
Water Gas Shift Reaction Activity on Fe (110): A DFT Study

1
Shaanxi Key Laboratory of Low Metamorphic Coal Clean Utilization, School of Chemistry and Chemical Engineering, Yulin University, Yulin 719000, China
2
State Key Laboratory of High-Efficiency Utilization of Coal and Green Chemical Engineering, School of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, China
3
School of Chemical Engineering, Northwestern University, Xi’an 710069, China
*
Authors to whom correspondence should be addressed.
†
These authors contribute equally.
Catalysts 2022, 12(1), 27; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010027
Submission received: 7 December 2021
/
Revised: 22 December 2021
/
Accepted: 23 December 2021
/
Published: 27 December 2021
(This article belongs to the Special Issue Catalysts in C1 Chemistry)
Abstract
:Metal Fe is one of the phases existing on iron-based catalysts for a high-temperature water gas shift reaction (WGSR), but research on the activity of metal Fe in WGSR is almost not reported. In this work, the density functional theory (DFT) method was used to systematically study the reaction activity and mechanisms of WGSR on metal Fe (110), including the dissociation of H2O, the transformation of CO and the formation of H2, as well as the analysis of surface electronic properties. The results show that (1) the direct dissociation of H2O occurs easily on Fe (110) and the energy barrier is less than 0.9 eV; (2) the generation of CO2 is difficult and its energy barrier is above 1.8 eV; (3) H migrates easily on the Fe surface and the formation of H2 also occurs with an energy barrier of 1.47 eV. Combined with the results of Fe3O4, it can be concluded that the active phase should be Fe3O4 with O vacancy defects, and the iron-rich region plays an important role in promoting the formation of H2 in WGSR.
1. Introduction
WGSR is an important reaction for H2 preparation in the chemical industry, and can convert CO and H2O into H2 and CO2 [1,2,3]. It is also used to reduce CO concentration and increase the H2/CO ratio under certain environmental conditions. WGSR can be divided into low-temperature (190–250 °C) WGSR and high-temperature (300–450 °C) WGSR under different catalysts according to the temperature needed for the reaction [1]. The high-temperature WGSR catalyst is mainly ferric oxide, which is widely used in industry due to its low price and excellent catalytic performance [4,5,6]. Generally, the active phase of the iron oxide catalyst is supposed to be Fe3O4 [7,8]. However, in practice, there is a very complex phase transformation of the catalyst, which inevitably contains some metal Fe or even iron carbide (χ-Fe5C2, θ-Fe3C and so on) [9]. In addition, due to the magnetic properties of Fe-based catalysts, existing experimental characterization techniques can hardly make a detailed analysis of their structure and surface information [10]. As a result, the current understanding of high-temperature WGSR on an Fe-based catalyst is inadequate, including the activity of each phase and the main reaction mechanisms [11].
Experimentally, it has been confirmed that Fe3O4 is the active phase of WGSR through the use of offline and in-situ XRD, offline and in-situ Mossbauer spectroscopy, in situ XAS, in-situ XPS and other characterization methods [9,12]. These in-situ spectroscopic methods can be used to detect the dynamic change in composition of bulk phase catalysts. In-situ Mossbauer spectra [13] were used to observe the redistribution of tetrahedral and octahedral iron complexes in the medium caused by changing the temperature and pressure in the high-temperature WGSR. The researchers found that the activity of the WGSR was related to the Fe2+/Fe3+ ratio of the iron oxides. Although many experimental methods can well observe and track changes in the bulk phase catalyst, the means that could better detect the surface information are very scarce. In addition, the experimental technologies are very limited for studying the mechanisms of WGSR, because the experimental characterization is difficult to capture and it is difficult to observe the reactants’ transformation at the extreme-speed time scale [14].
Computational simulation has shown strong advantages in studying the surface structure and properties of materials and the mechanisms of chemical reactions. In particular, first-principle calculation can often obtain knowledge and understanding that cannot be obtained in experiments in the aspects of solid material structure simulation, property prediction and in-depth understanding of reaction mechanisms [15,16,17,18]. Most of the existing theoretical research on iron-based catalysts for WGSR are focused on Fe3O4 (111) [19,20,21,22] and (001) [23,24,25]. Among them, the work of the Diebold group [26,27,28] and Freund group [29,30,31] is the most systematic and representative, and they have performed a significant amount of systematic study on the surface stability of Fe3O4 and some small molecule (H2O, CO, etc.) adsorption on the surfaces including experimental and theoretical calculation. In addition, Chen [32] used DFT to study the WGSR on Fe3O4 (111)-Fetet1, investigated the redox mechanism and the combination mechanism, finding that the combination mechanism is more favorable and the rate-control step of the whole reaction is the formation of H2 on the surface. Huang et al. [33] systematically calculated the WGSR mechanism including the redox mechanism, the joint mechanism and the coupling mechanism, and obtained the potential energy surface of these reactions on the Fe3O4 (111)-Feoct2. They concluded that the most favorable route is the redox path, and the speed-control step of the whole reaction is the desorption of adsorbed CO2* with a barrier of 1.04 eV. In addition, there are many studies on the influence of additives on the WGSR reactivity of Fe3O4. For example, Rim et al. [34] studied the Au additive by DFT and found that Au addition provided favorable adsorption sites for CO, which significantly promote CO and OH co-adsorption and improve the activity of WGSR by association mechanism. Thus, although there is a great deal of existing research on iron-based catalysts of WGSR, it has rarely examined the influence of the metal Fe surface on WGSR activity. However, Fe-based catalysts experience complex phase changes, including metal Fe formation, in the reaction process. Therefore, studying the WGSR performance on metal Fe is necessary.
In this paper, the most commonly exposed α-Fe (110) crystal plane was selected as the research object, and the redox mechanism and combined mechanism of WGSR on α-Fe (110) were studied by DFT; these were compared with the elementary reaction on the Fe3O4 surface in order to understand the activity and possible reaction mechanisms of WGSR. This study is helpful for us to know and understand the real active sites and main reaction mechanisms of WGSR on iron-based catalysts.
2. Results and Discussion
2.1. The Processes of H2O Dissociation and CO2 Formation
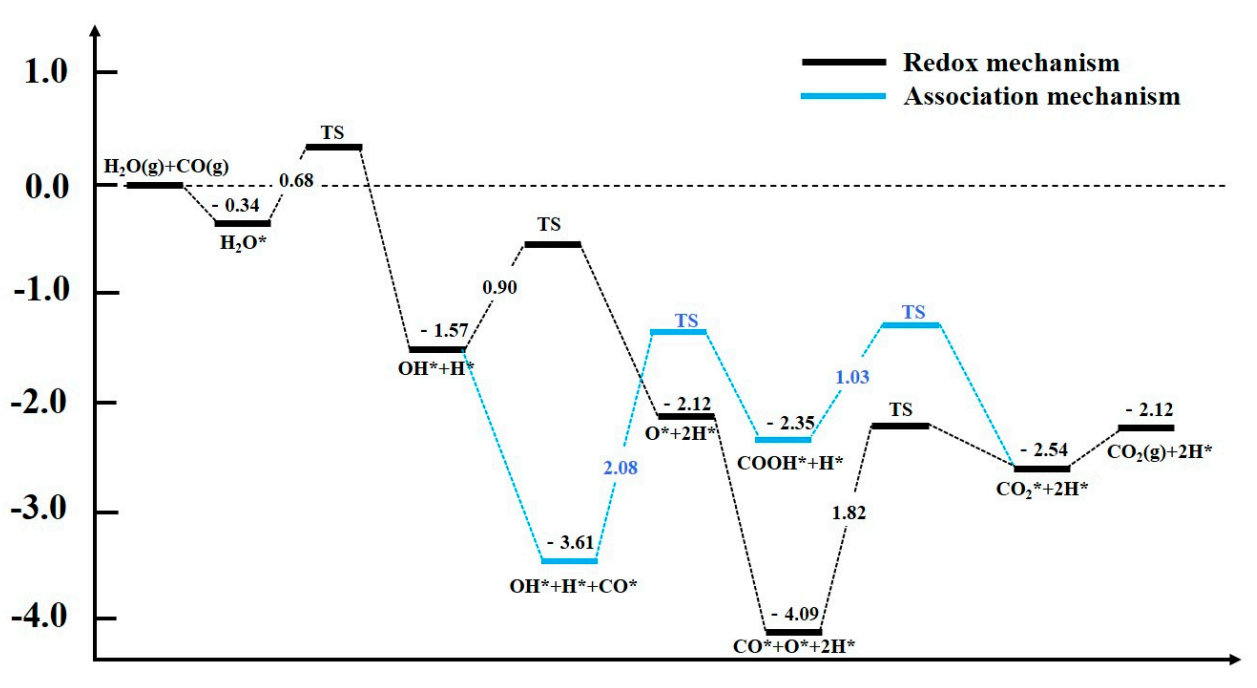
Firstly, the processes of H2O adsorption and dissociation on Fe (110) were studied. The most stable adsorption structures and energy change during the adsorption and dissociation processes are shown in Figure 1. The most stable adsorption configuration of H2O is one in which the O atom is adsorbed on top of the surface Fe atom, and the two H atoms are horizontally parallel to the surface with an adsorption energy of −0.35 eV. Among them, the H atom on one side keeps approaching the surface Fe near it, and then dissociates into *OH and *O, which need to overcome the energy barrier of 0.67 eV and unleash 1.22 eV heat. The process of the adsorbed *OH further dissociating into the O atom and H atom requires overcoming an energy barrier of 0.90 eV and releasing heat of 0.56 eV. The energy barriers in the two-step dissociation process of H2O on Fe (110) are 0.67 eV and 0.90 eV. Therefore, H2O is prone to complete dissociation on Fe (110) and the products are *O and two *H.
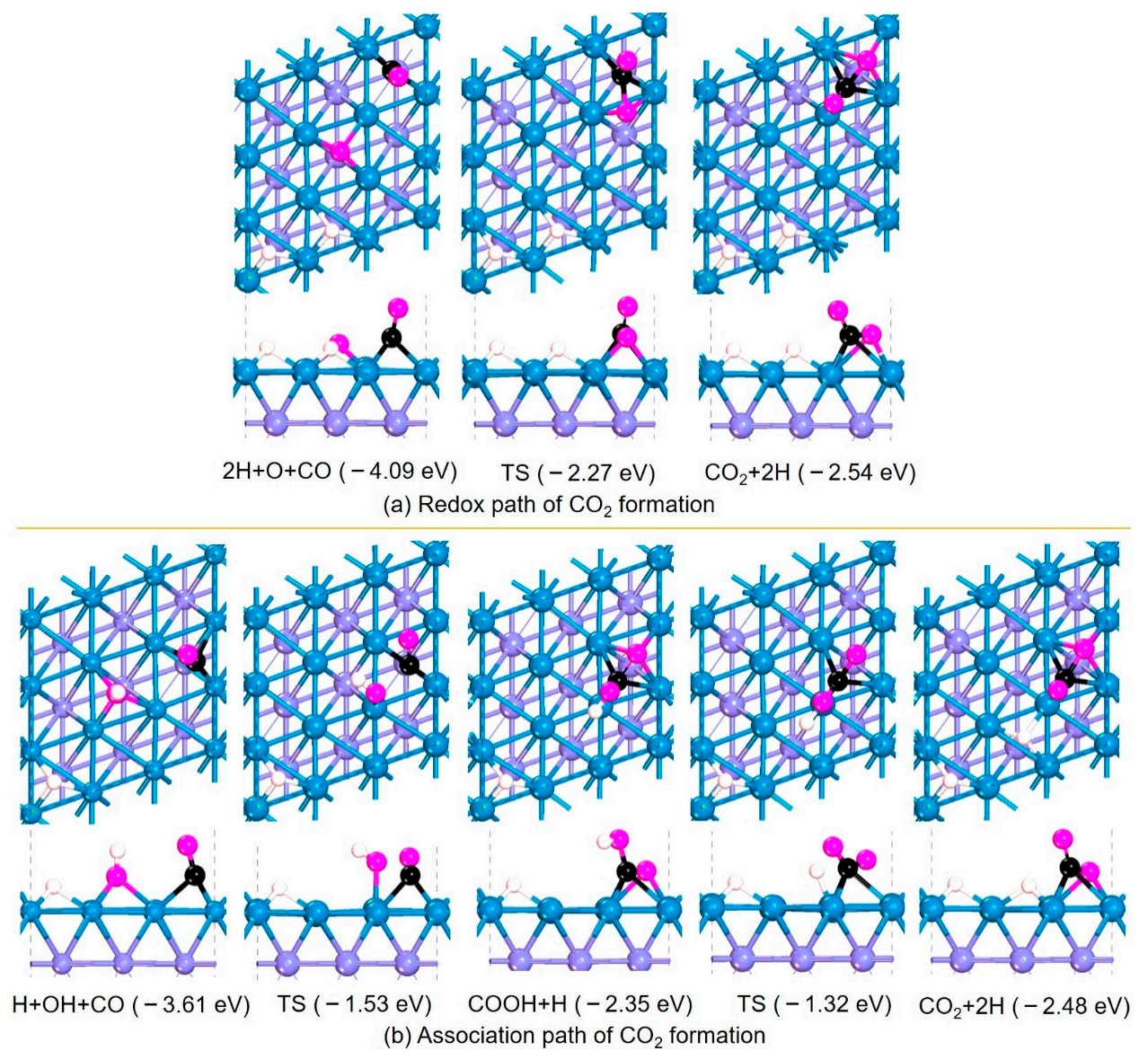
Similarly, the reaction process of the dissociated OH* and *O reacting with CO to produce CO2 was systematically studied and analyzed. Two reaction mechanisms, the redox mechanism and combined mechanism, were investigated and the results are shown in Figure 2. For the redox mechanism, the reaction process of *O and *CO co-adsorbed on Fe (110) reacting to form *CO2 only needs to overcome a barrier of 1.82 eV and absorb the heat of 1.55 eV, indicating that it is difficult for the redox mechanism to occur. For the association mechanism, *CO interacting with *OH adsorbed on adjacent Fe sites overcomes an energy barrier of 2.08 eV to generate *COOH and absorbs heat of 1.26 eV, indicating that it is also difficult for the formation of *COOH on Fe (110) to happen. However, if *COOH is generated, the energy barrier for dehydrogenation to generate *CO2 is 1.03 eV and the reaction releases heat of 0.13 eV. Therefore, the effective energy barrier for the combined mechanism is 2.29 eV and the total reaction process absorbs heat of 1.13 eV, which is almost impossible to occur.
The potential energy surface of both the H2O dissociation and the *OH/*O reacting with *CO to CO2 generation by different mechanisms on Fe (110) is shown in Figure 3. It can be found that the dissociation of H2O occurs relatively easily, and the dissociated *OH can be successively converted into adsorbed *O and 2 *H. However, whether CO reacts with adsorbed *O to generate CO2 through the redox mechanism, or CO2 through synthesis of *COOH and dehydrogenation by the association mechanism, its effective energy barrier is above 1.80 eV, and the whole CO2 generation process requires absorbing heat of more than 0.90 eV. Therefore, H2O can easily dissociate and oxidize the Fe (110) surface, but the activity of WGSR is poor, mainly reflected in the fact that it is difficult for *O or *OH adsorbed on the surface to react with adsorbed *CO.
Combined with our previous published work [35,36] that studied in detail the reaction processes of WGSR on two Fe3O4 surfaces by three mechanisms, the energy barrier and the corresponding reaction heat for each elementary reaction involved in the reaction processes of H2O dissociation and CO2 generation of WGSR on Fe (110) and two Fe3O4 surfaces are listed and compared as shown in Table 1. For the dissociation of H2O, the dissociation of H2O into OH and H is very easy on both Fe3O4 and Fe (110) with an energy barrier of less than 0.8 eV, and especially for Fe3O4 surfaces with an energy barrier of less than 0.5 eV. The dissociation of adsorbed *OH is more difficult than that of H2O. The direct dissociation energy barrier of *OH on the Fe3O4 surface is about 1.20 eV, and it is endothermic. Meanwhile, the energy barrier is 0.90 eV and the process is exothermic on the surface of Fe. For the process of dissociated *OH and *O reacting with CO to form CO2, the energy barriers of the redox mechanism and association mechanism on both Fe3O4 surfaces are low, and the two mechanisms could coexist in practice. On Fe (110), both dissociated *OH and *O require quite high energy barriers to react with adsorbed *CO, with effective energy barriers of 2.08 eV and 1.82 eV, respectively. Therefore, Fe3O4 has better activity in the process of CO transforming into CO2 in the WGSR, while Fe (110) occurs with more difficulty.
2.2. Migration of H and H2 Formation on Fe (110)
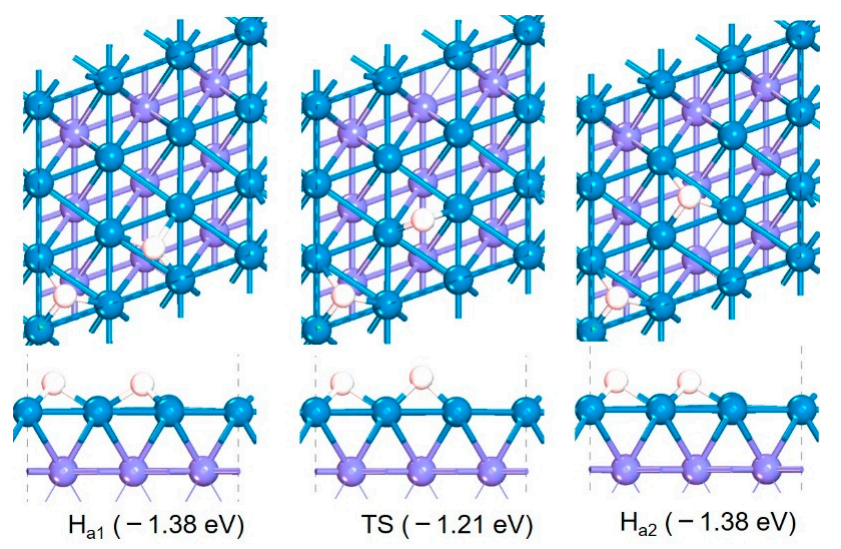
The migration processes and energy change of adsorbed H on Fe (110) are shown in Figure 4. When the coverage of *H is not quite high, H is absorbed on the three coordination holes of surface Fe sites. *H migrates from one Fe hole site (Ha1) to another Fe hole site adjacent to it (Feb) with an energy barrier of 0.17 eV and reaction heat of 0.0 eV. Therefore, the migration of H on Fe (110) is very easy, and the free migration of H on the Fe surface can easily occur.
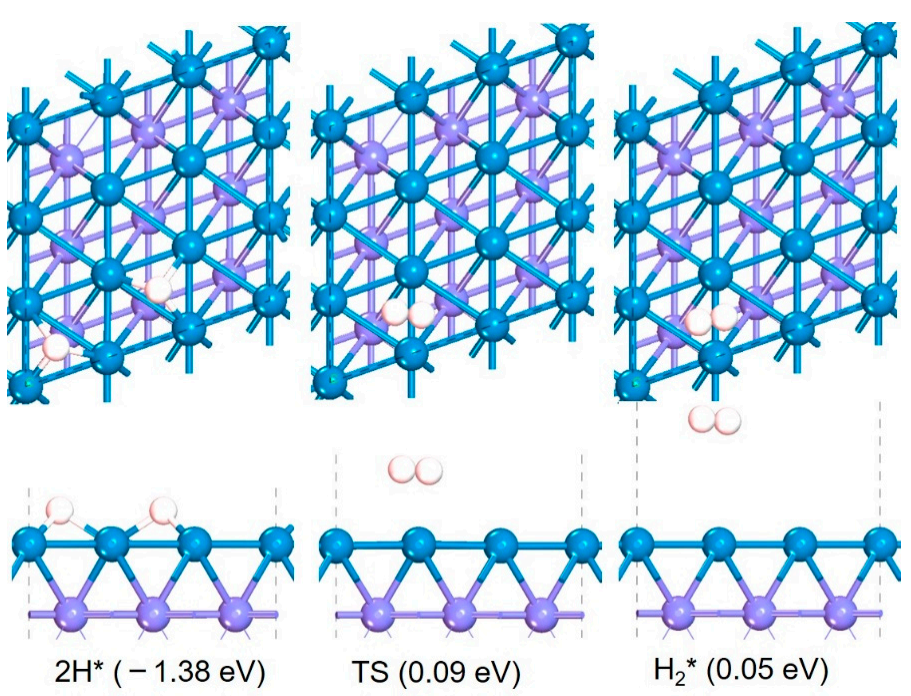
After calculating the migration capacity of H, the generation capacity of H2 on Fe (110) was also studied. Here, the most favorable H2 generation path on Fe (110), that is, the reaction process of H2 generation from two adjacent H atoms adsorbed on the surface, was calculated as shown in Figure 5. The H atom adsorbed from one Fe hole site migrates and attacks another *H adjacent to itself to generate adsorbed H2, which needs to overcome an energy barrier of 1.47 eV, and the reaction is endothermic with 1.43 eV. Therefore, when H and H2 interact with Fe (110), it is more favorable to H2 dissociation both thermally and dynamically. However, when the concentration of H on the surface is relatively high, it is also occurs for H2 generation.
Similarly, the energy barrier and the corresponding reaction heat of H2 formation on Fe (110) and Fe3O4 (111) and (001) are listed in Table 2 for comparative discussion. As can be seen from the table, the energy barriers of H2 formation on the two most frequently exposed Fe3O4 surfaces are relatively high, both close to 2.0 eV, and the reaction needs to absorb a small amount of heat. Thus, it is difficult to generate H2 on the intact Fe3O4 surface. On the surface of metal Fe, the energy barrier of H2 is lower than that of the regular Fe3O4 surface, and H2 especially is easily generated at high coverage. Therefore, for WGSR on an Fe-based catalyst, the orderly surface of the Fe3O4 phase is not the active surface for H2 formation, while the surface of Fe3O4 with some iron-rich defect regions may be the active region for H2 generation, and the adsorbed H migrates into these iron-rich or partially carbonized defect regions to form H2.
2.3. Electron Property Analysis of Adsorbed H on Fe (110)
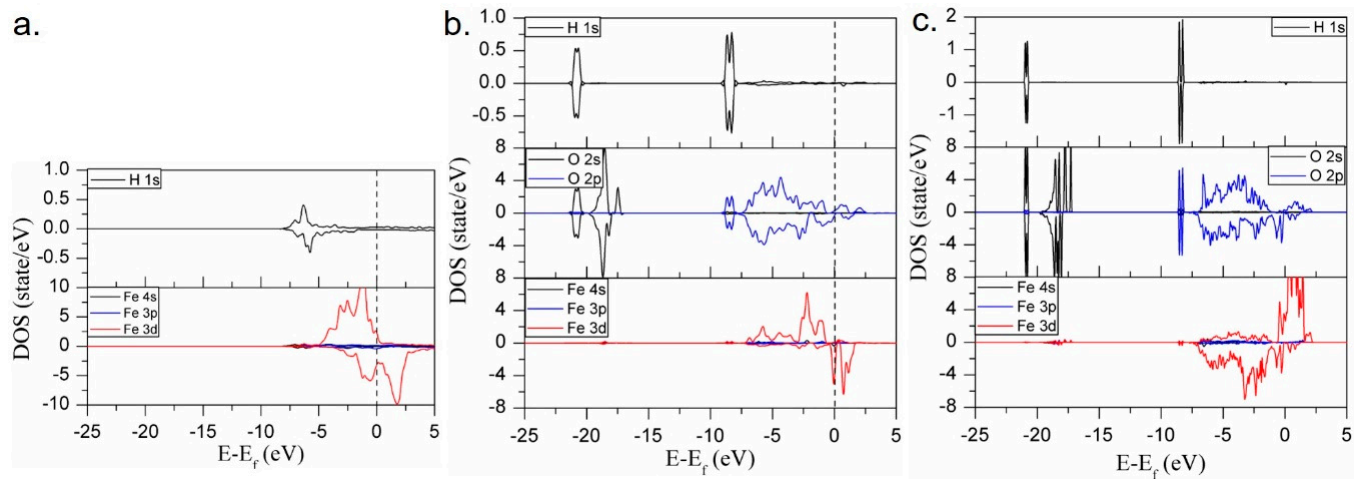
By comparing the H2 generation capacity of Fe (110) with that of Fe3O4 (111)-Fetet1 and Fe3O4 (001)-B, it can be found that the energy barrier for H2 generation of H adsorbed on O atoms on the Fe3O4 surface is very high, while the energy barrier of H adsorbed on Fe is low. Therefore, we want to know whether the strength of the interaction between the H atom and different surface sites could lead to the difference in H2 generation ability. Therefore, the local electron densities of state (LDOSs) of adsorbed *H interacting with the adsorption sites of Fe (110) were calculated, and the results are shown in Figure 6a. Similarly, the LDOS results of H interacting with Fe3O4 surfaces are compared and analyzed in Figure 6b,c. It can be seen from the figure that when H is adsorbed with the O site of Fe3O4 (001)-B or (111)-Fetet1, the energy level of the H 1s orbital is close to that of the O 2s and 2p orbitals; thus, the energy of their bonding orbital interaction is very low, and H bonding to O has a strong covalent bonding component. When H is adsorbed with Fe sites of Fe (110), the energy matching between the H 1s and Fe 3d orbitals is poor. The electron interaction between H and Fe belongs to weak metal bond interaction and there is almost no strong covalent bond interaction, so it can be seen that the interaction between H and Fe is weak. Thus, the electron interaction between H and Fe belongs to weak metal bond interaction and there is almost no strong covalent bond interaction.
In addition, the Bader charge transfer between the adsorbed H atom and Fe (110) was calculated and compared with the results of Fe3O4 (111)-Fetet1 and Fe3O4 (001)-B, and the results are shown in Table 3. When H was adsorbed on the O site of Fe3O4, the H atom lost most of its electrons (about 0.65–0.67 e) and transferred them to the O atom. When H adsorbs to the Fe atom of the metal Fe, H will gain about 0.36 electrons from the Fe atom. Therefore, from Bader charge, it can also be concluded that H has strong interaction with O sites of Fe3O4, while the interaction with the Fe surface is relatively weak, which results in the fact that when H interacts with different sites of the surface, the O-H bond is the most difficult to break, and the Fe-H bond is easy to break, therefore the energy barrier to be overcome should be reduced in turn.
3. Calculation Method and Model
3.1. Methods
Vienna Ab-initio Simulation Package (VASP) software [37,38] was used to complete the calculations. The interaction between electrons and ion nuclei is described by the projection additive wave (PAW) [39] pseudo-potential method, and the cut-off energy is 400 eV. In order to accurately describe the magnetic properties of the system, spin polarization is considered, which is crucial to accurately describe the electronic properties of the system [40]. The exchange-correlation energy adopts the generalized gradient approximation (GGA) of PBE [41]. The self-consistent convergence standards of force and the electron are −0.02 eV/A and 10−5 eV, respectively. The Gaussian method is used for electron broadening with 0.05 eV. The pseudopotentials of all primitives adopt the standard pseudopotentials of each element. The G-point centered Monkhorst–Pack [42] method is adopted for k-point sampling in the Brillouin area, and the specific k-point value scheme is 3 × 3 × 1.
In this work, the CI-NEB [43,44] method is employed to seek the transition states, and each transition state is verified by frequency analysis. The reaction energy barrier is defined as Equation (1) below:
Ea = ETS − EIS
The reaction heat is defined as Equation (2).
where EIS, ETS and EFS represent the total energy of the initial state, transition state and final state of an elementary reaction, respectively. Our previous study indicated that the zero-point energy correction has little effect on the adsorption energy and dissociation barrier of CO and H2O on the Fe surface [45]. Therefore, this work directly uses uncorrected energy values for analysis and discussion.
Er = EFS − EIS
For plotting the reaction potential surface, the energy of slab (Eslab) plus the total energy of reactant molecules (H2O (EH2O) and CO (ECO)) in the gas phase was taken as the zero point of the potential surface. The energy of the initial, final and transition states in all reaction paths were the energy of the relative reference state.
3.2. Calculation Model
Fe (110) is the most stable surface of metal Fe and also the most easily exposed surface, so it was selected as the research object. The slab models are shown in Figure 7. In order to eliminate the reactants or product repulsion due to the close distribution between molecules, P (3 × 3) supercell expansion for Fe (110) was used. The slab keeps the top three atomic layers relaxed and the remaining two atomic layers fixed to represent the bulk phase in all the calculations.
4. Conclusions
The whole reaction paths of WGSR on Fe (110) were calculated in detail, and the difficulty degree of the redox mechanism and association mechanism were calculated and compared. WGSR includes three processes: (1) dissociation of H2O; (2) CO conversion to CO2; (3) formation of H2. The first H2O dissociation step can be carried out easily on both Fe3O4 and metal Fe (110), and the *OH dissociation on Fe (110) is easier than that of Fe3O4 surfaces. For the process of CO converting to CO2, the energy barrier of that on Fe (110) is more than 1.8 eV, so it is difficult for this to occur on Fe (110). The migration barrier of adsorbed H on Fe is very low, and the barrier of H2 generated by adjacent *H is about 1.4 eV. When the *H coverage increases, the barrier will decrease.
Combined with the previous calculation results of the Fe3O4 surface, it can be concluded that the real active phase of WGSR is Fe3O4, but the regular surfaces of Fe3O4 are not good active surfaces, and the really good active surface is the defect surface with the iron-rich area. In WGSR, the dissociation of H2O and the conversion of CO takes place normally in the surface region with both O and Fe atoms coexisting, and where the energy barrier for the dissociation of H2O and the conversion of CO is very low. However, it is difficult to form H2 in the oxygen-rich region, and the energy barrier for H2 formation of adsorbed H is low only in the Fe-enriched region. Through the analysis of the electronic properties of the interaction between H and the adsorption sites, it was found that the O-H interaction is the strongest, and the Fe-H bond is weak, which results in the O-H bond being quite difficult to break, and the Fe-H bond being easy to break. Therefore, in order to improve the H2 generation activity, we can increase surface defects to weaken the interaction between adsorbed *H and directly adsorbed sites and thus reduce the energy barrier of H2 generation.
Author Contributions
X.L. and Z.M. contributed equally to this work. X.L. conducted the computation and data arrangement. Z.M.: analysis of data and writing—original draft preparation. X.G.: writing—reviewing and editing. M.B.: writing—reviewing and editing. Y.M. (Yajun Ma): writing—reviewing and editing and funding acquisition. Y.M. (Yu Meng): supervision of this work and writing—reviewing and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (No.22162028), Foundation of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (No.2022-K48), Young Talent fund of the Association for Science and Technology in Yulin, China (No. 20200211), Natural Science Foundation of Shaanxi Science and Technology Department (No.2021JQ-828), Scientific Research Program funded by Yulin National High Tech Industrial Development Zone Government (No.CXY-2020-07), the Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (Grant. YLU-DNL Fund 2021017) and Graduate Innovation Fund project of Yulin University (No.2021YLYCX07).
Acknowledgments
We also acknowledge the computational resources for the project that were supplied by Shaanxi Province Science and Technology Resources Open Sharing Platform Project (No.2019PT-18).
Conflicts of Interest
There are no conflicts to declare.
References
- Newsome, D.S. The water-gas shift reaction. Catal. Rev. 1980, 21, 275–318. [Google Scholar] [CrossRef]
- Pal, D.; Chand, R.; Upadhyay, S.; Mishra, P. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Gokhale, A.A.; Dumesic, A.J.A.; Mavrikakis, M. On the mechanism of low-temperature water gas shift reaction on copper. J. Am. Chem. Soc. 2008, 130, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.K.; Kim, S.J.; Dong, J.; Smirniotis, P.G.; Jasinski, J.B. Long-term WGS stability of Fe/Ce and Fe/Ce/Cr catalysts at high and low steam to CO ratios—XPS and Mössbauer spectroscopic study. Appl. Catal. A Gen. 2012, 415, 101–110. [Google Scholar] [CrossRef]
- Puig-Molina, A.; Cano, F.M.; Janssens, T.V.W. The Cu promoter in an Iron−Chromium−Oxide Based Water−Gas shift catalyst under industrial conditions studied by in-Situ XAFS. J. Phys. Chem. C 2010, 114, 15410–15416. [Google Scholar] [CrossRef]
- Kundu, M.L.; Sengupta, A.C.; Maiti, G.C.; Sen, B.; Ghosh, S.K.; Kuznetsov, V.I.; Kustova, G.N.; Yurchenko, E.N. Characterization of chromia-promoted γ-iron oxide catalysts and their CO conversion efficiency. J. Catal. 1988, 112, 375–383. [Google Scholar] [CrossRef]
- Rhodes, C.; Williams, B.P.; King, F.; Hutchings, G.J. Promotion of Fe3O4/Cr2O3 high temperature water gas shift catalyst. Catal. Commun. 2002, 3, 381–384. [Google Scholar] [CrossRef]
- Lee, D.-W.; Lee, M.S.; Lee, J.Y.; Kim, S.; Eom, H.-J.; Moon, D.J.; Lee, K.-Y. The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions. Catal. Today 2013, 210, 2–9. [Google Scholar] [CrossRef]
- Patlolla, A.; Carino, E.V.; Ehrlich, S.N.; Stavitski, E.; Frenkel, A.I. Application of Operando XAS, XRD, and Raman Spectroscopy for Phase Speciation in Water Gas Shift Reaction Catalysts. ACS Catal. 2012, 2, 2216–2223. [Google Scholar] [CrossRef]
- Liu, X.-W.; Zhao, S.; Meng, Y.; Peng, Q.; Dearden, A.K.; Huo, C.-F.; Yang, Y.; Li, Y.-W.; Wen, X.-D. Mössbauer spectroscopy of iron carbides: From prediction to experimental confirmation. Sci. Rep. 2016, 6, 26184. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Wachs, I.E. Iron-Based catalysts for the high-temperature water–gas shift (HT-WGS) reaction: A review. ACS Catal. 2016, 6, 722–732. [Google Scholar] [CrossRef]
- Reddy, G.K.; Boolchand, P.; Smirniotis, P.G. Unexpected behavior of copper in modified ferrites during high temperature WGS reaction aspects of Fe3+↔ Fe2+ Redox chemistry from Mössbauer and XPS studies. J. Phys. Chem. C 2012, 116, 11019–11031. [Google Scholar] [CrossRef]
- Cherkezova-Zheleva, Z.; Mitov, I. In situ Mössbauer investigation of iron oxide catalyst in water gas shift reaction–Impact of oxyreduction potential and temperature. J. Phys. Conf. Ser. 2010, 217, 012044. [Google Scholar] [CrossRef]
- Day, M.; Tachibana, S.; Bell, J.; Lijewski, M.; Beckner, V.; Cheng, R.K. A combined computational and experimental characterization of lean premixed turbulent low swirl laboratory flames: I. Methane flames. Combust. Flame 2012, 159, 275–290. [Google Scholar] [CrossRef]
- Meng, Y.; Liu, X.-W.; Bai, M.; Guo, W.-P.; Cao, D.-B.; Yang, Y.; Li, Y.-W.; Wen, X.-D. Prediction on morphologies and phase equilibrium diagram of iron oxides nanoparticles. Appl. Surf. Sci. 2019, 480, 478–486. [Google Scholar] [CrossRef]
- Bruix, A.; Margraf, J.T.; Andersen, M.; Reuter, K. First-principles-based multiscale modelling of heterogeneous catalysis. Nat. Catal. 2019, 2, 659–670. [Google Scholar] [CrossRef]
- Van der Ven, A.; Thomas, J.; Puchala, B.; Natarajan, A. First-Principles statistical mechanics of multicomponent crystals. Annu. Rev. Mater. Res. 2018, 48, 27–55. [Google Scholar] [CrossRef]
- Oba, F.; Kumagai, Y. Design and exploration of semiconductors from first principles: A review of recent advances. Appl. Phys. Express 2018, 11, 060101. [Google Scholar] [CrossRef]
- Yang, T.; Wen, X.D.; Cao, D.B.; Li, Y.W.; Wang, J.G.; Huo, C.F. Structures and energetics of H2O adsorption on the Fe3O4 (111) surface. J. Fuel Chem. Technol. 2009, 37, 506–512. [Google Scholar] [CrossRef]
- Grillo, M.E.; Finnis, M.W.; Ranke, W. Surface structure and water adsorption on Fe3O4(111): Spin-density functional theory and on-site Coulomb interactions. Phys. Rev. B 2008, 77, 075407. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, G.S.; Novotný, Z.; Jacobson, P.; Schmid, M.; Diebold, U. Room Temperature Water Splitting at the Surface of Magnetite. J. Am. Chem. Soc. 2011, 133, 12650–12655. [Google Scholar] [CrossRef]
- Huang, D.-M.; Cao, D.-B.; Li, Y.-W.; Jiao, H. Density Function Theory Study of CO Adsorption on Fe3O4(111) Surface. J. Phys. Chem. B 2006, 110, 13920–13925. [Google Scholar] [CrossRef]
- Mulakaluri, N.; Pentcheva, R.; Scheffler, M. Coverage-Dependent Adsorption Mode of Water on Fe3O4(001): Insights from First Principles Calculations. J. Phys. Chem. C 2010, 114, 11148–11156. [Google Scholar] [CrossRef]
- Mulakaluri, N.; Pentcheva, R.; Wieland, M.; Moritz, W.; Scheffler, M. Partial Dissociation of Water on Fe3O4(001): Adsorbate Induced Charge and Orbital Order. Phys. Rev. Lett. 2009, 103, 176102. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Liu, X.-Y.; Bai, M.-M.; Chen, J.; Ma, Y.-J.; Wen, X.-D. Adsorption or deoxidation of H2 interacted with Fe3O4 surface under different H coverage: A DFT study. Appl. Surf. Sci. 2020, 502, 144097. [Google Scholar] [CrossRef]
- Stanka, B.; Hebenstreit, W.; Diebold, U.; Chambers, S. Surface reconstruction of Fe3O4(001). Surf. Sci. 2000, 448, 49–63. [Google Scholar] [CrossRef]
- Bliem, R.; McDermott, E.; Ferstl, P.; Setvin, M.; Gamba, O.; Pavelec, J.; Schneider, M.A.; Schmid, M.; Diebold, U.; Blaha, P.; et al. Subsurface cation vacancy stabilization of the magnetite (001) surface. Science 2014, 346, 1215–1218. [Google Scholar] [CrossRef] [Green Version]
- Arndt, B.; Bliem, R.; Gamba, O.; van der Hoeven, J.E.; Noei, H.; Diebold, U.; Parkinson, G.; Stierle, A. Atomic structure and stability of magnetite Fe3O4(001): An X-ray view. Surf. Sci. 2016, 653, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Lemire, C.; Meyer, R.; Henrich, V.; Shaikhutdinov, S.; Freund, H.-J. The surface structure of Fe3O4(111) films as studied by CO adsorption. Surf. Sci. 2004, 572, 103–114. [Google Scholar] [CrossRef]
- Ritter, M.; Weiss, W. Fe3O4(111) surface structure determined by LEED crystallography. Surf. Sci. 1999, 432, 81–94. [Google Scholar] [CrossRef]
- Pentcheva, R.; Wendler, F.; Meyerheim, H.L.; Moritz, W.; Jedrecy, N.; Scheffler, M. Jahn-Teller stabilization of a “polar” metal oxide surface: Fe3O4 (001). Phys. Rev. Lett. 2005, 94, 126101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ni, G.; Han, B.; Zhou, C.G.; Wu, J.P. Mechanism of Water Gas Shift Reaction on Fe3O4 (111) Surface. Ac. Chim. Sin. 2011, 69, 393–398. [Google Scholar]
- Huang, L.; Han, B.; Zhang, Q.; Fan, M.; Cheng, H. Mechanistic study on water gas shift reaction on the Fe3O4 (111) reconstructed surface. J. Phys. Chem. C 2015, 119, 28934–28945. [Google Scholar] [CrossRef]
- Rim, K.T.; Eom, D.; Chan, S.-W.; Flytzani-Stephanopoulos, M.; Flynn, G.W.; Wen, X.-D.; Batista, E.R. Scanning tunneling microscopy and theoretical study of water adsorption on Fe3O4: Implications for catalysis. J. Am. Chem. Soc. 2012, 134, 18979–18985. [Google Scholar] [CrossRef]
- Meng, Y.; Liu, X.Y.; Chen, J.; Ma, Y.J.; Zhao, S. First-principle study on the reaction mechanism of water-gas shift on the Fe3O4 (001)-B surface. J. Fuel Chem. Technol. 2020, 48, 601–609. [Google Scholar]
- Liu, X.; Ma, Z.; Meng, Y.; Ma, Y.-J.; Wen, X.-D. First-principles study on the mechanism of water-gas shift reaction on the Fe3O4 (111)-Fetet1. Mol. Catal. 2021, 516, 111998. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for Ab Initio Total-Energy calculations using a Plane-wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. First-principles study of the adsorption of atomic H on Ni (111), (100) and (110). Surf. Sci. 2000, 459, 287–302. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Errata: Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef] [Green Version]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; p. 385. [Google Scholar]
- Henkelmann, G.; Uberuaga, B.P.; Jnsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Tian, X.-X.; Li, Y.-W.; Wang, J.; Beller, M.; Jiao, H. Coverage-Dependent CO adsorption and dissociation mechanisms on iron surfaces from dft computations. ACS Catal. 2014, 4, 1991–2005. [Google Scholar] [CrossRef]
Figure 1.
Structures and energies of H2O direct dissociation to O* + 2H*.

Figure 2.
Structures and energies of CO2 formation process on Fe (110).

Figure 3.
The comparison of CO2 formation potential energy surface on Fe (110).

Figure 4.
Structures and energies of adsorbed *H transferring process on Fe (110).

Figure 5.
The structures and corresponding energy changes of H2 formation on Fe (110).

Figure 6.
Density of state of H interacting with different adsorption sites: (a) Fe sites of Fe (110); (b) O sites of Fe3O4 (111)-Fetet1 and (c) O sites of Fe3O4 (001)-B.
Figure 6.
Density of state of H interacting with different adsorption sites: (a) Fe sites of Fe (110); (b) O sites of Fe3O4 (111)-Fetet1 and (c) O sites of Fe3O4 (001)-B.

Figure 7.
The computed slab model of Fe (110): (a) top view; (b) side view.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Reaction energy and barrier comparison of H2O dissociation and CO2 formation on Fe (110) and Fe3O4 (111)-Fetet1 and (001)-B.
Table 1.
Reaction energy and barrier comparison of H2O dissociation and CO2 formation on Fe (110) and Fe3O4 (111)-Fetet1 and (001)-B.
| Fe3O4 | Fe-110 | ||||||
|---|---|---|---|---|---|---|---|
| 001 | 111 | ||||||
| Ea | ΔE | Ea | ΔE | Ea | ΔE | ||
| H2O(g) + * → H2O* | - | −0.70 | - | −0.86 | - | −0.34 | |
| H2O* + * → HO* + H* | 0.32 | 0.07 | 0.14 | −0.20 | 0.68 | −1.23 | |
| Red. | HO* + * → H* + O* | 0.95 | 0.42 | 1.21 | 0.96 | 0.90 | −0.55 |
| CO(g) + * → CO* | - | −0.53 | - | 0.74 | - | −1.94 | |
| CO* + O* → CO2** | 0.22 | −2.32 | 0.11 | −1.77 | 1.82 | 1.55 | |
| CO2** → CO2 (g) + 2* | - | 0.06 | - | 0.22 | - | 0.38 | |
| Ass. | HO* + CO* → COOH*+* | 0.10 | −0.47 | 0.46 | −0.47 | 2.08 | 1.33 |
| COOH* + *→ CO2* + H* | 0.06 | −0.86 | 0.14 | −0.61 | 1.03 | −0.13 | |
| CO2** → CO2 (g) + 2* | - | 0.06 | - | 0.22 | - | 0.37 | |
| Reg. | CO + Os* ↔ CO2* | 0.60 | 0.27 | 0.85 | −0.80 | ||
| CO2*→ CO2 (g) + * | - | 0.10 | - | 0.04 | |||
| H2O + Ov* ↔ H2O* | - | −0.66 | - | −0.45 | |||
| H2O + Ov* ↔ OH* + H* | 0.10 | −1.38 | 0.79 | −0.64 | |||
Table 2.
The energy barrier and reaction heat (eV) of H2 formation on Fe (110) and Fe3O4 (111)-Fetet1 and (001)-B.
Table 2.
The energy barrier and reaction heat (eV) of H2 formation on Fe (110) and Fe3O4 (111)-Fetet1 and (001)-B.
| Path | Fe3O4 | Fe-110 | ||
|---|---|---|---|---|
| (001) | (111) | |||
| 2H ↔ H2* | ΔE (eV) | 0.04 | 0.11 | 1.43 |
| Ea (eV) | 1.98 | 1.97 | 1.47 | |
Table 3.
Bader charge transfer for H interacting on Fe (110) and Fe3O4 surfaces.
| Fe3O4 (111) | Fe3O4 (001) | Fe (110) | ||||
|---|---|---|---|---|---|---|
| Δq (e) | H-1 | H-2 | H-1 | H-2 | H-1 | H-2 |
| −0.69 | −0.67 | −0.65 | −0.64 | 0.36 | 0.36 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, X.; Ma, Z.; Gao, X.; Bai, M.; Ma, Y.; Meng, Y. Water Gas Shift Reaction Activity on Fe (110): A DFT Study. Catalysts 2022, 12, 27. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010027
AMA Style
Liu X, Ma Z, Gao X, Bai M, Ma Y, Meng Y. Water Gas Shift Reaction Activity on Fe (110): A DFT Study. Catalysts. 2022; 12(1):27. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010027
Chicago/Turabian StyleLiu, Xiaoyan, Zeyu Ma, Xinhua Gao, Miaomiao Bai, Yajun Ma, and Yu Meng. 2022. "Water Gas Shift Reaction Activity on Fe (110): A DFT Study" Catalysts 12, no. 1: 27. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010027
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.