Asymmetric Synthesis of Both Enantiomers of Dimethyl 2-Methylsuccinate by the Ene-Reductase-Catalyzed Reduction at High Substrate Concentration

 , , ,
, , ,
Abstract
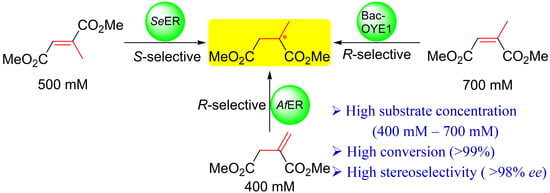
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Material
3.2. General Protein Expression Procedure
3.3. Purification of ERs
3.4. General Procedure for the Preparation of Dimethyl Mesaconate (3), Diethyl Citraconate (5), Diethyl Mesaconate (6) and Diethyl 2-Methylsuccinate (8)
3.5. Analytical Scale Reaction of ERs towards 2–7 with Wet Cells
3.6. Enzyme Activity Assay
3.7. Effect of pH and Temperature on Purified ERs
3.8. Determination of Kinetic Parameters
3.9. Preparative Scale Synthesis of Dimethyl 2-Methylsuccinate (1)
3.9.1. (R)-Dimethyl 2-Methylsuccinate ((R)-1) Using Bac-OYE1
3.9.2. (S)-Dimethyl 2-Methylsuccinate ((S)-1) Using SeER
3.9.3. (R)-Dimethyl 2-Methylsuccinate ((R)-1) Using AfER
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Novick, S.J.; Dellas, N.; Garcia, R.; Ching, C.; Bautista, A.; Homan, D.; Alvizo, O.; Entwistle, D.; Kleinbeck, F.; Schlama, T.; et al. Engineering an Amine Transaminase for the Efficient Production of a Chiral Sacubitril Precursor. ACS Catal. 2021, 11, 3762–3770. [Google Scholar] [CrossRef]
- Zhou, X.; Fang, W.; Tan, S.; Lin, X.; Xun, T.; Yang, B.; Liu, S.; Liu, Y. Aspernigrins with anti-HIV-1 activities from the marine-derived fungus Aspergillus niger SCSIO Jcsw6F30. Bioorg. Med. Chem. Lett. 2016, 26, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.J.; Davies, S.G. Asymmetric synthesis of moiramide B. Chem. Commun. 1996, 1797–1798. [Google Scholar] [CrossRef]
- Pohlmann, J.; Lampe, T.; Shimada, M.; Nell, P.G.; Pernerstorfer, J.; Svenstrup, N.; Brunner, N.A.; Schiffer, G.; Freiberg, C. Pyrrolidinedione derivatives as antibacterial agents with a novel mode of action. Bioorg. Med. Chem. Lett. 2005, 15, 1189–1192. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, C.; Huang, J.; Zhang, J.; Liu, D.; Huang, J.; Proksch, P.; Lin, W. Violaceimides A–E, sulfur-containing metabolites from a sponge-associated fungus Aspergillus violaceus. Tetrahedron Lett. 2018, 59, 3157–3160. [Google Scholar] [CrossRef]
- Ismail, K.A.; Bergmeier, S.C. Structure–activity studies with ring E analogues of methyllycaconitine. Synthesis and evaluation of enantiopure isomers of selective antagonist at the α3 nicotinic receptor. Eur. J. Med. Chem. 2002, 37, 469–474. [Google Scholar] [CrossRef]
- Mangan, D.; Miskelly, I.; Moody, T.S. A Three-Enzyme System Involving an Ene-Reductase for Generating Valuable Chiral Building Blocks. Adv. Synth. Catal. 2012, 354, 2185–2190. [Google Scholar] [CrossRef]
- Jacobsen, J.; Achenbach, B.; Reinsch, H.; Smolders, S.; Lange, F.D.; Friedrichs, G.; Vos, D.; Stock, N. The first water-based synthesis of Ce(Ⅳ)-MOFs with saturated chiral and achiral C4-dicarboxylate linkers. Dalton Trans. 2019, 48, 8433–8441. [Google Scholar] [CrossRef] [PubMed]
- Alonso, B.; Ocejo, M.; Carrillo, L.; Vicario, J.L.; Reyes, E.; Uria, U. Using heteroaryl-lithium reagents as hydroxycarbonyl anion equivalents in conjugate addition reactions with (S,S)-(+)-pseudoephedrine as chiral auxiliary; enantioselective synthesis of 3-substituted pyrrolidines. J. Org. Chem. 2013, 78, 614–627. [Google Scholar] [CrossRef]
- Kovalenko, V.N.; Kulinkovich, O.G. The resolution of trans-2,2-dichloro-3-methylcyclopropanecarboxylic acid via crystallization of its salts with (+)- and (−)-α-phenylethylamine, and the transformation of the resulting enantiomers into (R)- and (S)-dimethyl 2-methylsuccinates. Tetrahedron Asymmetry 2011, 22, 26–30. [Google Scholar] [CrossRef]
- Pongrácz, P.; Bartal, B.; Kollár, L.; Mika, L.T. Rhodium-catalyzed hydroformylation in γ-valerolactone as a biomass-derived solvent. J. Organomet. Chem. 2017, 847, 140–145. [Google Scholar] [CrossRef]
- Frank, D.J.; Franzke, A.; Pfaltz, A. Asymmetric hydrogenation using rhodium complexes generated from mixtures of monodentate neutral and anionic phosphorus ligands. Chem. Eur. J. 2013, 19, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Vaghi, L.; Cirilli, R.; Pierini, M.; Rizzo, S.; Terraneo, G.; Benincori, T. PHANE-TetraPHOS, the First D2 Symmetric Chiral Tetraphosphane. Synthesis, Metal Complexation, and Application in Homogeneous Stereoselective Hydrogenation. Eur. J. Org. Chem. 2021, 2021, 2367–2374. [Google Scholar] [CrossRef]
- Sen, A.; Kumar, R.; Pandey, S.; Vipin, K.R.; Kumar, P.; Kumar, V.; Chikkali, S.H. Mechanistically Guided One Pot Synthesis of Phosphine-Phosphite and Its Implication in Asymmetric Hydrogenation. Eur. J. Org. Chem. 2022, 2022, e202101447. [Google Scholar] [CrossRef]
- Parmeggiani, F.; Brenna, E.; Colombo, D.; Gatti, F.G.; Tentori, F.; Tessaro, D. “A Study in Yellow”: Investigations in the Stereoselectivity of Ene-Reductases. ChemBioChem 2022, 23, e202100445. [Google Scholar] [CrossRef]
- Hollmann, F.; Opperman, D.J.; Paul, C.E. Biocatalytic Reduction Reactions from a Chemist’s Perspective. Angew. Chem. Int. Ed. 2021, 60, 5644–5665. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.C.E.; Holtmann, D. Enzymatic reductions for the chemist. Green Chem. 2011, 13, 2285–2313. [Google Scholar] [CrossRef]
- Winkler, C.K.; Faber, K.; Hall, M. Biocatalytic reduction of activated C=C-bonds and beyond: Emerging trends. Curr. Opin. Chem. Biol. 2018, 43, 97–105. [Google Scholar] [CrossRef]
- Stuermer, R.; Hauer, B.; Hall, M.; Fabe, K. Asymmetric bioreduction of activated C=C bonds using enoate reductases from the old yellow enzyme family. Curr. Opin. Chem. Biol. 2007, 11, 203–213. [Google Scholar] [CrossRef]
- Williams, R.E.; Bruce, N.C. ‘New uses for an Old Enzyme’ the Old Yellow Enzyme family of flavoenzymes. Microbiology 2002, 148, 1607–1614. [Google Scholar] [CrossRef] [Green Version]
- Toogood, H.S.; Scrutton, N.S. Discovery, Characterisation, Engineering and Applications of Ene Reductases for Industrial Biocatalysis. ACS Catal. 2018, 8, 3532–3549. [Google Scholar] [CrossRef] [PubMed]
- Brenna, E.; Gatti, F.G.; Manfredi, A.; Monti, D.; Parmeggiania, F. Steric Effects on the Stereochemistry of Old Yellow Enzyme-Mediated Reductions of Unsaturated Diesters: Flipping of the Substrate within the Enzyme Active Site Induced by Structural Modifications. Adv. Synth. Catal. 2012, 354, 2859–2864. [Google Scholar] [CrossRef]
- Yanto, Y.; Winkler, C.K.; Lohr, S.; Hall, M.; Faber, K.; Bommarius, A.S. Asymmetric Bioreduction of Alkenes Using Ene Reductases YersER and KYE1 and Effects of Organic Solvents. Org. Lett. 2011, 13, 2540–2543. [Google Scholar] [CrossRef]
- Nett, N.; Duewel, S.; Schmermund, L.; Benary, G.E.; Ranaghan, K.; Mulholland, A.; Opperman, D.J.; Hoebenreich, S. A robust and stereocomplementary panel of ene-reductase variants for gram-scale asymmetric hydrogenation. Mol. Catal. 2021, 502, 111404. [Google Scholar] [CrossRef]
- Iqbal, N.; Rudroff, F.; Brige, A.; Beeumen, J.V.; Mihovilovic, M.D. Asymmetric bioreduction of activated carbon-carbon double bonds using Shewanella yellow enzyme (SYE-4) as novel enoate reductase. Tetrahedron 2012, 68, 7619–7623. [Google Scholar] [CrossRef] [PubMed]
- Durchschein, K.; Wallner, S.; Macheroux, P.; Schwab, W.; Winkler, T.; Kreis, W.; Faber, K. Nicotinamide-Dependent Ene Reductases as Alternative Biocatalysts for the Reduction of Activated Alkenes. Eur. J. Org. Chem. 2012, 2012, 4963–4968. [Google Scholar] [CrossRef]
- Mueller, N.J.; Stueckler, C.; Hauer, B.; Baudendistel, N.; Housden, H.; Bruce, N.C.; Faber, K. The Substrate Spectra of Pentaerythritol Tetranitrate Reductase, Morphinone Reductase,N-Ethylmaleimide Reductase and Estrogen-Binding Protein in the Asymmetric Bioreduction of Activated Alkenes. Adv. Synth. Catal. 2010, 352, 387–394. [Google Scholar] [CrossRef]
- Domínguez, B.; Schell, U.; Bisagni, S.; Kalthoff, T. Reduction of Activated Carbon-Carbon Double Bonds using Highly Active and Enantioselective Double Bond Reductases. Johnson Matthey Technol. Rev. 2016, 60, 243–249. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, X.; Ren, J.; Feng, J.; Zhang, T.; Wu, Q.; Zhu, D. Enzymatic hydrogenation of diverse activated alkenes. Identification of two Bacillus old yellow enzymes with broad substrate profiles. J. Mol. Catal. B Enzym. 2014, 105, 118–125. [Google Scholar] [CrossRef]
- Durchschein, K.; Wallner, S.; Macheroux, P.; Schwab, W.; Winkler, T.; Kreis, W.; Faber, K. Unusual C=C bond isomerization of an α, β-unsaturated γ-butyrolactone catalysed by flavoproteins from the old yellow enzyme family. ChemBioChem 2012, 13, 2346–2351. [Google Scholar] [CrossRef] [Green Version]
- Rüthlein, E.; Classen, T.; Dobnikar, L.; Schölzel, M.; Pietruszka, J. Finding the Selectivity Switch A Rational Approach towards Stereocomplementary Variants of the Ene Reductase YqjM. Adv.Synth. Catal. 2015, 357, 1775–1786. [Google Scholar] [CrossRef]
- Schmida, R.; Antoulas, S.; Ruttimannh, A.; Schmida, M.; Vecchia, M.; Weiserb, H. Synthesis of All Four Stereoisomers of (E)-Vitamin K, (Phylloquinone), Analysis of Their Diastereoisomeric and Enantiomeric Purities and Determination of Their Biopotencies. Helv. Chim. Acta 1990, 73, 1276–1299. [Google Scholar] [CrossRef]
- Alpdagtas, S.; Sevil, Y.; Handan, A.K.; Liu, S.; Binay, B. Discovery of an acidic, thermostable and highly NADP+ dependent formate dehydrogenase from Lactobacillus buchneri NRRL B-30929. Biotechnol. Lett. 2018, 40, 1135–1147. [Google Scholar] [CrossRef]
- Klimovica, K.; Grigorjeva, L.; Maleckis, A.; Popelis, J.; Jirgensons, A. C-Quaternary Vinylglycinols by Metal-Catalyzed Cyclization of Allylic Bistrichloroacetimidates. Synlett 2011, 2011, 2849–2851. [Google Scholar]
- Bernasconi, M.; Muller, M.A.; Pfaltz, A. Asymmetric hydrogenation of maleic acid diesters and anhydrides. Angew. Chem. Int. Ed. 2014, 126, 5489–5492. [Google Scholar] [CrossRef]
- Muler, A.; Sturmer, R.; Hauer, B.; Rosche, B. Stereospecific alkyne reduction: Novel activity of old yellow enzymes. Angew. Chem. Int. Ed. 2007, 46, 3316–3318. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, Z.; Wu, J.; Tu, S.; Ding, K. Directed orthogonal self-assembly of homochiral coordination polymers for heterogeneous enantioselective hydrogenation. Angew. Chem. Int. Ed. 2010, 122, 3709–3712. [Google Scholar] [CrossRef]
- Li, M.; Cui, Y.; Xu, Z.; Chen, X.; Feng, J.; Wang, M.; Yao, P.; Wu, Q.; Zhu, D. Asymmetric synthesis of N-substitutedγ-amino esters and γ-lactams containing α,γ-stereogenic centers via a stereoselective enzymatic cascade. Adv. Synth. Catal. 2022, 364, 372–379. [Google Scholar] [CrossRef]
- Padhi, S.K.; Bougioukou, D.J.; Stewart, J.D. Site-saturation mutagenesis of tryptophan 116 of saccharomyces pastorianus old yellow enzyme uncovers stereocomplementary variants. J. Am. Chem. Soc. 2009, 131, 3271–3280. [Google Scholar] [CrossRef]
- Hall, M.; Stueckler, C.; Hauer, B.; Stuermer, R.; Friedrich, T.; Breuer, M.; Kroutil, W. Asymmetric bioreduction of activated C=C bonds using zymomonas mobilis ncr enoate reductase and old yellow enzymes OYE 1–3 from yeasts. Eur. J. Org. Chem. 2008, 2008, 1511–1516. [Google Scholar] [CrossRef]
- Cheallaigh, A.N.; Mansell, D.J.; Toogood, H.S.; Tait, S.; Lygidakis, A.; Gardiner, J.M. Chemoenzymatic synthesis of the intermediates in the peppermint monoterpenoid biosynthetic pathway. J. Nat. Prod. 2018, 81, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Spiegelhauer, O.; Mende, S.; Dickert, F.; Knauer, S.H.; Ullmann, G.M.; Dobbek, H. Cysteine as a modulator residue in the active site of xenobiotic reductase a: A structural, thermodynamic and kinetic study. J. Mol. Biol. 2010, 398, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Walton, A.Z.; Sullivan, B.; Patterson-Orazem, A.C.; Stewart, J.D. Residues controlling facial selectivity in an alkene reductase and semirational alterations to create stereocomplementary variants. ACS Catal. 2014, 4, 2307–2318. [Google Scholar] [CrossRef] [PubMed]
- Breithaupt, C.; Strassner, J.; Breitinger, U.; Huber, R.; Macheroux, P.; Schaller, A.; Clausen, T. X-Ray structure of 12-oxophytodienoate reductase 1 provides structural insight into substrate binding and specificity within the family of oYE. Structure 2001, 9, 419–429. [Google Scholar] [CrossRef]
- Miura, K.; Tomioka, Y.; Suzuki, H.; Yonezawa, M.; Hishinuma, T.; Mizugaki, M. Molecular cloning of the nemA gene encoding N-ethylmaleimide reductase from Escherichia coli. Biol. Pharm. Bull. 1997, 20, 110–112. [Google Scholar] [CrossRef]
- French, C.E.; Bruce, N.C. Purification and characterization of morphinone reductase from Pseudomonas putida M10. Biochem. J. 1994, 301, 97–103. [Google Scholar] [CrossRef]
- Gao, X.; Ren, J.; Wu, Q.; Zhu, D. Biochemical characterization and substrate profiling of a new NADH-dependent enoate reductase from Lactobacillus casei. Enzyme Microb. Tech. 2012, 51, 26–34. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, M.; Xu, L.; Wei, M. Identification and characterization of a novel Old Yellow Enzyme from Bacillus subtilis str.168. J. Mol. Cataly. B Enzym. 2016, 130, 18–24. [Google Scholar] [CrossRef]



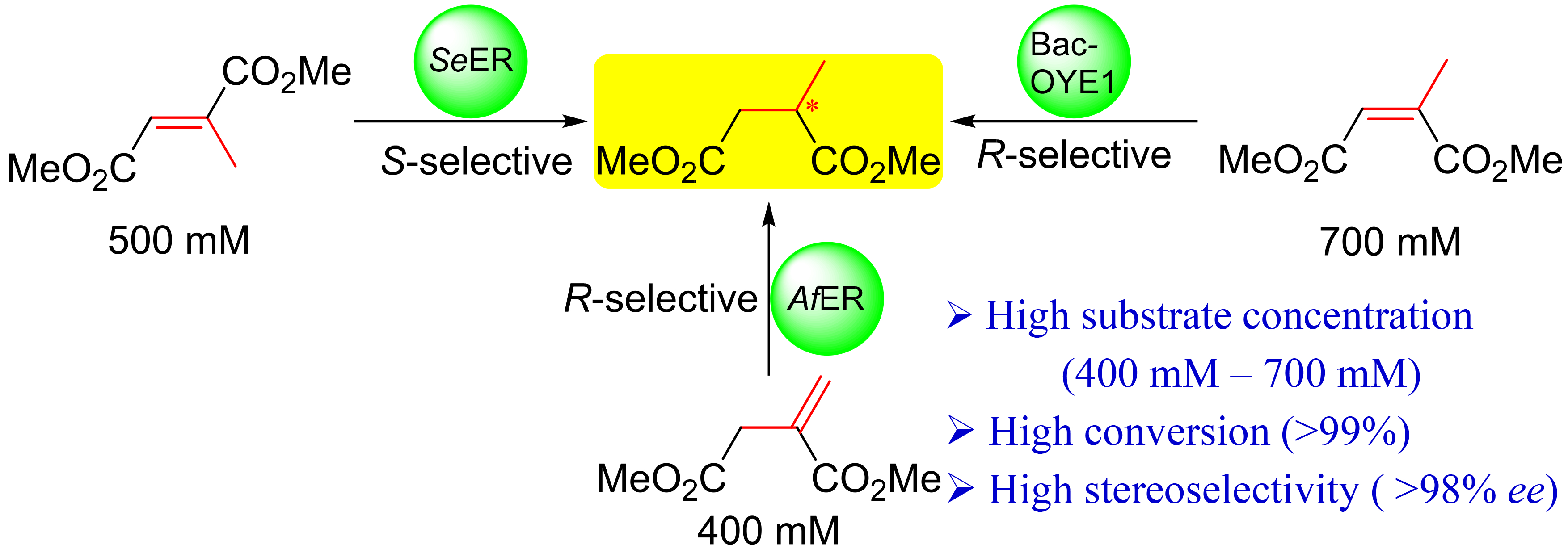{kind=link}
{kind=link}
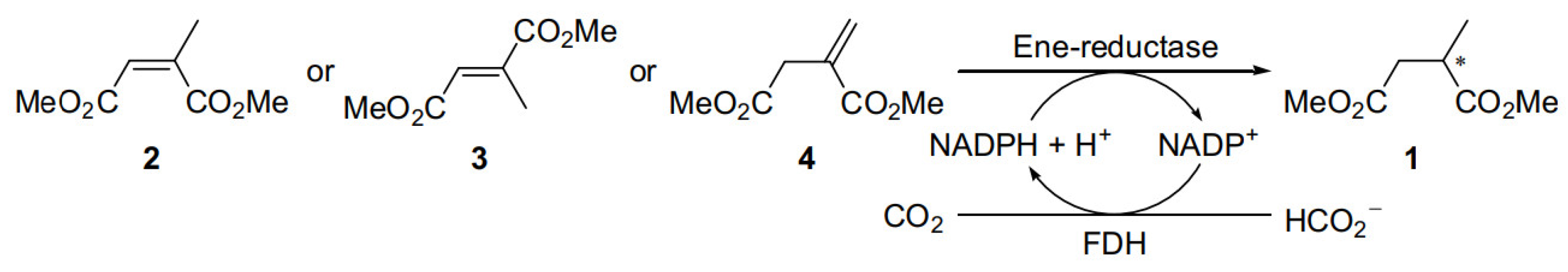{kind=link}
| ERs | 2 | 3 | 4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Conc. (mM) | Conv. (%) 2 | ee (%) 2 | Conc. (mM) | Conv. (%) 2 | ee (%) 2 | Conc. (mM) | Conv. (%) 2 | ee (%) 2 | |
| Bac-OYE1 | 700 | >99 | 99R | 300 | >99 | 99S | 50 | 97 | 99R |
| YqjM | 500 | 75 | 99R | 300 | >99 | 99S | 400 | 99 | 99R |
| BzER | 500 | >99 | 99R | 300 | >99 | 99S | 30 | >99 | 99R |
| AfER | 500 | 62 | 99R | 300 | >99 | 99S | 400 | >99 | 99R |
| SeER | 50 | 58 | 54R | 500 | 99 | 98S | 50 | 27 | 99R |
| Enzyme | Specific Activity (U/mg) | ||
|---|---|---|---|
| 2 | 3 | 4 | |
| Bac-OYE1 | 2.17 | 0.36 | 0.95 |
| YqjM | 1.06 | 0.05 | 0.61 |
| BzER | 0.55 | 0.10 | 0.53 |
| AfER | 0.25 | 0.14 | 1.43 |
| SeER | 0.03 | 0.38 | 0.02 |
| Enzyme |  |  |  | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (mM−1 min−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1· min−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1· min−1) | |
| Bac-OYE1 | 44.5 | 1.57 | 2.12 | 6.0 | 0.13 | 1.30 | 63.4 | 0.75 | 0.71 |
| YqjM | 34.4 | 0.35 | 0.61 | - 2 | - 2 | - 2 | 76.4 | 0.33 | 0.26 |
| BzER | 30.6 | 0.27 | 0.53 | 5.5 | 0.08 | 0.87 | 69.7 | 0.38 | 0.33 |
| AfER | 82.8 | 0.26 | 0.19 | 3.6 | 0.10 | 1.67 | 42.5 | 1.10 | 1.55 |
| SeER | - 2 | - 2 | - 2 | 10.0 | 0.30 | 1.80 | - 2 | - 2 | - 2 |
| Sub. | Prod. | Conc. | Vol. (mL) | ERs | Conv. (%) 2 | Yield (%) 3 | ee (%) 2 |
|---|---|---|---|---|---|---|---|
| 3 | (S)-1 | 500 mM (3.95 g, 79.1 g/L) | 50 | SeER | >99 | 80 | 98 |
| 2 | (R)-1 | 700 mM (11.07 g, 110.7 g/L) | 100 | Bac-OYE1 | >99 | 86 | 99 |
| 4 | 400 mM (3.16 g, 63.3 g/L) | 50 | AfER | >99 | 77 | 99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Li, J.; Cui, Y.; Wang, M.; Feng, J.; Yao, P.; Wu, Q.; Zhu, D. Asymmetric Synthesis of Both Enantiomers of Dimethyl 2-Methylsuccinate by the Ene-Reductase-Catalyzed Reduction at High Substrate Concentration. Catalysts 2022, 12, 1133. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101133
Li J, Li J, Cui Y, Wang M, Feng J, Yao P, Wu Q, Zhu D. Asymmetric Synthesis of Both Enantiomers of Dimethyl 2-Methylsuccinate by the Ene-Reductase-Catalyzed Reduction at High Substrate Concentration. Catalysts. 2022; 12(10):1133. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101133
Chicago/Turabian StyleLi, Jiacheng, Jianjiong Li, Yunfeng Cui, Min Wang, Jinhui Feng, Peiyuan Yao, Qiaqing Wu, and Dunming Zhu. 2022. "Asymmetric Synthesis of Both Enantiomers of Dimethyl 2-Methylsuccinate by the Ene-Reductase-Catalyzed Reduction at High Substrate Concentration" Catalysts 12, no. 10: 1133. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101133