
Closed-Form Formulation of the Thermodynamically Consistent Electrochemical Model Considering Electrochemical Co-Oxidation of CO and H2 for Simulating Solid Oxide Fuel Cells


Abstract
:1. Introduction
2. SOFC Electrochemical Model
2.1. Derivation of Basic Governing Equations
2.2. Simplified 1D Transport of Gaseous Species in the GDL
2.3. Closed-Form Determination of Relative Reactant’s Utilisation Ratios
3. Material and Methods
3.1. Determination of Calibration Parameters
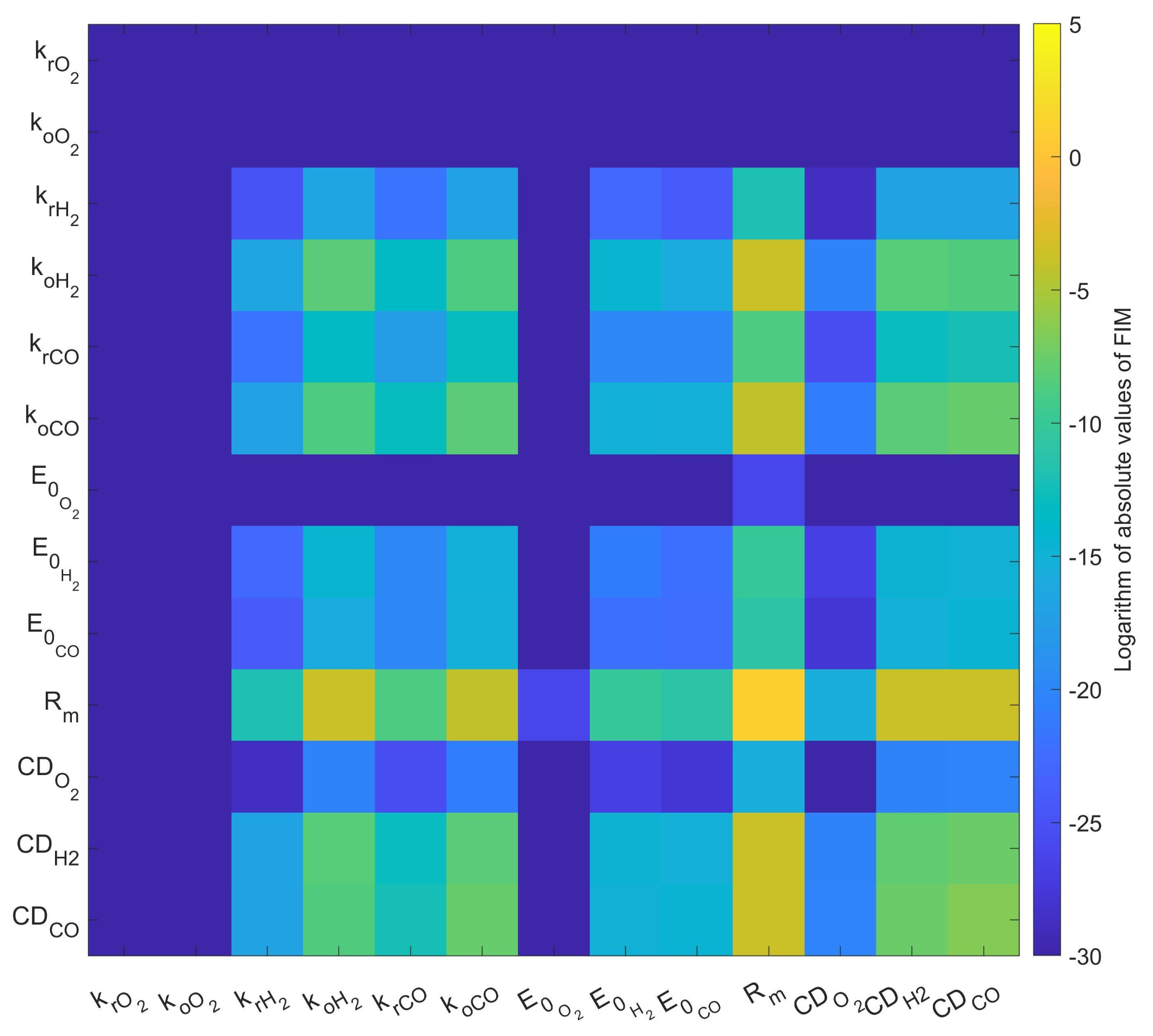
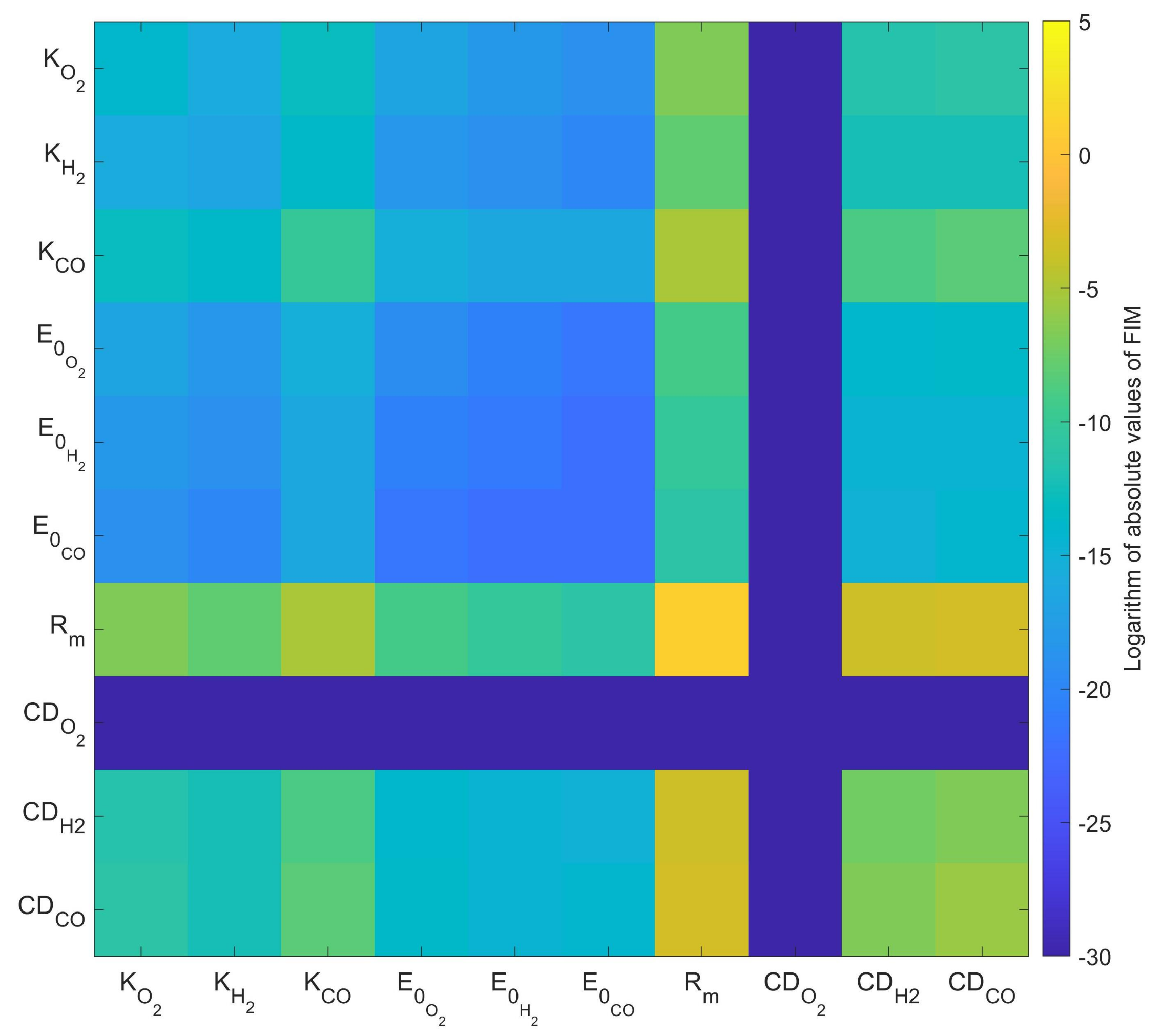
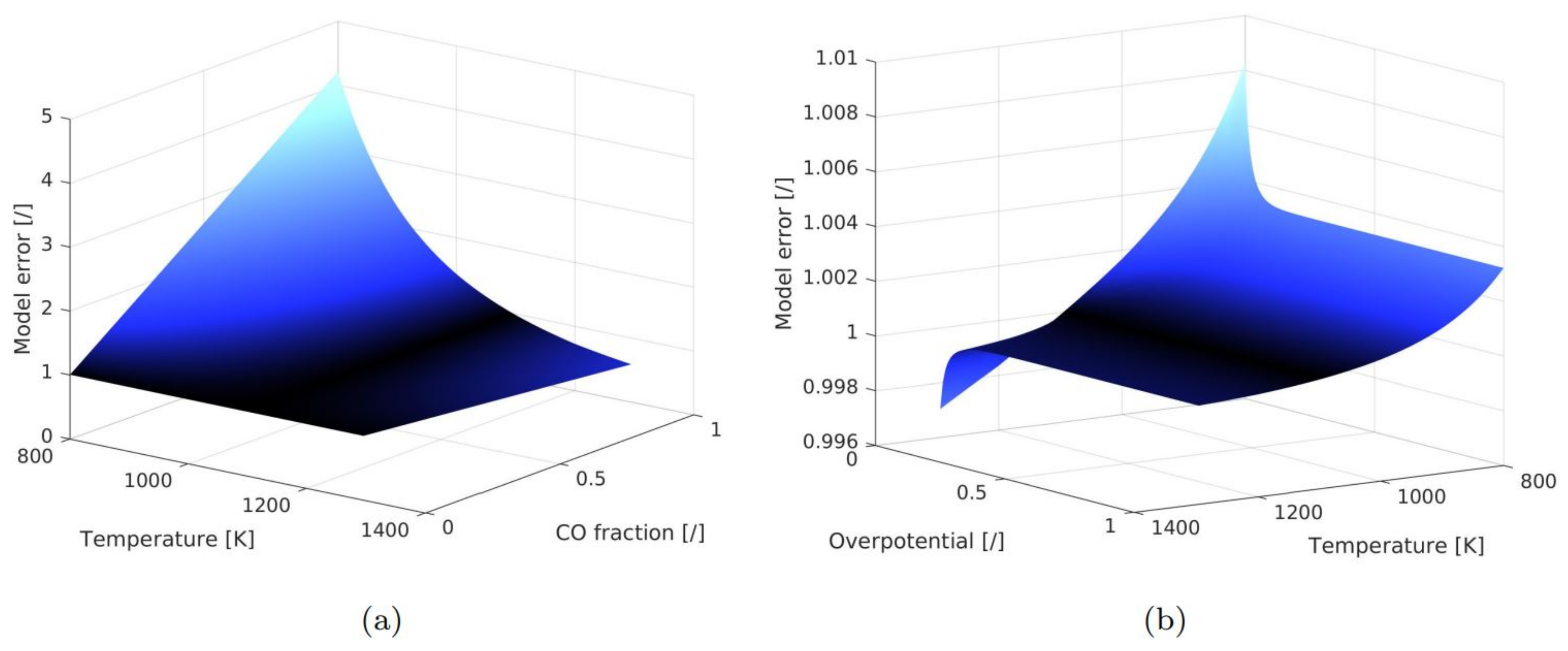
3.2. Parameter Sensitivity Analysis and Error of Calibration
3.3. Experimental Data
3.4. Calibration Procedure
4. Results and Discussion
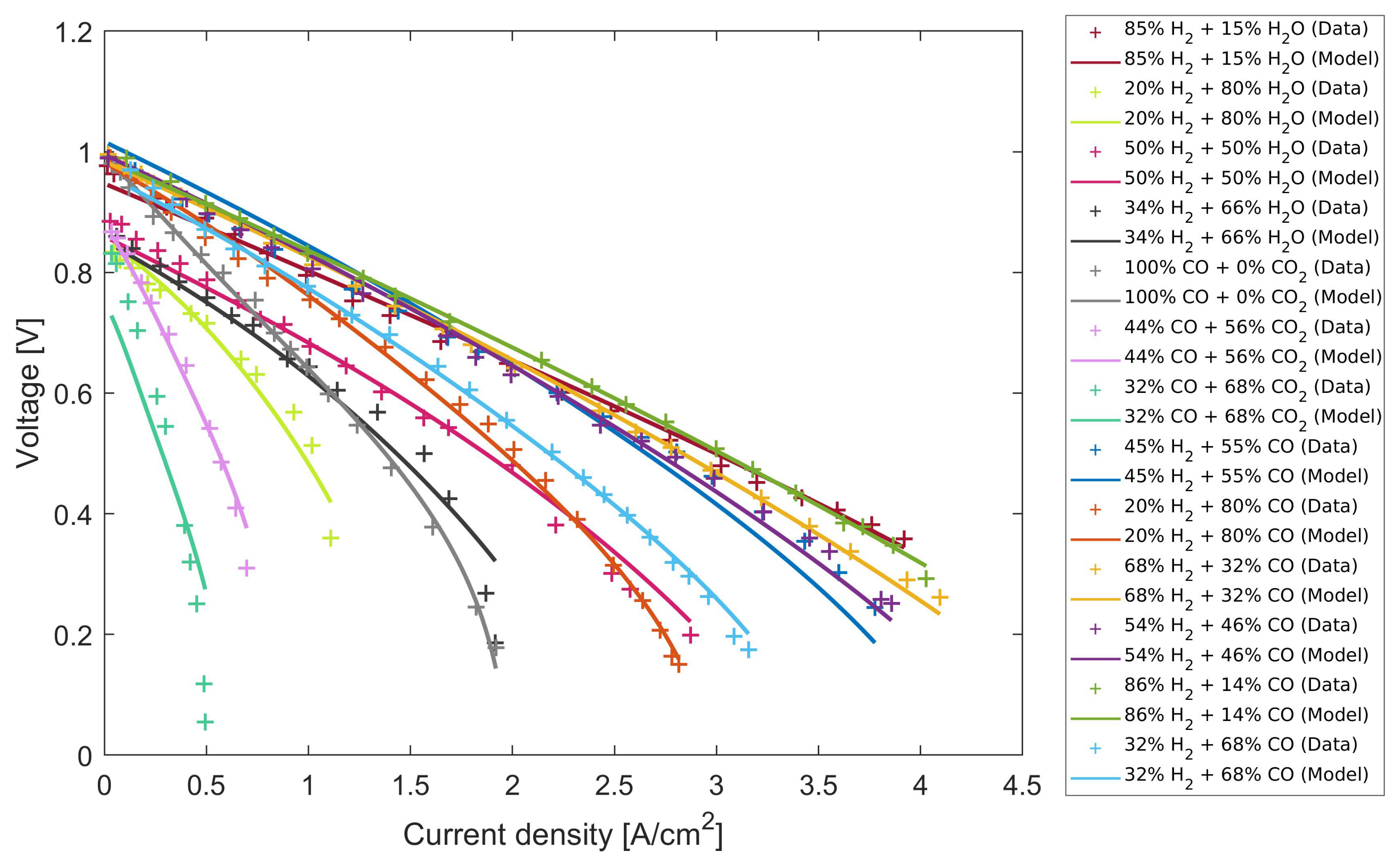
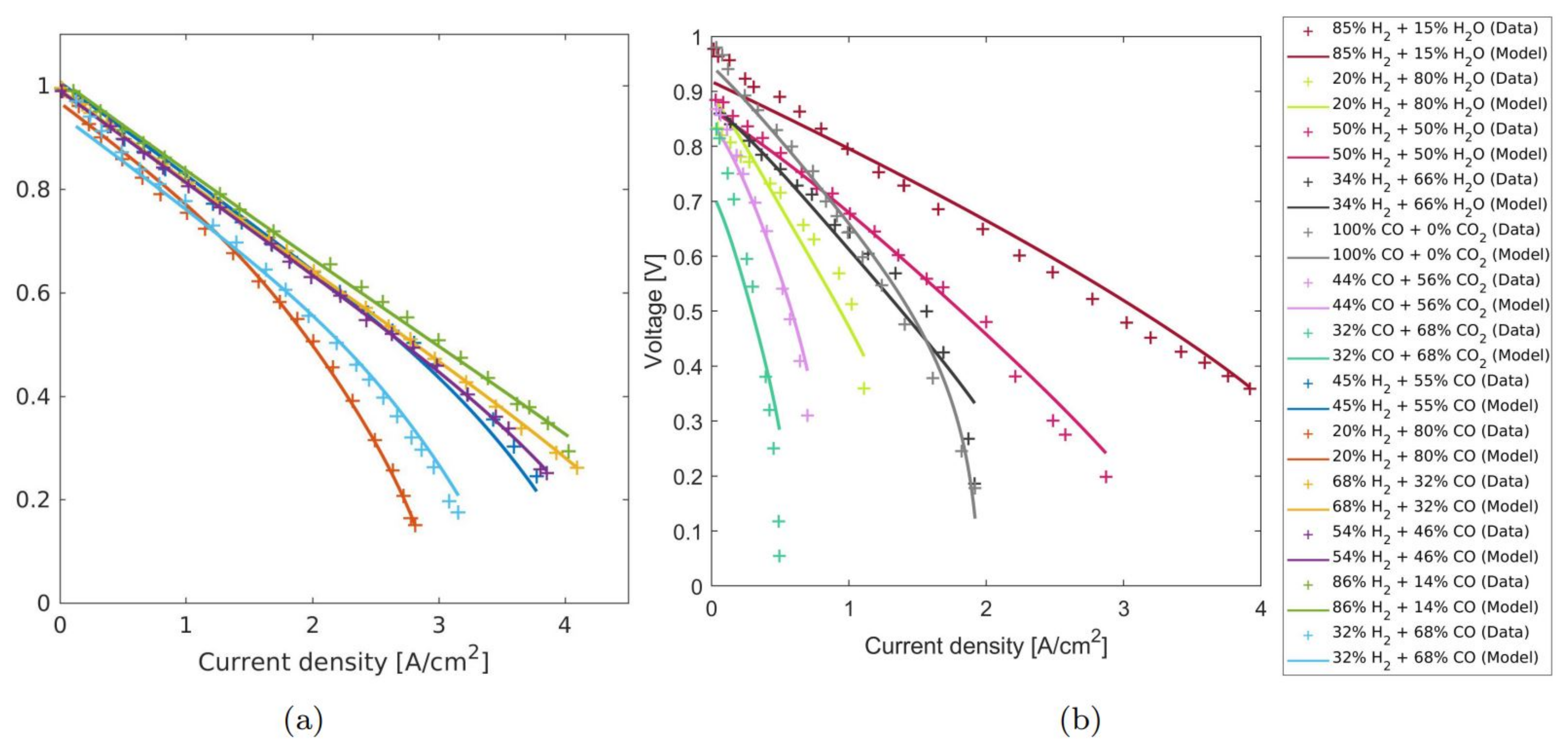
4.1. Calibrating the Model to Experimental Data
4.2. Parameter Sensitivity and the Standard Error of Parameter Values
Validation with Known Values of Calibration Parameters from the Literature
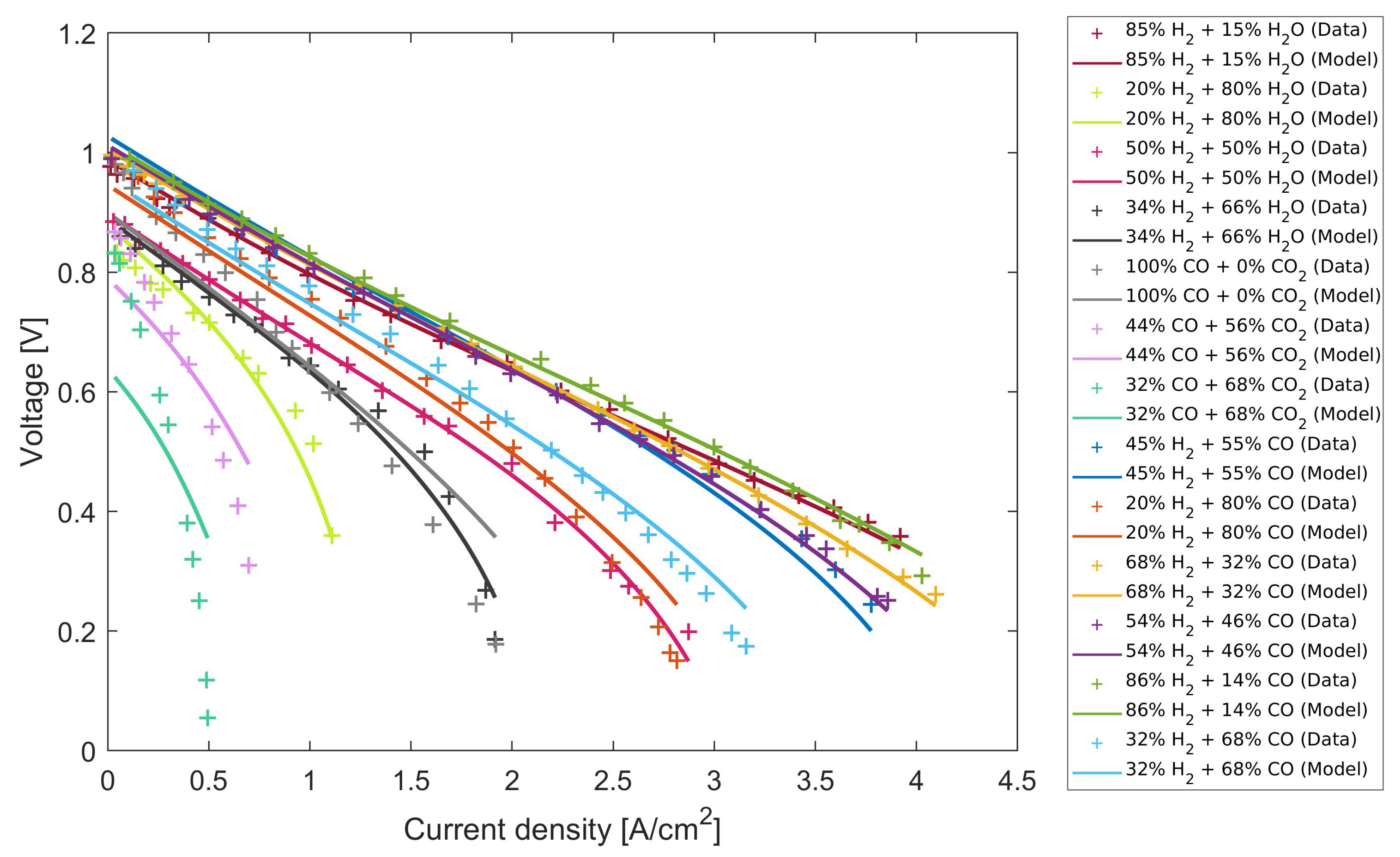
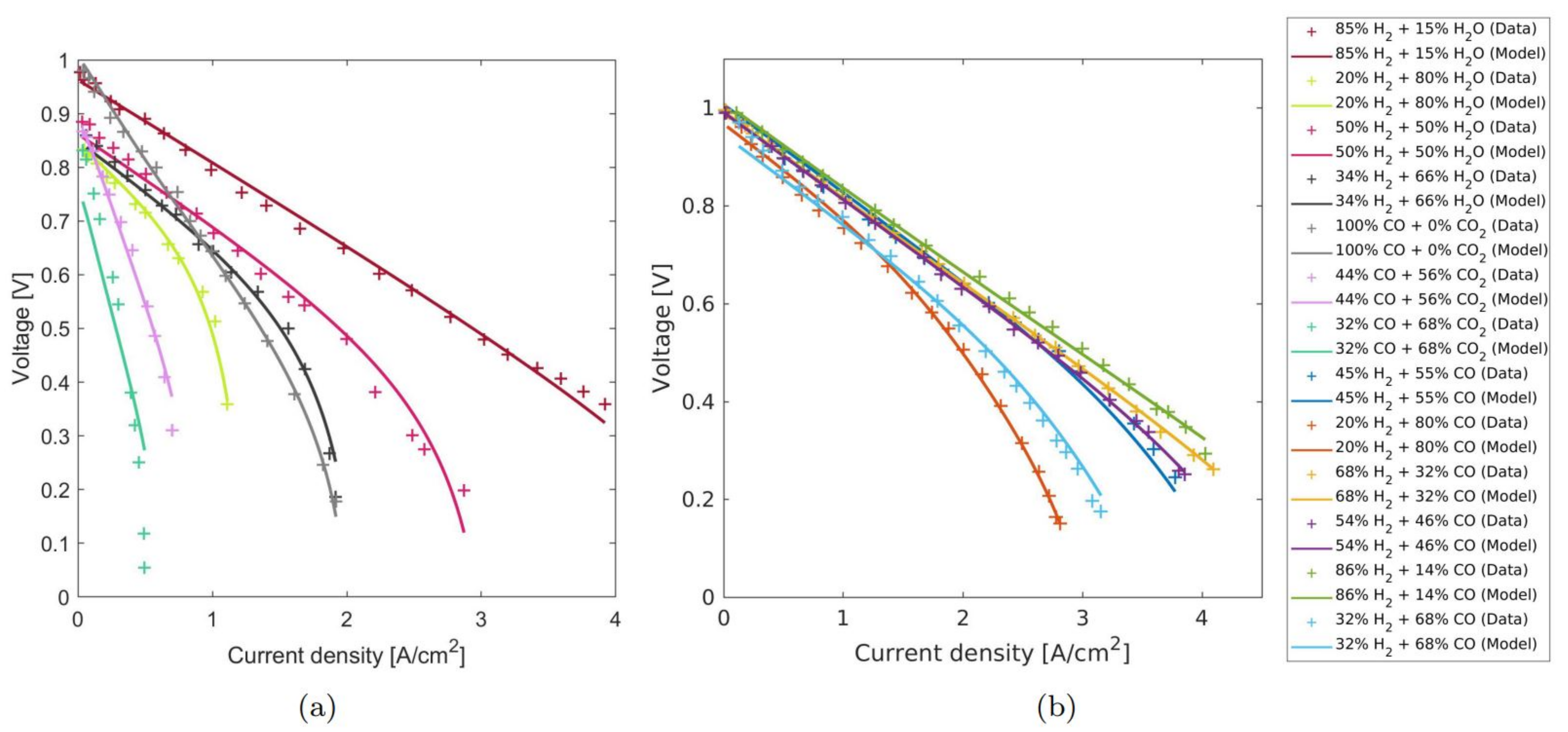
4.3. Extrapolation Validation
4.4. Comparison with Other Known Models from the Literature
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| a | Anode |
| BV | Butler–Volmer |
| c | Cathode |
| FC | Fuel Cell |
| GDL | Gas Diffusion Layer |
| LT | Low Temperature |
| o | Oxidation reaction |
| PEM | Proton Exchange Membrane |
| r | Reduction reaction |
| R | Reactants |
| ref | Reference |
| SoF | State of Function |
| SoH | State of Health |
| SoOC | State of Operational Conditions |
| TC | Thermodynamically Consistent |
| A | Energy needed to arrive at the transition state—cathode |
| B | Energy needed to arrive at the transition state—carbon monoxide |
| C | Concentration |
| Specific concentration | |
| Combined diffusive parameter | |
| CO | Carbon Monoxide |
| D | Energy needed to arrive at the transition state—hydrogen |
| E | Energy |
| g | Specific Gibbs free energy |
| H | Hydrogen |
| HO | Water |
| i | Current density |
| I | Current |
| Intrinsic exchange current | |
| j | Molar flux |
| k | Reaction rate |
| K | Lumped reaction rate |
| O | Oxygen |
| R | Resistance |
| s | Specific Entropy |
| T | Temperature |
| U | Voltage |
| Open circuit voltage | |
| Z | Number of electrons transferred in the electrochemical reaction |
| Charge transfer coefficient | |
| Width | |
| Change, difference | |
| Over-potential | |
| Stoichiometric ratio | |
| Kinetic reaction orders | |
| Basic charge | |
| F | Faraday constant |
| Boltzmann constant | |
| Gas constant |
Appendix A. Estimation of the Modelling Error for the Untypical Operating Conditions

Appendix B. Values of the Model Parameters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Units | Description | Source |
|---|---|---|---|---|
| d | 20 | m | Thickness of interlayer | [45] |
| d | 10 | m | Thickness of interlayer | [45] |
| S | 1.1 | cm | Effective electrode area | [45] |
| 0.54 | unitless | Porosity | [45] | |
| [ here. 4.89–9] | unitless | Tortuosity | [45] | |
| AV | 70 × 10 | mm | EASA-to-volume-ratio | [59] |
| K | 1.66 × 10 | atm | H_2 equilibrium constant | [32] |
| K | 1.07 × 10 | atm | CO equilibrium constant | [32] |
| K | 2.1 × 10 RT/F | A/cm | Reaction rate H | [32] |
| K | 0.84 × 10 RT/F | A/cm | Reaction rate CO | [32] |
| K | 0.25 × 10RT/F | A/cm | Reaction rate O | [32] |
| E | 130,000 | units RT | Activation energy H | [32] |
| E | 120,000 | units RT | Activation energy CO | [32] |
| E | 120,000 | units RT | Activation energy O | [32] |
| D | [3.858–5.677] | cms | Difussion coefficient H | [45] |
| D | 0.958 | cms | Difussion coefficient CO | [45] |
| D | 1.9844 | cms | Difussion coefficient O | [58] |
References
- Bridgwater, A. The technical and economic feasibility of biomass gasification for power generation. Fuel 1995, 74, 631–653. [Google Scholar] [CrossRef]
- Samiran, N.A.; Jaafar, M.N.M.; Ng, J.H.; Lam, S.S.; Chong, C.T. Progress in biomass gasification technique—With focus on Malaysian palm biomass for syngas production. Renew. Sustain. Energy Rev. 2016, 62, 1047–1062. [Google Scholar] [CrossRef]
- Di Blasi, C. Combustion and gasification rates of lignocellulosic chars. Prog. Energy Combust. Sci. 2009, 35, 121–140. [Google Scholar] [CrossRef]
- de Lasa, H.; Salaices, E.; Mazumder, J.; Lucky, R. Catalytic Steam Gasification of Biomass: Catalysts, Thermodynamics and Kinetics. Chem. Rev. 2011, 111, 5404–5433. [Google Scholar] [CrossRef]
- Devi, L.; Ptasinski, K.J.; Janssen, F.J. A review of the primary measures for tar elimination in biomass gasification processes. Biomass Bioenergy 2003, 24, 125–140. [Google Scholar] [CrossRef]
- Holladay, J.; Hu, J.; King, D.; Wang, Y. An overview of hydrogen production technologies. Catal. Today 2009, 139, 244–260. [Google Scholar] [CrossRef]
- Kravos, A.; Seljak, T.; Rodman Oprešnik, S.; Katrašnik, T. Operational stability of a spark ignition engine fuelled by low H2 content synthesis gas: Thermodynamic analysis of combustion and pollutants formation. Fuel 2020, 261, 116457. [Google Scholar] [CrossRef]
- Marcantonio, V.; Del Zotto, L.; Ouweltjes, J.P.; Bocci, E. Main issues of the impact of tar, H2S, HCl and alkali metal from biomass-gasification derived syngas on the SOFC anode and the related gas cleaning technologies for feeding a SOFC system: A review. Int. J. Hydrog. Energy 2021, 47, 517–539. [Google Scholar] [CrossRef]
- Arriagada, J.; Olausson, P.; Selimovic, A. Artificial neural network simulator for SOFC performance prediction. J. Power Sources 2002, 112, 54–60. [Google Scholar] [CrossRef]
- Entchev, E.; Yang, L. Application of adaptive neuro-fuzzy inference system techniques and artificial neural networks to predict solid oxide fuel cell performance in residential microgeneration installation. J. Power Sources 2007, 170, 122–129. [Google Scholar] [CrossRef]
- Huo, H.B.; Zhu, X.J.; Hu, W.Q.; Tu, H.Y.; Li, J.; Yang, J. Nonlinear model predictive control of SOFC based on a Hammerstein model. J. Power Sources 2008, 185, 338–344. [Google Scholar] [CrossRef]
- Bellman, R.E. Dynamic Programming; Princeton University Press: Princeton, NJ, USA, 1957. [Google Scholar]
- Kravos, A.; Ritzberger, D.; Tavčar, G.; Hametner, C.; Jakubek, S.; Katrašnik, T. Thermodynamically consistent reduced dimensionality electrochemical model for proton exchange membrane fuel cell performance modelling and control. J. Power Sources 2020, 454, 227930. [Google Scholar] [CrossRef]
- Kulikovsky, A.A. A Physically–Based Analytical Polarization Curve of a PEM Fuel Cell. J. Electrochem. Soc. 2013, 161, F263–F270. [Google Scholar] [CrossRef]
- Gu, W.; Baker, D.R.; Liu, Y.; Gasteiger, H.A. Proton exchange membrane fuel cell (PEMFC) down-the-channel performance model. In Handbook of Fuel Cells; American Cancer Society: Atlanta, GA, USA, 2010. [Google Scholar] [CrossRef]
- Mueller, F.; Brouwer, J.; Jabbari, F.; Samuelsen, S. Dynamic Simulation of an Integrated Solid Oxide Fuel Cell System Including Current-Based Fuel Flow Control. J. Fuel Cell Sci. Technol. 2005, 3, 144–154. [Google Scholar] [CrossRef]
- Wu, C.C.; Chen, T.L. Dynamic Modeling of a Parallel-Connected Solid Oxide Fuel Cell Stack System. Energies 2020, 13, 501. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Khor, K.; Xia, Z. A complete polarization model of a solid oxide fuel cell and its sensitivity to the change of cell component thickness. J. Power Sources 2001, 93, 130–140. [Google Scholar] [CrossRef]
- Dolenc, B.; Boskoski, P.; Pohjoranta, A.; Noponen, M.; Juricic, D. Hybrid Approach to Remaining Useful Life Prediction of Solid Oxide Fuel Cell Stack. ECS Trans. 2017, 78, 2251–2264. [Google Scholar] [CrossRef]
- DiGiuseppe, G. An Electrochemical Model of a Solid Oxide Fuel Cell Using Experimental Data for Validation of Material Properties. In Proceedings of the ASME 2010 8th International Fuel Cell Science, Engineering and Technology Conference: Volume 2, Brooklyn, NY, USA, 14–16 June 2010; pp. 193–203. [Google Scholar] [CrossRef]
- Siegel, J.B.; Wang, Y.; Stefanopoulou, A.G.; McCain, B.A. Comparison of SOFC and PEM Fuel Cell Hybrid Power Management Strategies for Mobile Robots. In Proceedings of the 2015 IEEE Vehicle Power and Propulsion Conference (VPPC), Montreal, QC, Canada, 19–22 October 2015; pp. 1–6. [Google Scholar] [CrossRef]
- Dolenc, B.; Boskoski, P.; Stepancic, M.; Pohjoranta, A.; Juricic, D. State of health estimation and remaining useful life prediction of solid oxide fuel cell stack. Energy Convers. Manag. 2017, 148, 993–1002. [Google Scholar] [CrossRef]
- Bianchi, F.R.; Bosio, B.; Baldinelli, A.; Barelli, L. Optimization of a Reference Kinetic Model for Solid Oxide Fuel Cells. Catalysts 2020, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Das, T.; Narayanan, S.; Mukherjee, R. Model Based Characterization of Transient Response of a Solid Oxide Fuel Cell System. In ASME International Mechanical Engineering Congress and Exposition, Seattle, WA, USA, 11–15 November 2017, Proceedings of the Volume 6: Energy Systems: Analysis, Thermodynamics and Sustainability; ASME: New York, NY, USA, 2007; pp. 655–664. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Wee, D.; Ghoniem, A.F. An improved one-dimensional membrane-electrode assembly model to predict the performance of solid oxide fuel cell including the limiting current density. J. Power Sources 2009, 186, 417–427. [Google Scholar] [CrossRef]
- Tabish, A.; Patel, H.; Chundru, P.; Stam, J.; Aravind, P. An SOFC anode model using TPB-based kinetics. Int. J. Hydrog. Energy 2020, 45, 27563–27574. [Google Scholar] [CrossRef]
- Zhu, H.; Kee, R.; Janardhanan, V.; Deutschmann, O.; Goodwin, D. Modeling Elementary Heterogeneous Chemistry and Electrochemistry in Solid-Oxide Fuel Cells. J. Electrochem. Soc. 2005, 152, A2427. [Google Scholar] [CrossRef] [Green Version]
- Audasso, E.; Bianchi, F.R.; Bosio, B. 2D Simulation for CH4 Internal Reforming-SOFCs: An Approach to Study Performance Degradation and Optimization. Energies 2020, 13, 4116. [Google Scholar] [CrossRef]
- Li, P.W.; Chyu, M.K. Electrochemical and Transport Phenomena in Solid Oxide Fuel Cells. J. Heat Transf. 2005, 127, 1344–1362. [Google Scholar] [CrossRef]
- Nerat, M.; Juricic, D. A comprehensive 3-D modeling of a single planar solid oxide fuel cell. Int. J. Hydrog. Energy 2016, 41, 3613–3627. [Google Scholar] [CrossRef]
- Zhu, H.; Kee, R.J. A general mathematical model for analyzing the performance of fuel-cell membrane-electrode assemblies. J. Power Sources 2003, 117, 61–74. [Google Scholar] [CrossRef]
- Suwanwarangkul, R.; Croiset, E.; Entchev, E.; Charojrochkul, S.; Pritzker, M.; Fowler, M.; Douglas, P.; Chewathanakup, S.; Mahaudom, H. Experimental and modeling study of solid oxide fuel cell operating with syngas fuel. J. Power Sources 2006, 161, 308–322. [Google Scholar] [CrossRef]
- Suwanwarangkul, R.; Croiset, E.; Pritzker, M.; Fowler, M.; Douglas, P.; Entchev, E. Modelling of a cathode-supported tubular solid oxide fuel cell operating with biomass-derived synthesis gas. J. Power Sources 2007, 166, 386–399. [Google Scholar] [CrossRef]
- Andersson, M.; Yuan, J.; Sundén, B. SOFC modeling considering hydrogen and carbon monoxide as electrochemical reactants. J. Power Sources 2013, 232, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Nagel, F.P.; Schildhauer, T.J.; Biollaz, S.M.; Wokaun, A. Performance comparison of planar, tubular and Delta8 solid oxide fuel cells using a generalized finite volume model. J. Power Sources 2008, 184, 143–164. [Google Scholar] [CrossRef]
- Ni, M. Modeling of SOFC running on partially pre-reformed gas mixture. Int. J. Hydrog. Energy 2012, 37, 1731–1745. [Google Scholar] [CrossRef] [Green Version]
- Ni, M. The effect of electrolyte type on performance of solid oxide fuel cells running on hydrocarbon fuels. Int. J. Hydrog. Energy 2013, 38, 2846–2858. [Google Scholar] [CrossRef] [Green Version]
- Bao, C.; Wang, Y.; Feng, D.; Jiang, Z.; Zhang, X. Macroscopic modeling of solid oxide fuel cell (SOFC) and model-based control of SOFC and gas turbine hybrid system. Prog. Energy Combust. Sci. 2018, 66, 83–140. [Google Scholar] [CrossRef]
- Hornbostel, K. Modeling of Solid Oxide Fuel Cell Performance with Coal Gasification. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 2016. [Google Scholar]
- Bao, C.; Shi, Y.; Croiset, E.; Li, C.; Cai, N. A multi-level simulation platform of natural gas internal reforming solid oxide fuel cell–gas turbine hybrid generation system: Part I. Solid oxide fuel cell model library. J. Power Sources 2010, 195, 4871–4892. [Google Scholar] [CrossRef]
- Petruzzi, L.; Cocchi, S.; Fineschi, F. A global thermo-electrochemical model for SOFC systems design and engineering. J. Power Sources 2003, 118, 96–107. [Google Scholar] [CrossRef]
- Bao, C.; Zhang, X. One-Dimensional Macroscopic Model of Solid Oxide Fuel Cell Anode with Analytical Modeling of H2/CO Electrochemical Co-oxidation. Electrochim. Acta 2014, 134, 426–434. [Google Scholar] [CrossRef]
- Bao, C.; Yixiang, S.; Li, C.; Cai, N.; Su, Q. Mathematical Modeling of Solid Oxide Fuel Cells at High Fuel Utilization Based on Diffusion Equivalent Circuit Model. AIChE J. 2009, 56, 1363–1371. [Google Scholar] [CrossRef]
- Bao, C.; Jiang, Z.; Zhang, X. Mathematical modeling of synthesis gas fueled electrochemistry and transport including H2/CO co-oxidation and surface diffusion in solid oxide fuel cell. J. Power Sources 2015, 294, 317–332. [Google Scholar] [CrossRef]
- Jiang, Y.; Virkar, A.V. Fuel Composition and Diluent Effect on Gas Transport and Performance of Anode-Supported SOFCs. J. Electrochem. Soc. 2003, 150, A942. [Google Scholar] [CrossRef]
- Kravos, A.; Ritzberger, D.; Hametner, C.; Jakubek, S.; Katrašnik, T. Methodology for efficient parametrisation of electrochemical PEMFC model for virtual observers: Model based optimal design of experiments supported by parameter sensitivity analysis. Int. J. Hydrog. Energy 2021, 46, 13832–13844. [Google Scholar] [CrossRef]
- O’Hayre, R.; Cha, S.; Colella, W.; Prinz, F. Fuel Cell Fundamentals; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Mench, M. Fuel Cell Engines; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Atkins, P.; De Paula, J.; Keeler, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Fornasiero, P.; Graziani, M. Renewable Resources and Renewable Energy: A Global Challenge, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Kulikovsky, A. Analytical Modelling of Fuel Cells; Elsevier B.V.: Amsterdam, The Netherlands, 2010. [Google Scholar] [CrossRef]
- U.S. Patent; Trademark Office. Official Gazette of the United States Patent and Trademark Office: Patents; Number v. 1168, nos. 3-4; U.S. Department of Commerce, Patent and Trademark Office: Washington, DC, USA, 1994. [Google Scholar]
- Kravos, A.; Kregar, A.; Katrašnik, T. Hybrid Methodology for Efficient on the Fly (Re)Parametrization of Proton Exchange Membrane Fuel Cells Electrochemical Model for Diagnostics and Control Applications. ECS Trans. 2020, 98, 13–24. [Google Scholar] [CrossRef]
- Kay, S.M. Fundamentals of Statistical Signal Processing; Prentice Hall PTR: Hoboken, NJ, USA, 1993. [Google Scholar]
- Storn, R.; Price, K. Differential Evolution—A Simple and Efficient Heuristic for Global Optimization over Continuous Spaces. J. Glob. Optim. 1997, 11, 341–359. [Google Scholar] [CrossRef]
- MATLAB. Optimization Toolbox Release; The MathWorks, Inc.: Natick, MA, USA, 2018. [Google Scholar]
- Minh, N.Q.; Takahashi, T. Chapter 8—Electrode reaction. In Science and Technology of Ceramic Fuel Cells; Minh, N.Q., Takahashi, T., Eds.; Elsevier Science Ltd.: Oxford, UK, 1995; pp. 199–232. [Google Scholar] [CrossRef]
- Roberts, R. American Institute of Physics Handbook: 2s. Molecular Diffusion of Gases; A McGraw-Hill Classic Handbook Reissue; McGraw-Hill: New York, NY, USA, 1972. [Google Scholar]
- Marrero-López, D.; Ruiz-Morales, J.; Peña-Martínez, J.; Canales-Vázquez, J.; Núñez, P. Preparation of thin layer materials with macroporous microstructure for SOFC applications. J. Solid State Chem. 2008, 181, 685–692. [Google Scholar] [CrossRef]






| No. | ||||
|---|---|---|---|---|
| 1 | 0.86 | 0.14 | 0.00 | 0.00 |
| 2 | 0.68 | 0.32 | 0.00 | 0.00 |
| 3 | 0.54 | 0.46 | 0.00 | 0.00 |
| 4 | 0.45 | 0.55 | 0.00 | 0.00 |
| 5 | 0.32 | 0.68 | 0.00 | 0.00 |
| 6 | 0.20 | 0.80 | 0.00 | 0.00 |
| 7 | 0.00 | 0.32 | 0.00 | 0.68 |
| 8 | 0.00 | 0.44 | 0.00 | 0.56 |
| 9 | 0.00 | 1.00 | 0.00 | 0.00 |
| 10 | 0.20 | 0.00 | 0.80 | 0.00 |
| 11 | 0.34 | 0.00 | 0.66 | 0.00 |
| 12 | 0.50 | 0.00 | 0.50 | 0.00 |
| 13 | 0.85 | 0.00 | 0.15 | 0.00 |
| Parameter | Value | Units | Description |
|---|---|---|---|
| K | 1.9284 × 10 RT/F | A/cm | Reaction rate H |
| K | 0.7534 × 10 RT/F | A/cm | Reaction rate CO |
| K | 0.21697 × 10 RT/F | A/cm | Reaction rate O |
| E | 136,167 | units RT | Activation energy H |
| E | 118,311 | units RT | Activation energy CO |
| E | 113,149 | units RT | Activation energy O |
| D | 6.3843 | cms | Difussion coefficient H |
| D | 1.2438 | cms | Difussion coefficient CO |
| D | 0.7457 | cms | Difussion coefficient O |
| Composition [%] | [43] | [42] | [44] | [25] | [39] | [26] | Our Model |
|---|---|---|---|---|---|---|---|
| 85 H2-15 H2O | 0.1099 | 0.1516 | 0.1587 | / | 0.1755 | 0.1361 | 0.02175 |
| 50 H2-50 H2O | 0.1189 | 0.1010 | 0.1074 | 0.1529 | 0.1901 | 0.1928 | 0.04116 |
| 34 H2-66 H2O | 0.1228 | 0.09958 | 0.1761 | 0.1399 | 0.2150 | 0.2114 | 0.02212 |
| 20 H2-80 H2O | 0.1483 | 0.1813 | 0.3999 | 0.1625 | 0.1499 | 0.2041 | 0.04556 |
| 97 CO-3 CO2 | 0.1507 | 0.1572 | 0.1285 | / | 0.2267 | / | 0.02556 |
| 44 CO-56 CO2 | 0.2158 | 0.1731 | 0.1663 | / | 0.1634 | / | 0.03037 |
| 32 CO-68 CO2 | 0.1491 | 0.2629 | 0.3245 | / | 0.3714 | / | 0.1208 |
| 86 H2-14 CO | 0.1541 | 0.1224 | 0.1593 | / | 0.3025 | / | 0.04165 |
| 68 H2-32 CO | / | 0.3464 | 0.1532 | / | 0.2076 | / | 0.03675 |
| 54 H2-46 CO | / | / | / | / | 0.2298 | / | 0.03629 |
| 45 H2-55 CO | 0.1794 | 0.1314 | 0.1339 | / | 0.3478 | / | 0.01652 |
| 32 H2-68 CO | / | 0.1780 | 0.3140 | / | 0.1771 | / | 0.02496 |
| 20 H2-80 CO | 0.2914 | 0.1753 | 0.2229 | / | 0.2648 | / | 0.02117 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kravos, A.; Katrašnik, T. Closed-Form Formulation of the Thermodynamically Consistent Electrochemical Model Considering Electrochemical Co-Oxidation of CO and H2 for Simulating Solid Oxide Fuel Cells. Catalysts 2022, 12, 56. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010056
Kravos A, Katrašnik T. Closed-Form Formulation of the Thermodynamically Consistent Electrochemical Model Considering Electrochemical Co-Oxidation of CO and H2 for Simulating Solid Oxide Fuel Cells. Catalysts. 2022; 12(1):56. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010056
Chicago/Turabian StyleKravos, Andraž, and Tomaž Katrašnik. 2022. "Closed-Form Formulation of the Thermodynamically Consistent Electrochemical Model Considering Electrochemical Co-Oxidation of CO and H2 for Simulating Solid Oxide Fuel Cells" Catalysts 12, no. 1: 56. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010056