
A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst

Department of Chemical Engineering, College of Engineering, King Faisal University, Al Ahsa 31982, Saudi Arabia
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(10), 1174; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101174
Submission received: 12 September 2022
/
Revised: 25 September 2022
/
Accepted: 30 September 2022
/
Published: 5 October 2022
(This article belongs to the Special Issue Reactivity and Structural Dynamics of Catalysts)
Abstract
:Methanol steam reforming (MSR) is a promising technology for on-board hydrogen production in fuel cell applications. Although traditional Cu-based catalysts demonstrate high catalytic activity and selectivity towards CO2 relative to CO, which is produced via methanol decomposition, they suffer from poor thermal stability and rapid coke formation. Nickel phosphides have been widely investigated in recent years for many different catalytic reactions owing to their remarkable activity and selectivity, as well as their low cost. In this work, we present a mechanistic study of methanol decomposition and MSR pathways on Ni2P using density functional theory (DFT) calculations. DFT-predicted enthalpic barriers indicate that MSR may compete with methanol decomposition on Ni2P, in contrast to other transition metals (e.g., Pt, Pd, and Co) which primarily decompose methanol into CO. The formaldehyde intermediate (CH2O*) can react with co-adsorbed hydroxyl (OH*) from water dissociation to produce H2COOH* which then undergoes subsequent dehydrogenation steps to produce CO2 via H2COOH*→ HCOOH* → HCOO* → CO2. We also examined the conversion of CO into CO2 via the water–gas shift (WGS) reaction, but we ruled out this pathway because it exhibits high activation barriers on Ni2P. These findings suggest that Ni2P is a promising new catalyst for MSR.
1. Introduction
Proton-exchange membrane (PEM) fuel cells are widely considered to have significant potential for the future of energy [1,2]. Although PEM fuel cells are highly energy-efficient and environmentally friendly, mechanical and safety challenges associated with hydrogen storage hinder their range of applications [3,4,5]. To overcome these challenges, methanol steam reforming (MSR) has emerged as a promising technology for on-board hydrogen production from liquid hydrocarbons [6,7,8]. Methanol has a high hydrogen to carbon ratio with no carbon–carbon bonds and can be easily stored and handled [9,10]. Hydrogen can be produced via methanol decomposition:
The formation of carbon monoxide poisons at the Pt-based anodes renders this pathway unsuitable for on-board fuel cell applications [11,12]. Therefore, this reaction must be followed by the water–gas shift (WGS) reaction:
to convert CO into CO2 [13,14]. Alternatively, a high yield of hydrogen can be directly produced from methanol via steam reforming:
while minimizing the production of CO, thus making MSR an attractive process for hydrogen fuel cell applications.
CH3OH → 2H2 + CO
CO + H2O → H2 + CO2
CH3OH + H2O → 3H2 + CO2
Methanol and other alcohols, such as ethanol and propanol, steam reforming reactions have been studied extensively over the past two decades [9,15,16,17,18,19,20,21,22]. Cu/ZnO catalysts are considered the traditional catalysts for MSR, which selectively produce CO2 (>99%) instead of CO with high catalytic activity [5]. However, they suffer from poor thermal stability due to metal sintering at the operating temperatures, in addition to coking and rapid deactivation [22,23,24]. Thus, finding new and more stable catalysts for MSR has become increasingly important. Subsequent studies have focused on Groups 8–10 metal catalysts [4,20,21], particularly Pd/ZnO and PdZn alloys [18,19]. Generally, Groups 8–10 metals predominantly produce CO via methanol decomposition and have poor selectivity towards CO2. Pd/ZnO alloys have shown higher selectivity towards CO2 via MSR but with lower catalytic activity, especially when PdZn alloys are formed by previously reducing the catalysts [25,26,27,28]. Catalyst performance improvement is focused on enhancing H2 production rate, CO2 selectivity, and long-term thermal stability.
Transition metal phosphides (TMPs) are a versatile class of catalysts [29] that have been recently investigated for many different reactions, such as hydrodesulfurization [30], hydrodenitrogenation [31], hydrodeoxygenation [32], and the water–gas shift reaction [33,34]. They exhibit unique selectivity and resistance to coking. For example, nickel phosphides (Ni2P and Ni12P5) have remarkable selectivity towards cleaving the hindered C–O bonds (a near 50 times increase in selectivity) with an increasing P:Ni ratio in the hydrodeoxygenation of biomass-derived species when compared to pure Ni, which only cleaves the unhindered bonds [35,36]. The water–gas shift reaction was also investigated on Ni2P using a combination of experimental and theoretical methods. These studies show that Ni2P resistance to coke formation surpass those of pure Ni or even Cu, the traditional WGS reaction catalyst [33,34]. The unique characteristics of nickel phosphides were attributed to the role of phosphorus atoms in altering the electronic nature and geometry of Ni atoms, as well as disrupting and dispersing the Ni ensembles at the surface.
The mechanism of MSR on Cu-based catalysts is relatively well established [37,38]. Scheme 1 depicts the main possible reaction network of methanol decomposition, MSR, and WGS reactions examined in this study. On Cu-based catalysts, it has been proposed that methanol first undergoes O–H bond cleavage to form surface methoxy (CH3O*) followed by C–H bond cleavage to form formaldehyde (CH2O*), a key intermediate in determining the selectivity [14,16]. It can either undergo further dehydrogenation and decompose to CO or, when water is fed under steam reforming conditions, it can react with OH* to form H2COOH* which eventually produces CO2. Another possible pathway to form CO2 is through methanol decomposition to CO* followed by the WGS reaction because the operating temperature of MSR overlaps with that of the low-temperature WGS reaction. Prior studies indicate that Cu predominantly produces CO2 via MSR and/or WGS depending on the operating conditions, while Groups 8–10 metals (e.g., Pd and Co) predominantly produce CO via methanol decomposition [4,5,15,25]. Here, we use density functional theory (DFT) calculations to examine the mechanisms of methanol decomposition, MSR, and the WGS reaction over Ni2P catalyst. We show that methanol decomposition and MSR pathways both have relatively similar barriers on Ni2P, in contrast to other transition metals (except Cu) where methanol decomposition into CO is more dominant.
2. Results and Discussion
2.1. Structures and Energetics of Adsorbed Intermediates
The optimized structures and calculated binding energies of various intermediates involved in the MSR reaction on Ni2P(001) are discussed in this section. Multiple configurations have been examined for each intermediate to determine the most stable adsorption mode, and the binding energies (ΔEads) were calculated using Equation (4). Table 1 lists all of the intermediates examined in this study, and their binding energies; Figure 1 shows the optimized structure for each intermediate.
Water (H2O; Figure 1b), methanol (CH3OH; Figure 1e), and formic acid (HCOOH; Figure 1l) show weak adsorption energies (around −24 kJ mol−1), and they bind to the metal atop site (M1) through their oxygen atoms (the oxygen from HCO* in the case of HCOOH). The phosphorus atop site (P1) was attempted for these three species, but they eventually desorb from the surface after geometric convergence, indicating that these three species bind weakly to Ni sites only. Formaldehyde (CH2O; Figure 1h), another saturated species and a key intermediate, also binds weakly to the metal three-fold hollow site (M3) in a bidentate fashion, with the oxygen atom sitting above a bridge site and the carbon atom on an atop site. Its weak binding energy of −25 kJ mol−1 is consistent with the weak binding energy of formaldehyde on Cu [16] relative to other transition metals such as Co [15], Pd [14], and Pt [39]. The binding strength of CH2O* on the surface plays an important role in determining the selectivity towards CO2 relative to CO, as shown previously [15]. In the MSR pathway, CH2O* has to partially desorb from the surface (with only the O atom bound to the surface), then it can react with an adjacent OH* to form H2COOH*, which subsequently dehydrogenates to form CO2 (Scheme 1). Stronger CH2O* binding hinders its partial desorption and subsequent reaction with OH*, leading to CO* formation via the methanol decomposition route. Therefore, metals that bind CH2O* weakly such as Cu increase the selectivity towards CO2, while metals that bind CH2O* more strongly such as Pt, Pd, and Co predominantly produce CO. Similar findings were also reported on α-MoC(100) and α-MoC(111) surfaces, in which weaker CH2O* binding on the α-MoC(100) surface leads to a facile reaction with OH* compared with the α-MoC(111) surface that strongly binds CH2O* [40].
All other unsaturated species typically bind to the surface much more strongly. Hydroxyl (OH*; Figure 1c), methoxy (CH3O*; Figure 1g), and hydroxymethoxy (H2COOH*; Figure 1k), preferentially adsorb on the metal three-fold hollow sites (M3) through their oxygen atoms in a monodentate fashion with binding energies of −294, −204, and −194 kJ mol−1, respectively. Dioxymethylene (H2COO*; Figure 1m) binds very strongly to the Ni2P(001) surface; it binds to the M3 site, but in a bidentate fashion, which is consistent with previous studies on other transition metals and metal carbides [14,15,16,40]. CH2OH*, CHO*, COOH*, HCOO*, and O* all bind to the bridging sites and exclusively to Ni atoms, except atomic oxygen, which binds to the Ni-P bridge site (Figure 1d); it is the only species in which the phosphorus atom directly participates in binding. Carbon monoxide (CO*; Figure 1j) binds to the metal atop site (M1) with an adsorption energy of −109 kJ mol−1. Overall, these findings show that the trends in adsorbates binding energy on Ni2P(001) are similar to those reported on other transition metals and metal carbides, and ΔEads values calculated here on Ni2P are in range with those reported on Cu.
2.2. Reaction Pathways
Here, we describe the reaction pathways for methanol decomposition, methanol steam reforming, and the water–gas shift reaction on Ni2P(001) surface based on the reaction network displayed on Scheme 1. All elementary steps examined in this work are listed in Table 2 along with their forward activation barriers and reaction energies. Multiple configurations have been examined on different surface sites for each elementary step to find the most stable transition state. These ΔHact and ΔHrxn values listed on Table 2, however, denote differences between a transition state and the precursor reactant, or the product and the precursor reactant for that elementary step. They do not include diffusion of reactants or products from less stable into more stable adsorption sites or vice versa before and after the reaction. Diffusion steps are usually assumed to be fast and have low barriers [15].
2.2.1. Water Dissociation
Water dissociates on the Ni2P(001) surface with an activation enthalpic barrier (ΔHact) of 91 kJ mol−1 and is nearly thermoneutral with ΔHrxn = 5 kJ mol−1 (Table 2; reaction 17). This reaction starts with an H2O* adsorbed on a metal atop site (M1) through the oxygen atom and proceeds to cleave the O–H bond on a metal three-fold hollow site (M3), then OH* and O* diffuse into two adjacent M3 sites (Figure S1; Supporting Information). Further dissociation of OH* into O* and H* (reaction 18) is highly unfavorable with an activation barrier of 161 kJ mol−1, but O* can be produced via the disproportion of two OH* (reaction 19) with an activation barrier of 63 kJ mol−1. In comparison with Cu, the barrier of H2O dissociation on Ni2P is lower by more than 30 kJ mol−1 [14,41]. An adjacent H2O molecule can also stabilize the O–H bond activation in water via hydrogen bonding as shown previously [15,42]. H2O-assisted water dissociation (reaction 20) occurs with two adjacent H2O molecules stabilized by a hydrogen bond (Figure S1; Supporting Information). This reaction has an enthalpic barrier of 68 kJ mol−1 on Ni2P(001), 23 kJ mol−1 lower than that of direct water dissociation (reaction 17). Therefore, hydrogen bonding can promote O–H bond activation in water.
2.2.2. Methanol Decomposition
Adsorbed CH3OH* can react with a vacant site on the Ni2P(001) surface to undergo C–H bond cleavage which has a barrier of 114 kJ mol−1, and it is strongly endothermic with a reaction energy of 63 kJ mol−1 (Table 2; reaction 1). Alternatively, CH3OH* undergoes O–H bond cleavage with an activation barrier of 98 kJ mol−1 and a reaction energy of 9 kJ mol−1 (Table 2; reaction 2), suggesting that O–H bond activation is more facile than C–H bond activation in methanol on Ni2P. To compare the different reaction pathways depicted on Scheme 1, we examined the effective enthalpy (ΔH҂) calculated using Equation (5) which is defined as the energy of forming the transition state (or adsorbed species) and a stoichiometric amount of gas-phase H2 (each H atom removed is desorbed from the surface) relative to gas-phase methanol and water (if the reaction involves water). Figure 2 shows the DFT-predicted effective enthalpy diagram for the reactions network displayed on Scheme 1. Here, we consider the most stable adsorption state for each intermediate discussed in Section 2.1.
Methanol has an adsorption enthalpy of −14 kJ mol−1, and there are two possible pathways for its dehydrogenation to formaldehyde (CH2O*). It can undergo C–H bond activation to form CH2OH* and H* with a ΔH҂ value of 100 kJ mol−1 (Figure 2). This H* atom is then desorbed from the surface to form ½ H2(g), leaving CH2OH* on the surface with an effective enthalpy of 61 kJ mol−1. The O–H bond cleavage in CH2OH* requires an activation barrier of 146 kJ mol−1 to form CH2O*. Another route to formaldehyde which is more favorable is through O–H bond cleavage in methanol with a barrier of 84 kJ mol−1 to form surface methoxy (CH3O*), which then undergoes C–H bond cleavage with a barrier of 75 kJ mol−1 to form CH2O*. This is consistent with the preference of formaldehyde formation through CH3O* on Cu instead of CH2OH*, which is more favorable on Pd [14,43,44].
Formaldehyde (CH2O*) is an important intermediate in determining the selectivity towards CO or CO2. Subsequent dehydrogenation steps lead to the decomposition to CO. C–H bond cleavage in CH2O* requires an effective activation barrier of 78 kJ mol−1 (Figure 3a) to form CHO*, which further decomposes into CO* with a barrier of 89 kJ mol−1 (Figure 2). CO* can desorb from the surface as the final product or it can be converted into CO2 through the WGS reaction discussed in Section 2.2.4. Next, we examine the reaction of CH2O* with co-adsorbed OH* to form CO2.
2.2.3. Methanol Steam Reforming
Instead of further dehydrogenation of CH2O* which yields CO, CH2O* can react with co-adsorbed OH* from water dissociation to form hydroxymethoxy (H2COOH*). Prior DFT studies have shown that Cu has a unique selectivity towards H2COOH* formation relative to CH2O* decomposition (by >50 kJ mol−1) compared to other transition metals [14,16]. Here, on Ni2P(001), we also observe a similar preference for H2COOH* formation in terms of intrinsic barriers and reaction energies shown in Table 2. H2COOH* formation (reaction 7) is highly exothermic (−40 kJ mol−1), and it has an intrinsic activation enthalpy (ΔHact) of 5 kJ mol−1 compared to 17 kJ mol−1 for CH2O* dehydrogenation (reaction 5). CH2O adsorbs weakly on the surface as shown in Table 1 (ΔEads = −25 kJ mol−1), and it partially desorbs from the surface with only the oxygen atom bounding to a metal atop site (Figure 3b) before it reacts with an adjacent OH*. This reaction requires an effective enthalpic barrier (ΔH҂) of 82 kJ mol−1, only 4 kJ mol−1 higher than that of CH2O* dehydrogenation (Figure 2). This small difference in barriers is within the margin of error expected from DFT calculations, indicating that both pathways may compete on Ni2P.
We have also examined the possible H2COOH* decomposition pathways to CO2 (Figure 2). Dehydrogenation of H2COOH* proceeds by either breaking the O–H bond or the C–H bond, but O–H activation requires a much higher barrier (by 58 kJ mol−1) than C–H bond activation to form formic acid (HCOOH*), in agreement with previous findings on Cu [14,16]. After HCOOH* is formed, O–H bond cleavage becomes more favorable with an effective barrier of 87 kJ mol−1 compared to 111 kJ mol−1 for C–H bond cleavage. The final product of methanol steam reforming (CO2*) is then produced by dehydrogenation of HCOO* with a barrier of 94 kJ mol−1, which is the highest effective barrier in the MSR pathway. The highest effective barrier in the methanol decomposition pathway, on the other hand, is for the CO* formation from CHO* which requires an effective barrier of 89 kJ mol−1. This small difference of 5 kJ mol−1 is again within the margin of error expected from DFT calculations, suggesting that both pathways, methanol decomposition and MSR, may compete on Ni2P.
2.2.4. Water-Gas Shift Reaction
Previous studies have suggested that the water-gas shift (WGS) reaction may play a role in dictating the CO/CO2 selectivity during MSR [38,45]. The resulting CO* from the methanol decomposition route can desorb from the surface as the final product with a desorption energy of 109 kJ mol−1 (Table 1). Alternatively, it can be converted into CO2 through the reaction with O* or OH*. Here, we examined two different mechanisms for the WGS reaction on Ni2P: the carboxyl mechanism (CO + OH → COOH → CO2 + H) and the redox mechanism (CO + O → CO2). In the carboxyl mechanism, the product of methanol decomposition (CO*) reacts with an adjacent hydroxyl (OH*) to form COOH* with a ΔH҂ value of 67 kJ mol−1 (Figure 2). COOH* then undergoes a dehydrogenation reaction to form the product CO2* with a large barrier of 164 kJ mol−1. Instead, CO* may react directly with a co-adsorbed O* atom to form CO2 in the redox mechanism with an effective barrier of 155 kJ mol−1. Given the large barriers for both pathways compared with the MSR pathway or even with the energy required to desorb CO* from the surface (109 kJ mol−1), we conclude that the WGS reaction may not contribute to CO2 formation on Ni2P.
3. Computational Methods
Periodic plane-wave DFT calculations were performed using the Vienna ab initio simulation package (VASP) [46,47,48,49], as implemented in the computational catalysis interface (CCI) [50]. Plane waves were constructed using projector augmented-wave (PAW) potentials [51,52] with an energy cutoff of 396 eV. The revised Perdew−Burke−Ernzerhof (RPBE) form of the generalized gradient approximation was used to describe exchange and correlation energies [53,54,55]. Gaseous species were modeled within 15 × 15 × 15 Å unit cells of vacuum. The unit cell of bulk Ni2P (space group P62m) was built based on crystallographic data [56], and the lattice parameters were then optimized using DFT as described in more detail in our previous work [35]. The optimized bulk lattice parameters (a = b = 5.87 Å and c = 3.37 Å) were consistent with previous DFT and experimental studies (a = b = 5.86 Å and c = 3.38 Å) [57,58]. Calculations were not run spin-polarized because Ni2P was previously shown not to have spin-polarized electronic structures [35,57].
The Ni2P(001) surface with the Ni-rich termination (Figure 4a) was used for all calculations because it has the lowest surface formation energy [35]. The system was modeled as a 2 × 2 periodic lattice with two repeating units and a 10 Å of vacuum in the z-direction (Figure 4b). The atoms in the bottom unit (two atomic layers) were fixed in their bulk positions while all other atoms were relaxed. A 3 × 3 × 1 Monkhorst−Pack sampling of the first Brillouin zone (k-point mesh) was used during geometric convergence [59,60]. Wave functions were converged to within 10−6 eV, and structures were relaxed until all forces on unconstrained atoms were <0.05 eVÅ−1.
Transition-state structures were obtained for each elementary reaction by using the nudged elastic band (NEB) method and the dimer method [61,62,63]. The NEB method was carried out using 8 images, and wave functions were converged to within 10−4 eV. The maximum force on each atom was converged to <0.5 eV Å−1. These convergence criteria provided an estimate of the reaction path and initial transition-state structures and reaction modes. The dimer algorithm was then used with wave functions converged to within 10−6 eV, and the maximum force on each atom was converged to <0.05 eV Å−1.
Frequency calculations were performed on gas-phase molecules and all optimized adsorbed species to determine zero-point vibrational energies; vibrational, translational, and rotational enthalpy; and free energy. These terms were then used, together with electronic energies (E0, provided by VASP), to estimate enthalpies for the reactants, products, and transition states at 573 K (the typical temperature for MSR). The adsorbates binding energy (ΔEads) relative to the gas-phase species is defined as:
Thus, more negative values indicate stronger binding. DFT-derived intrinsic activation enthalpy (ΔHact) and enthalpy of reaction (ΔHrxn) denote differences between a transition state, or the product and the precursor reactant for that elementary step. Effective enthalpy barriers (ΔH҂) are defined as the enthalpy of forming the transition state and a stoichiometric amount of gas-phase H2 from gas-phase methanol and water (if the reaction involves water):
where λ is the number of H2 molecules evolved during the reaction from the dehydrogenation steps. Further details of the computational methods (Section S1) and the energy correction terms (Table S1) are reported in the Supporting Information (SI).
4. Conclusions
Methanol decomposition, methanol steam reforming, and the water–gas shift reaction pathways over the Ni2P catalyst were investigated using DFT. Calculated effective enthalpy barriers (ΔH҂) indicate that methanol decomposition occurs through CH3OH* → CH3O* → CH2O* → CHO* → CO*. In MSR, formaldehyde (CH2O*) can react with a co-adsorbed OH* to produce H2COOH*, which then undergoes subsequent dehydrogenation steps to produce CO2 via H2COOH*→ HCOOH* → HCOO* → CO2. Effective barriers for both pathways are within 5 kJ mol−1, suggesting that they both may compete on Ni2P, unlike other transition metals where methanol decomposition is much more dominant. We also examined CO2 formation via the WGS reaction; however, the high calculated activation barriers preclude this pathway from contributing to CO2 formation on Ni2P. These findings, taken together, suggest that Ni2P with its unique selectivity and resistance to coke formation is a promising alternative for Cu in methanol steam reforming.
Supplementary Materials
The following Supporting Information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12101174/s1, Section S1: Details of density functional calculations of thermochemical properties; Table S1: Electronic energy, ZPVE, and vibrational, translational, and rotational enthalpy and free energy at 573 K in eV. Figure S1: Reactants, transition state, and products structures for all reactions listed on Table 2.
Author Contributions
Conceptualization, A.A. and Z.A.; methodology, A.A.; software, A.A.; validation, A.A. and Z.A.; formal analysis, A.A.; investigation, A.A. and Z.A.; resources, A.A.; data curation, A.A.; writing—original draft preparation, A.A.; writing—review and editing, A.A. and Z.A.; visualization, A.A. and Z.A.; supervision, A.A.; project administration, A.A.; funding acquisition, A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia (Grant No. 1136).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Manoharan, Y.; Hosseini, S.E.; Butler, B.; Alzhahrani, H.; Senior, B.T.F.; Ashuri, T.; Krohn, J. Hydrogen Fuel Cell Vehicles; Current Status and Future Prospect. Appl. Sci. 2019, 9, 2296. [Google Scholar] [CrossRef] [Green Version]
- Kurnia, J.C.; Chaedir, B.A.; Sasmito, A.P.; Shamim, T. Progress on Open Cathode Proton Exchange Membrane Fuel Cell: Performance, Designs, Challenges and Future Directions. Appl. Energy 2021, 283, 116359. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shuai, K.; Xu, B. Review on Copper and Palladium Based Catalysts for Methanol Steam Reforming to Produce Hydrogen. Catalysts 2017, 7, 183. [Google Scholar] [CrossRef]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for Methanol Steam Reforming—A Review. Appl. Catal. B 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Rivard, E.; Trudeau, M.; Zaghib, K. Hydrogen Storage for Mobility: A Review. Materials 2019, 12, 1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and Physical Solutions for Hydrogen Storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-Storage Materials for Mobile Applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef]
- Agrell, J.; Birgersson, H.; Boutonnet, M. Steam Reforming of Methanol over a Cu/ZnO/Al2O3 Catalyst: A Kinetic Analysis and Strategies for Suppression of CO Formation. J. Power Sources 2002, 106, 249–257. [Google Scholar] [CrossRef]
- Agrell, J. Production of Hydrogen from Methanol over Cu/ZnO Catalysts Promoted by ZrO2 and Al2O3. J. Catal. 2003, 219, 389–403. [Google Scholar] [CrossRef]
- Levitan, D.; Rozenblit, A.; Laborde, M.; Giunta, P. Self-Sustained Oscillations in the Potential of a CO-Poisoned PEM Fuel Cell: A Model Based on Physical Principles. J. Electroanal. Chem. 2021, 880, 114924. [Google Scholar] [CrossRef]
- Bellows, R.J.; Marucchi-Soos, E.P.; Buckley, D.T. Analysis of Reaction Kinetics for Carbon Monoxide and Carbon Dioxide on Polycrystalline Platinum Relative to Fuel Cell Operation. Ind. Eng. Chem. Res. 1996, 35, 1235–1242. [Google Scholar] [CrossRef]
- Denkwitz, Y.; Karpenko, A.; Plzak, V.; Leppelt, R.; Schumacher, B.; Behm, R. Influence of CO2 and H2 on the Low-Temperature Water–Gas Shift Reaction on Au/CeO2 Catalysts in Idealized and Realistic Reformate. J. Catal. 2007, 246, 74–90. [Google Scholar] [CrossRef]
- Gu, X.-K.; Li, W.-X. First-Principles Study on the Origin of the Different Selectivities for Methanol Steam Reforming on Cu(111) and Pd(111). J. Phys. Chem. C 2010, 114, 21539–21547. [Google Scholar] [CrossRef]
- Luo, W.; Asthagiri, A. Density Functional Theory Study of Methanol Steam Reforming on Co(0001) and Co(111) Surfaces. J. Phys. Chem. C 2014, 118, 15274–15285. [Google Scholar] [CrossRef]
- Lin, S.; Johnson, R.S.; Smith, G.K.; Xie, D.; Guo, H. Pathways for Methanol Steam Reforming Involving Adsorbed Formaldehyde and Hydroxyl Intermediates on Cu(111): Density Functional Theory Studies. Phys. Chem. Chem. Phys. 2011, 13, 9622–9631. [Google Scholar] [CrossRef]
- Sutton, J.E.; Panagiotopoulou, P.; Verykios, X.E.; Vlachos, D.G. Combined DFT, Microkinetic, and Experimental Study of Ethanol Steam Reforming on Pt. J. Phys. Chem. C 2013, 117, 4691–4706. [Google Scholar] [CrossRef]
- Smith, G.K.; Lin, S.; Lai, W.; Datye, A.; Xie, D.; Guo, H. Initial Steps in Methanol Steam Reforming on PdZn and ZnO Surfaces: Density Functional Theory Studies. Surf. Sci. 2011, 605, 750–759. [Google Scholar] [CrossRef]
- Lin, S.; Xie, D.; Guo, H. Pathways of Methanol Steam Reforming on Pdzn and Comparison with Cu. J. Phys. Chem. C 2011, 115, 20583–20589. [Google Scholar] [CrossRef]
- Papavasiliou, J.; Paxinou, A.; Słowik, G.; Neophytides, S.; Avgouropoulos, G. Steam Reforming of Methanol over Nanostructured Pt/TiO2 and Pt/CeO2 Catalysts for Fuel Cell Applications. Catalysts 2018, 8, 544. [Google Scholar] [CrossRef]
- Wu, H.; la Parola, V.; Pantaleo, G.; Puleo, F.; Venezia, A.; Liotta, L. Ni-Based Catalysts for Low Temperature Methane Steam Reforming: Recent Results on Ni-Au and Comparison with Other Bi-Metallic Systems. Catalysts 2013, 3, 563–583. [Google Scholar] [CrossRef] [Green Version]
- Köpfle, N.; Mayr, L.; Schmidmair, D.; Bernardi, J.; Knop-Gericke, A.; Hävecker, M.; Klötzer, B.; Penner, S. A Comparative Discussion of the Catalytic Activity and CO2-Selectivity of Cu-Zr and Pd-Zr (Intermetallic) Compounds in Methanol Steam Reforming. Catalysts 2017, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Twigg, M.V. Deactivation of Copper Metal Catalysts for Methanol Decomposition, Methanol Steam Reforming and Methanol Synthesis. Top. Catal. 2003, 22, 191–203. [Google Scholar] [CrossRef]
- Kurtz, M.; Wilmer, H.; Genger, T.; Hinrichsen, O.; Muhler, M. Deactivation of Supported Copper Catalysts for Methanol Synthesis. Catal. Lett. 2003, 86, 77–80. [Google Scholar] [CrossRef]
- Takezawa, N.; Iwasa, N. Steam Reforming and Dehydrogenation of Methanol: Difference in the Catalytic Functions of Copper and Group VIII Metals. Catal. Today 1997, 36, 45–56. [Google Scholar] [CrossRef]
- Iwasa, N.; Masuda, S.; Ogawa, N.; Takezawa, N. Steam Reforming of Methanol over Pd/ZnO: Effect of the Formation of PdZn Alloys upon the Reaction. Appl. Catal. A Gen. 1995, 125, 145–157. [Google Scholar] [CrossRef]
- Iwasa, N.; Mayanagi, T.; Nomura, W.; Arai, M.; Takezawa, N. Effect of Zn Addition to Supported Pd Catalysts in the Steam Reforming of Methanol. Appl. Catal. A Gen. 2003, 248, 153–160. [Google Scholar] [CrossRef]
- Iwasa, N. New Supported Pd and Pt Alloy Catalysts for Steam Reforming and Dehydrogenation of Methanol. Top. Catal. 2003, 22, 215–224. [Google Scholar] [CrossRef]
- Al-Ali, L.I.; Elmutasim, O.; al Ali, K.; Singh, N.; Polychronopoulou, K. Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds. Nanomaterials 2022, 12, 1435. [Google Scholar] [CrossRef]
- Oyama, S. Effect of Phosphorus Content in Nickel Phosphide Catalysts Studied by XAFS and Other Techniques. J. Catal. 2002, 210, 207–217. [Google Scholar] [CrossRef]
- Li, W.; Dhandapani, B.; Oyama, S.T. Molybdenum Phosphide: A Novel Catalyst for Hydrodenitrogenation. Chem. Lett. 1998, 27, 207–208. [Google Scholar] [CrossRef]
- Bui, P.; Cecilia, J.A.; Oyama, S.T.; Takagaki, A.; Infantes-Molina, A.; Zhao, H.; Li, D.; Rodríguez-Castellón, E.; Jiménez López, A. Studies of the Synthesis of Transition Metal Phosphides and Their Activity in the Hydrodeoxygenation of a Biofuel Model Compound. J. Catal. 2012, 294, 184–198. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Takahashi, Y.; Nakamura, K. Water–Gas-Shift Reaction on a Ni2P(001) Catalyst: Formation of Oxy-Phosphides and Highly Active Reaction Sites. J. Catal. 2009, 262, 294–303. [Google Scholar] [CrossRef]
- Yin, P.; Yang, Y.-S.; Chen, L.-F.; Xu, M.; Chen, C.-Y.; Zhao, X.-J.; Zhang, X.; Yan, H.; Wei, M. DFT Study on the Mechanism of the Water Gas Shift Reaction Over NixPy Catalysts: The Role of P. J. Phys. Chem. C 2020, 124, 6598–6610. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Hibbitts, D.D.; Flaherty, D.W. Mechanisms and Active Sites for C–O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts. ACS Catal. 2018, 8, 7141–7157. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Triezenberg, M.D.; Hibbitts, D.D.; Flaherty, D.W. In Situ Methods for Identifying Reactive Surface Intermediates during Hydrogenolysis Reactions: C–O Bond Cleavage on Nanoparticles of Nickel and Nickel Phosphides. J. Am. Chem. Soc. 2019, 141, 16671–16684. [Google Scholar] [CrossRef]
- Jiang, C.J.; Trimm, D.L.; Wainwright, M.S.; Cant, N.W. Kinetic Study of Steam Reforming of Methanol over Copper-Based Catalysts. Appl. Catal. A Gen. 1993, 93, 245–255. [Google Scholar] [CrossRef]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol–Steam Reforming on Cu/ZnO/Al2O3 Catalysts. Part 2. A Comprehensive Kinetic Model. Appl. Catal. A Gen. 1999, 179, 31–49. [Google Scholar] [CrossRef]
- Błoński, P.; López, N. On the Adsorption of Formaldehyde and Methanol on a Water-Covered Pt(111): A DFT-D Study. J. Phys. Chem. C 2012, 116, 15484–15492. [Google Scholar] [CrossRef]
- Li, J.; Wan, Q.; Lin, G.; Lin, S. DFT Study on the Catalytic Role of α-MoC(100) in Methanol Steam Reforming. Chin. J. Chem. Phys. 2022, 35, 639–646. [Google Scholar] [CrossRef]
- Gokhale, A.A.; Dumesic, J.A.; Mavrikakis, M. On the Mechanism of Low-Temperature Water Gas Shift Reaction on Copper. J. Am. Chem. Soc. 2008, 130, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; Auneau, F.; Delbecq, F.; Sautet, P. C–H versus O–H Bond Dissociation for Alcohols on a Rh(111) Surface: A Strong Assistance from Hydrogen Bonded Neighbors. ACS Catal. 2011, 1, 1430–1440. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Methanol Decomposition on Cu(111): A DFT Study. J. Catal. 2002, 208, 291–300. [Google Scholar] [CrossRef]
- Jiang, R.; Guo, W.; Li, M.; Fu, D.; Shan, H. Density Functional Investigation of Methanol Dehydrogenation on Pd(111). J. Phys. Chem. C 2009, 113, 4188–4197. [Google Scholar] [CrossRef]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol–Steam Reforming on Cu/ZnO/Al2O3. Part 1: The Reaction Network. Appl. Catal. A Gen. 1999, 179, 21–29. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kravchenko, P.; Plaisance, C.; Hibbitts, D. A New Computational Interface for Catalysis. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Larsson, E. An X-ray Investigation of Ni-P System and Crystal Structures of NiP and NiP2. Ark. Kemi 1965, 23, 335. [Google Scholar]
- Ren, J.; Wang, J.; Li, J.; Li, Y. Density Functional Theory Study on Crystal Nickel Phosphides. J. Fuel Chem. Technol. 2007, 35, 458–464. [Google Scholar] [CrossRef]
- Rundqvist, S. X-ray Investigations of Mn3P, Mn2P, and Ni2P. Acta Chem. Scand 1962, 16, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special Points for Brillouin-Zone Integrations”—a Reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
Scheme 1.
Possible reaction pathways to CO and CO2 examined in this study over Ni2P catalysts. Asterisk (*) denotes a surface site.
Scheme 1.
Possible reaction pathways to CO and CO2 examined in this study over Ni2P catalysts. Asterisk (*) denotes a surface site.

Figure 1.
(a–p) The most stable adsorption geometries for all MSR intermediates examined in this study. Shown beneath each image are the adsorption mode (see Section 3 for more details) and the binding energy calculated using Equation (4) in kJ mol−1.
Figure 1.
(a–p) The most stable adsorption geometries for all MSR intermediates examined in this study. Shown beneath each image are the adsorption mode (see Section 3 for more details) and the binding energy calculated using Equation (4) in kJ mol−1.

Figure 2.
DFT-predicted reaction enthalpy diagram for methanol decomposition (black and red), methanol steam reforming (green), and the water–gas shift reaction (blue) pathways. Energies are calculated using Equation (5) at 573 K and each platform represents the most stable adsorption state for each intermediate discussed in Section 2.1. H atoms removed are desorbed from the surface as H2(g) and are not displayed for clarity. Dashed lines show barriers for unfavorable bond activation.
Figure 2.
DFT-predicted reaction enthalpy diagram for methanol decomposition (black and red), methanol steam reforming (green), and the water–gas shift reaction (blue) pathways. Energies are calculated using Equation (5) at 573 K and each platform represents the most stable adsorption state for each intermediate discussed in Section 2.1. H atoms removed are desorbed from the surface as H2(g) and are not displayed for clarity. Dashed lines show barriers for unfavorable bond activation.

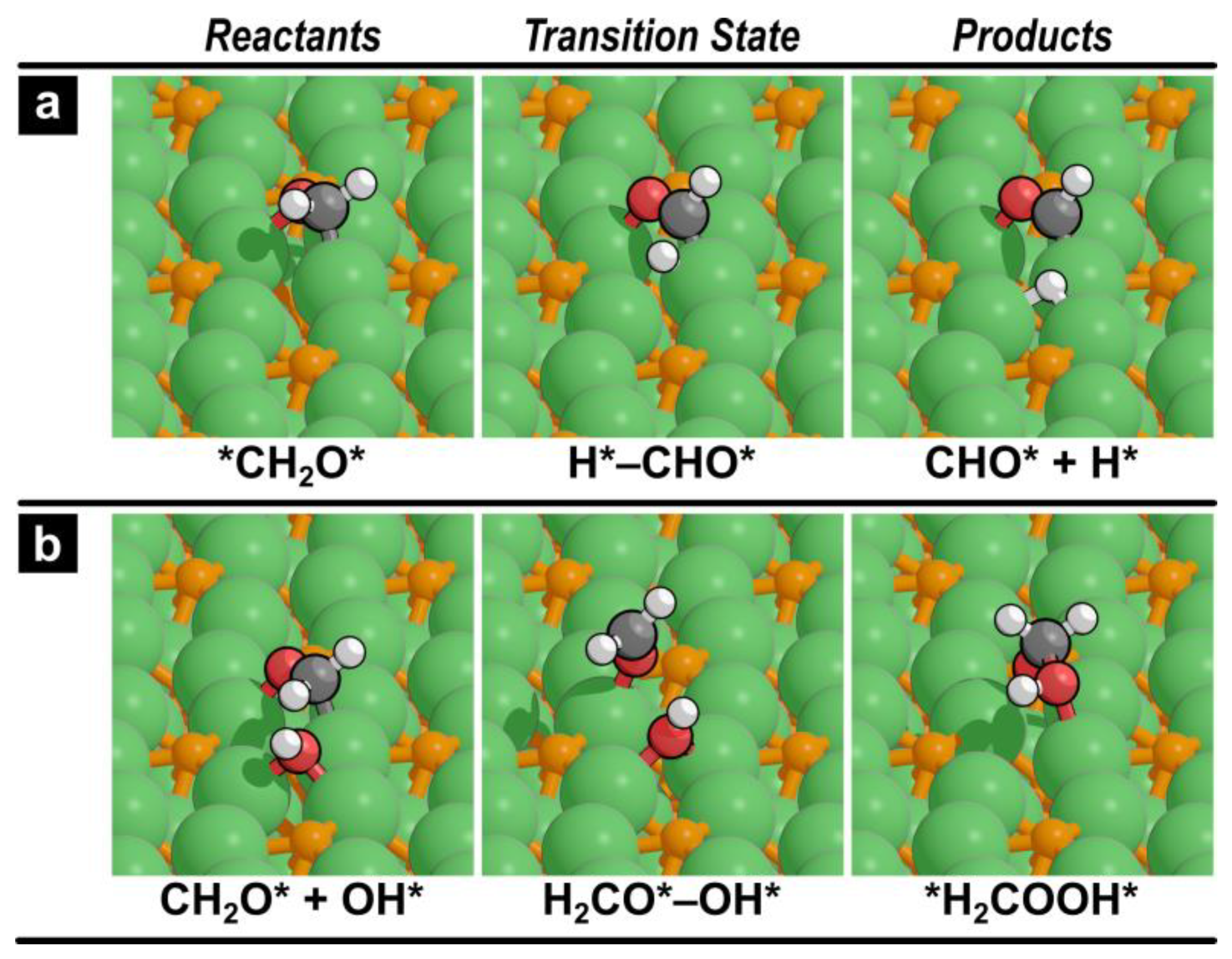
Figure 3.
Reactants, transition state, and product structures for (a) formaldehyde decomposition to CHO* (reaction 5) and (b) for formaldehyde reaction with OH* (reaction 7). Similar images for all other reactions can be found in Figure S1 in the Supporting Information.
Figure 3.
Reactants, transition state, and product structures for (a) formaldehyde decomposition to CHO* (reaction 5) and (b) for formaldehyde reaction with OH* (reaction 7). Similar images for all other reactions can be found in Figure S1 in the Supporting Information.

Figure 4.
(a) Top and (b) side views of the Ni2P(001) model used in this study. The adsorption sites are labeled as M1: metal atop, M2: metal bridge, M3: metal three-fold hollow, MP: metal-phosphorus bridge, and P1: phosphorus atop. The atoms in the bottom unit (two atomic layers) were fixed in their bulk positions while all other atoms were relaxed.
Figure 4.
(a) Top and (b) side views of the Ni2P(001) model used in this study. The adsorption sites are labeled as M1: metal atop, M2: metal bridge, M3: metal three-fold hollow, MP: metal-phosphorus bridge, and P1: phosphorus atop. The atoms in the bottom unit (two atomic layers) were fixed in their bulk positions while all other atoms were relaxed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated adsorbates binding energies ΔEads (Equation (4); kJ mol−1) and their most stable adsorption modes on Ni2P(001).
Table 1.
Calculated adsorbates binding energies ΔEads (Equation (4); kJ mol−1) and their most stable adsorption modes on Ni2P(001).
| Species | Adsorption Mode | ΔEads |
|---|---|---|
| kJ mol−1 | ||
| H* | M3 | −224 |
| H2O* | M1 | −23 |
| OH* | M3 | −294 |
| O* | MP | −489 |
| CH3OH* | M1 | −24 |
| CH2OH* | M2 | −133 |
| CH3O* | M3 | −204 |
| CH2O* | M3 | −25 |
| CHO* | M2 | −161 |
| CO* | M1 | −109 |
| H2COOH* | M3 | −194 |
| HCOOH* | M1 | −24 |
| H2COO* | M3 | −360 |
| COOH* | M2 | −188 |
| HCOO* | M2 | −273 |
| CO2* | Parallel | −1 |
Table 2.
Calculated forward activation enthalpy (ΔHact = HTS − Hreactants) and the reaction (ΔHrxn = Hproducts − Hreactants) for the elementary reactions considered in this study on Ni2P(001) at 573 K.
Table 2.
Calculated forward activation enthalpy (ΔHact = HTS − Hreactants) and the reaction (ΔHrxn = Hproducts − Hreactants) for the elementary reactions considered in this study on Ni2P(001) at 573 K.
| No. | Reaction | ΔHact | ΔHrxn |
|---|---|---|---|
| kJ mol−1 | kJ mol−1 | ||
| 1 | CH3OH → CH2OH + H | 114 | 63 |
| 2 | CH3OH → CH3O + H | 98 | 9 |
| 3 | CH2OH → CH2O + H | 82 | −8 |
| 4 | CH3O → CH2O + H | 73 | 41 |
| 5 | CH2O → CHO + H | 17 | −3 |
| 6 | CHO → CO + H | 19 | −60 |
| 7 | CH2O + OH → H2COOH | 5 | −40 |
| 8 | H2COOH → HCOOH + H | 33 | −38 |
| 9 | H2COOH → H2COO + H | 87 | 43 |
| 10 | HCOOH → COOH + H | 82 | 11 |
| 11 | HCOOH → HCOO + H | 61 | 20 |
| 12 | H2COO → HCOO + H | 13 | −64 |
| 13 | HCOO → CO2 + H | 39 | −33 |
| 14 | COOH → CO2 + H | 115 | −25 |
| 15 | CO + OH → COOH | 69 | 53 |
| 16 | CO + O → CO2 | 128 | 3 |
| 17 | H2O → OH + H | 91 | 5 |
| 18 | OH → O + H | 161 | 81 |
| 19 | OH + OH → H2O + O | 63 | 55 |
| 20 | 2H2O → H2O + OH + H | 68 | 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Almithn, A.; Alhulaybi, Z. A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst. Catalysts 2022, 12, 1174. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101174
AMA Style
Almithn A, Alhulaybi Z. A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst. Catalysts. 2022; 12(10):1174. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101174
Chicago/Turabian StyleAlmithn, Abdulrahman, and Zaid Alhulaybi. 2022. "A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst" Catalysts 12, no. 10: 1174. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12101174
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.