; Bogaerts, A. Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2021, 11, 590. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050590
AMA Style
Li S, Ahmed R, Yi Y, Bogaerts A. Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis. Catalysts. 2021; 11(5):590. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050590
Chicago/Turabian StyleLi, Shangkun, Rizwan Ahmed, Yanhui Yi, and Annemie Bogaerts. 2021. "Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis" Catalysts 11, no. 5: 590. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050590
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.