
Photocatalyzed Oxidative Decarboxylation Forming Aminovinylcysteine Containing Peptides †

Graduate School of Pharmaceutical Sciences, Tohoku University, 6-3 Aza-Aoba, Aramaki, Aoba-ku, Sendai 980-8578, Japan
*
Author to whom correspondence should be addressed.
†
This paper is dedicated to late Professor Jiro Tsuji.
Catalysts 2022, 12(12), 1615; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12121615
Submission received: 21 November 2022
/
Revised: 5 December 2022
/
Accepted: 6 December 2022
/
Published: 9 December 2022
(This article belongs to the Special Issue Theme Issue in Memory to Prof. Jiro Tsuji (1927–2022))
Abstract
:The formation of (2S,3S)-S-[(Z)-aminovinyl]-3-methyl-D-cysteine (AviMeCys) substructures was developed based on the photocatalyzed-oxidative decarboxylation of lanthionine-bearing peptides. The decarboxylative selenoetherification of the N-hydroxyphthalimide ester, generated in situ, proceeded under mild conditions at −40 °C in the presence of 1 mol% of eosin Y-Na2 as a photocatalyst and the Hantzsch ester. The following β-elimination of the corresponding N,Se-acetal was operated in a one-pot operation, led to AviMeCys substructures found in natural products in moderate to good yields. The sulfide-bridged motif, and also the carbamate-type protecting groups, such as Cbz, Teoc, Boc and Fmoc groups, were tolerant under the reaction conditions.
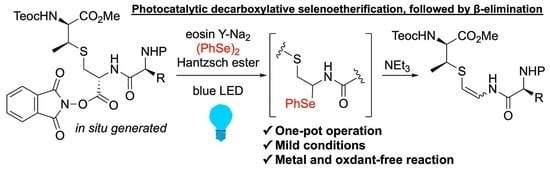
1. Introduction
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are one of the largest classes of natural products that exhibit various biological properties [1,2,3]. Among the diverse substructures found in RiPPs, cross-linked sulfides between two amino acid residues have been identified as an important chemical functionality for constrained conformation in the peptide backbone, providing high target specificity and biological stability [4,5,6,7]. The major components of RiPPs with thioether bonds are (2S,6R)-lanthionine (Lan) and (2S,3S,6R)-3-methyllanthionine (β-MeLan). Thioether-bridged units in Lan/β-MeLan are biosynthetically constructed through a conjugated addition of thiols of cysteine residues to dehydroalanine (Dha)/dehydrobutyrine (Dhb) [8,9]. Intriguingly, (Z)-thioenolates, which are generated by the oxidative decarboxylation of cysteine residues positioned at the C-terminal, also attack Dhb/Dha residues to produce S-[(Z)-aminovinyl]-D-cysteine (AviCys) and (2S,3S)-S-[(Z)-aminovinyl]-3-methyl-D-cysteine (AviMeCys), respectively. Owing to β-thioenamide units including sp2 α-carbons in the peptide backbone, AviCys/AviMeCys are attractive substructures for improving the structural rigidity and drug-like properties of cyclopeptides [10,11,12].
Biosynthesis-inspired approaches to obtain lanthionines have been accomplished through the stereoselective conjugated addition of thiols of cysteine derivatives to Dha/Dhb derivatives [13,14,15], whereas the synthesis of AviCys/AviMeCys motifs in a similar manner is a difficult task due to the chemical instability of thioenolates [16]. Thus, alternative methodologies for constructing AviCys/AviMeCys have been developed to date. There have been several reports on the synthesis of AviCys substructures by condensation using primary amides and α-thioaldehyde/acetals [17,18] and the β-addition of thiyl radicals to terminal ynamides [19]. However, the methodologies for constructing AviMeCys substructures in complex natural products are limited. One of the efficient approaches is a decarboxylative olefination of carboxylic acid derivatives, as in the palladium-catalyzed reaction of allyl β-ketoesters reported by Tsuji [20]. Recently, oxidative decarboxylation/decarbonylation using lanthionine units has been reported for the AviMeCys formation. VanNieuwenhze et al. reported Z-selective AviMeCys formation via nickel(0)-promoted decarbonylation of activated thioesters in short peptide fragments during the synthesis of D-ring in mersacidin (Scheme 1a) [21]. Furthermore, they realized the direct conversion from carboxylic acids through two procedures: (i) Curtius rearrangement using diphenylphosphoryl azide (DPPA), followed by the collapse of the resulting isocyanate, and (ii) oxidative decarboxylation using lead tetraacetate, followed by the elimination of the resulting acetate (Scheme 1b) [22]. Nevertheless, there is no report on the total synthesis of any AviMeCys-containing natural peptides, due to harsh conditions and an excess amount of toxic oxidants during oxidative decarboxylation/decarbonylation steps in the late-stages of the synthesis. Considering the tolerance to functional groups in RiPPs, we focused on photocatalytic reactions mediated by visible light, which have been widely used in the modification of amino acids and peptides [23,24,25]. We envisioned that a radical species, generated from a N-hydroxyphthalimide (NHPI) ester of the corresponding lanthionine by oxidative decarboxylation [26], can be readily trapped in the presence of oxidation-sensitive sulfides under mild conditions. The following β-elimination with a weak base would yield the desired β-thioenamide motifs, suppressing the retro-thio-Michael reaction (Scheme 1c). Herein, we report the β-thioenamide formation through the photocatalyzed oxidative decarboxylation of lanthionine derivatives. A series of reactions were conducted in a one-pot operation under mild conditions, providing AviMeCys units with functional group compatibility.
2. Results and Discussion
Our study began with the preparation of the lanthionine-containing peptides 1, as shown in Scheme 2. According to the procedure reported by VanNieuwenhze [27], the regioselective ring-opening of the N-{2-(trimethylsilyl)ethoxycarbonyl} (Teoc)-protected aziridine 2, which was prepared from D-threonine, with Fmoc-Cys-OH (3) was performed in the presence of indium(III) chloride, providing the lanthionine derivative 4 in a 54% yield. The subsequent protection of the carboxylic acid group with a methoxymethyl (MOM) group provided the MOM ester 5 in a 99% yield. The removal of the Fmoc group in 5 with 20% diethylamine/acetonitrile, followed by the coupling of the resulting amine with N-protected amino acids afforded the peptides 6a–f over two steps. Finally, the MOM group in 5 was removed under acidic conditions, yielding the carboxylic acids 1a–f.
Next, we surveyed the formation of the AviMeCys unit using 1a as the model lanthionine-bearing peptide as shown in Scheme 3. Given the immediate capture of the resulting radical species during the oxidative decarboxylation [28,29,30], diphenyl diselenide was selected as a radical trapping agent [31,32]. In addition, we envisioned that the resulting N,Se-acetal would be converted into the corresponding β-thioenamide without losing the β-methylcysteine unit due to high leaving activity of phenylselenolates [33,34,35]. According to the reported procedures [32,36,37], the NHPI ester 7a, prepared from 1a in situ, was treated with 1 mol% of [Ru(bpy)3](PF6)2 as the photocatalyst in the presence of the Hantzsch ester and diphenyl diselenide under 40 W blue light-emiting diode (LED) irradiation. The following β-elimination of the obtained selenoether 8a by treatment with triethylamine furnished the β-thioenamieds (Z)-9a and (E)-9a in 23% and 13% yields, respectively. The geometry of olefins in (Z)-9a and (E)-9a was determined by 1H nuclear magnetic resonance (NMR) spectroscopy through the coupling constants (3JH,H = 7.2 Hz for (Z)-9a, and 13.8 Hz for (E)-9a) of isolated compounds [19].
As a moderate yield was observed, we conducted the screening of photocatalysts, as shown in Table 1. When selenoetherification, followed by β-elimination was conducted in a one-pot operation, the combined yields of (Z)-9a and (E)-9a were slightly up to 49% (entry 1). After the optimization of metal and organic photosensitizers, eosin Y-Na2 [38] promoted the transformation to increase the yields by up to 59% (entries 2–4). Intriguingly, selenoetherification of 7a proceeded without eosin Y-Na2, albeit with slightly lower yields, suggesting that the formation of the electron donor-acceptor (EDA) complex between the NHPI and Hantzsch esters should promote the reaction (entry 5) [39,40]. No product was obtained in the absence of the blue light (entry 6).
To further improve the yield, we optimized the reaction conditions using eosin Y-Na2, and the results are summarized in Table 2. Using other solvents, such as CH2Cl2, MeCN, N,N-dimethylaniline (DMA) and dimethyl sulfoxide (DMSO), instead of N,N-dimethylformamide (DMF) was fruitless (entry 1 vs, entries 2–5). As reductants, 1-benzyl-1,4-dihydronicotinamide and γ-terpinene decreased the yield (entry 1 vs, entries 6 and 7). Notably, N,N-diisopropylethylamine (DIEA), widely used for photocatalytic reactions, involved the decomposition of the NHPI ester 7a because of its strong basicity (entry 8). The yield increased to 68% when the amount of the Hantzsch ester was reduced to 1.0 equiv (entry 9). Excess amounts of the Hantzsch ester may interfere with the capture of the resulting radical species by diphenyl diselenide [41,42]. Further reduction of the Hantzsch ester decreased the yield (entries 10 and 11). With a decrease in the reaction temperature at −40 °C, the yield was up to 74% (entry 12). In contrast, selenoetherification did not complete at −78 °C (entry 13). Thus, we determined that the optimized condition is observed in entry 12. Our developed methodology was performed on a 1.0 mmol scale, giving 9a in a moderate yield (51%, entry 14).
The substrate scope for our developed AviMeCys formation is shown in Scheme 4. Carbamate-type protecting groups, such as Cbz, Teoc, Boc and Fmoc groups, were tolerant under the reaction conditions, providing the corresponding β-thioenamides 9a–c in moderate to good yields (37–74%). AviMeCys substructures in natural products, such as 9d for cacaodin [43], 9e for mersacidin [44], and 9f for lexapeptide [45], were obtained from lanthionines 1d–1f in 58–68% yields.
A plausible reaction mechanism of the photocatalytic synthesis of AviMeCys is depicted in Scheme 5 according to the above results and previous reports on decarboxylative selenoetherification [31,46]. Given that the reaction proceeded without photocatalysts, we assumed the formation of an EDA complex between the NHPI and Hantzsch esters [39,40]. Photoirradiation induces intramolecular single-electron transfer, generating a phthalimide radical anion B with a dihydropyridine radical cation A. The resulting B undergoes decarboxylation to form a radical species D and a phthalimide anion C. The radical D is then captured by diphenyl diselenide to form a N,Se-acetal F with a seleno radical E. The β-elimination with the N,Se-acetal F in the presence of Et3N affords the corresponding AviMeCys (G) (Scheme 5a). A (Z)-isomer will be obtained with slight priority because of the electrostatic attraction between a sulfur atom and the amide moiety [17]. The resulting A, C, and E are converted into phthalimide, pyridine derivative and phenylselenol, respectively, through two possible pathways. Although the radical-quenching of A and E automatically occurs (Scheme 5c), photocatalysts may mediate this step to improve the yields (Scheme 5b).
3. Materials and Methods
3.1. General Techniques
All commercially available reagents were purchased from commercial suppliers and used as received. Dry THF and CH2Cl2 (Kanto Chemical Co., Inc., Tokyo, Japan) were obtained by passing commercially available pre-dried, oxygen-free formulations. DMF (for peptide synthesis) was purchased from Watanabe Chemical Industries, Ltd. (Hiroshima, Japan). Photocatalyzed oxidative decarboxylation was performed with a Kessil A160WE Tuna Blue (Dicon Fiberoptic Inc., Richmond, CA, USA), as shown in Figure S1.
All reactions were monitored by TLC carried out on Merck silica gel plates (0.2 mm, 60F-254) with UV light, and visualized by p-anisaldehyde/H2SO4/EtOH solution, phosphomolybdic acid–EtOH solution or ninhydrin/AcOH/BuOH solution. Column chromatography was carried out with silica gel 60 N (Kanto Chemical Co. 100–210 µm). Preparative TLC was performed on 0.75 mm Wakogel® B-5F PLC plates (FUJIFILM Wako Pure Chemical Co., Ltd., Osaka, Japan). 1H NMR spectra (400 and 600 MHz) and 13C{1H} NMR spectra (100 and 150 MHz) were recorded on JEOL JNM-AL400 and JEOL JNM-ECA600 spectrometers (JEOL Ltd., Tokyo, Japan) in the indicated solvent. Chemical shifts (δ) are reported in unit parts per million (ppm) relative to the signal for internal TMS (0.00 ppm for 1H) for solutions in CDCl3. NMR spectral data are reported as follows: CHCl3 (7.26 ppm for 1H) or CDCl3 (77.0 ppm for 13C), and DMSO (2.49 ppm for 1H) or DMSO-d6 (39.5 ppm for 13C), when internal standard is not indicated. Multiplicities are reported by using standard abbreviations, and coupling constants are given in hertz.
High-resolution mass spectra (HRMS) were recorded on Thermo Scientific Exactive Plus Orbitrap Mass Spectrometer (Thermo Fisher Scientific K.K., Tokyo, Japan) for ESI or JEOL JMS-AX500 (JEOL Ltd., Tokyo, Japan) for FAB. IR spectra were recorded on a JASCO FTIR-4100 spectrophotometer (JASCO Co., Tokyo, Japan). Only the strongest and/or structurally important absorption are reported as the IR data afforded in wavenumbers (cm−1). Optical rotations were measured on a JASCO P-1010 polarimeter (JASCO Co., Tokyo, Japan). Melting points were measured with Round Science Inc. RFS-10 (J-SCIENCE LAB Co., Ltd., Kyoto, Japan), and are not corrected.
3.2. Synthesis of the Lanthionine 5
3.2.1. 2-Methyl 1-(2-(Trimethylsilyl)ethyl) (2R,3R)-3-methylaziridine-1,2-dicarboxylate (2)
To a solution of D-threonine (5.00 g, 42.0 mmol, 1.0 equiv) in MeOH (150 mL) was added SOCl2 (15.3 mL, 210 mmol, 5.0 equiv) dropwise at 0 °C, and the mixture was stirred at the same temperature for 30 min. After being stirred at reflux in an oil bath for 12 h, the reaction mixture was cooled to room temperature, and concentrated in vacuo. The resulting crude methyl ester was used for the next reaction without further purification.
To a solution of the crude amine in dry CH2Cl2 (150 mL) were added Et3N (14.6 mL, 105 mmol, 2.5 equiv) and TrtCl (11.7 g, 42.0 mmol, 1.0 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 43 h, the reaction mixture was washed with 10% aqueous citric acid, saturated aqueous NaHCO3 and brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude N-Trt amine was used for the next reaction without further purification.
To a solution of the crude alcohol in dry THF (120 mL) were added Et3N (14.6 mL, 105 mmol, 2.5 equiv) and MsCl (3.6 mL, 46.2 mmol, 1.1 equiv) at 0 °C under an argon atmosphere, and the mixture was stirred at the same temperature for 30 min. After being stirred at reflux in an oil bath for 72 h, the reaction mixture was concentrated in vacuo to remove THF. The resulting residue was diluted with EtOAc, and the organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude aziridine was used for next reaction without further purification.
To a solution of the crude N-Trt aziridine in dry CH2Cl2 (150 mL) were added dry MeOH (2.6 mL, 63.0 mmol, 1.5 equiv) and TFA (6.5 mL, 84.0 mmol, 2.0 equiv) at 0 °C under an argon atmosphere. After being stirred at the same temperature for 1 h, the reaction mixture was basified by Et3N (20.5 mL, 147 mmol, 3.5 equiv). TeocOSu (10.9 g, 42.0 mmol, 1.0 equiv) was then added to the above mixture at 0 °C. After being stirred at room temperature for 19 h, the reaction mixture was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was suspended in CH2Cl2/MeOH. The suspension was filtered through a pad of Celite®, and the filtrate was concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 4:1) to afford the N-Teoc aziridine 2 (5.98 g, 23.0 mmol, 55% in 4 steps) as a colorless oil. [α]22D +64 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.18–4.22 (m, 2H), 3.79 (s, 3H), 3.15 (d, 1H, J = 6.8 Hz), 2.77–2.82 (m, 1H), 1.35 (d, 3H, J = 6.4 Hz), 1.00–1.04 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 167.7, 161.8, 65.3, 52.2, 39.7, 38.7, 17.4, 12.9, −1.5; IR (neat) 2955, 1756, 1729, 1442, 1425, 1285, 1252, 1201, 1181, 1081, 1038, 860, 838 cm−1; HRMS [ESI] m/z calcd for C11H21NO4SiNa [M+Na]+ 282.1132, found 282.1131.
3.2.2. Fmoc-Cys-OH (3)
To a solution of L-cystine (5.00 g, 20.8 mmol, 1.0 equiv) in 1,4-dioxane (90 mL) were added a solution of Na2CO3 (6.62 g, 62.4 mmol, 3.0 equiv) in water (60 mL) and a solution of FmocOSu (14.0 g, 41.6 mmol, 2.0 equiv) in 1,4-dioxane (90 mL) at 0 °C. After being stirred at the room temperature for 15 h, the reaction mixture was concentrated in vacuo to remove 1,4-dioxane. The aqueous layer was acidified with 6 M aqueous HCl until pH1, and extracted three times with CH2Cl2. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was suspended in Et2O. The white precipitate was filtered, and dried under vacuum to afford the N-Fmoc amine (12.7 g, 18.5 mmol, 89%) as a white solid. The spectral data of synthetic compound were in good agreement with those of reported [47]. Mp 153–154 °C [lit. 149–151 °C]; [α]20D −89 (c 1.2, MeOH) [lit. [α]24D −87.1 (c 1.0, MeOH)]; 1H NMR (400 MHz, DMSO-d6, rotamer mixture) δ 13.0 (s, 1H), 7.87 (d, 2H, J = 7.5 Hz), 7.78 (d, 1H, J = 7.5 Hz), 7.69 (d, 2H, J = 7.5 Hz), 7.37–7.41 (m, 2H), 7.30 (t, 2H, J = 7.5 Hz), 4.26–4.31 (m, 3H), 4.21 (dd, 1H, J = 12.6, 5.6 Hz), 3.16 (dd, 1H, J = 13.5, 3.9 Hz), 2.94 (dd, 1H, J = 13.5, 10.3 Hz); 13C{1H} NMR (100 MHz, DMSO-d6, rotamer mixture) δ 172.2, 156.0, 143.8, 143.7, 140.7, 127.6, 127.1, 125.3, 125.2, 120.1, 65.8, 53.0, 46.6, 39.1; IR (neat) 1696, 1515, 1448, 1331, 1228, 1049, 758, 739 cm−1; HRMS [FAB] m/z calcd for C36H33N2O8S2 [M+H]+ 685.1673, found 685.1690.
To a solution of the disulfide (14.0 g, 20.4 mmol, 1.0 equiv) in dry THF (70 mL) were added 1 M aqueous HCl (70 mL) and activated zinc dust (4.00 g, 61.2 mmol, 3.0 equiv) at 0 °C. After being stirred at the room temperature for 30 min, the reaction mixture was filtered through of a pad of Celite®. The filtrate was concentrated in vacuo, and the resulting residue was diluted with 1 M aqueous HCl. The aqueous layer was extracted with CH2Cl2. The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was suspended in CH2Cl2/hexane. The precipitate was filtered and dried under vacuum to afford the thiol 3 (10.7 g, 31.3 mmol, 77%) as a white solid. The spectral data of synthetic compound were in good agreement with those of reported [48]. Mp 119–123 °C; [α]20D −5.7 (c 0.93, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 12.9 (s, 1H), 7.88 (d, 2H, J = 7.5 Hz), 7.69–7.73 (m, 3H), 7.41 (t, 2H, J = 7.5 Hz), 7.32 (t, 2H, J = 7.5 Hz), 4.29–4.31 (m, 2H), 4.23 (t, 1H, J = 7.0 Hz), 4.14 (dt, 1H, J = 8.3, 4.3 Hz), 2.89–2.92 (m, 1H), 2.71–2.78 (m, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.8, 156.0, 143.8, 140.7, 127.6, 127.1, 125.2, 120.1, 65.7, 56.5, 46.6, 25.4; IR (neat) 3314, 1694, 1536, 1476, 1447, 1418, 1230, 1103, 1047, 756, 736, 620 cm−1; HRMS [FAB] m/z calcd for C18H18NO4S [M+H]+ 344.0951, found 344.0942.
3.2.3. The Lanthionine 4
To a solution of the aziridine 2 (4.62 g, 17.8 mmol, 2.0 equiv) in dry Et2O (90 mL) were added Fmoc-Cys-OH (3) (3.06 g, 8.91 mmol, 1.0 equiv) and InCl3 (788 mg, 3.56 mmol, 0.4 equiv) at room temperature under an argon atmosphere. After being stirred at the same temperature for 18 h, the reaction mixture was quenched with water. The organic layer was separated, and aqueous layer was extracted three times with EtOAc. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with CH2Cl2/MeCN = 1:1) to afford the lanthionine 4 (2.89 g, 4.79 mmol, 54%) as a white amorphous solid. [α]24D +2.3 (c 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, 2H, J = 7.5 Hz), 7.71–7.73 (m, 3H), 7.41 (t, 2H, J = 7.5 Hz), 7.31–7.33 (m, 3H), 4.21–4.34 (m, 4H), 4.09–4.15 (m, 1H), 4.04–4.06 (m, 2H), 3.65 (s, 3H), 3.21–3.24 (m, 1H), 2.95 (dd, 1H, J = 13.6, 4.7 Hz), 2.74 (dd, 1H, J = 13.5, 9.4 Hz), 1.21 (d, 3H, J = 7.0 Hz), 0.91–0.93 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 172.0, 170.8, 156.2, 155.9, 143.7, 140.7, 127.6, 127.0, 125.2, 120.0, 65.7, 62.2, 58.4, 54.0, 51.9, 46.6, 41.6, 32.1, 18.8, 17.3, −1.6; IR (neat) 3327, 3019, 2953, 1720, 1513, 1478, 1449, 1338, 1249, 1213, 1080, 1049, 859, 837, 758, 740 cm−1; HRMS [ESI] m/z calcd for C29H38N2O8NaSSi [M+Na]+ 625.2010, found 625.2007.
3.2.4. The MOM Ester 5
To a solution of the carboxylic acid 4 (2.89 g, 4.79 mmol, 1.0 equiv) in dry acetone (90 mL) were added KHCO3 (1.20 g, 12.0 mmol, 2.5 equiv) and MOMCl (437 μL, 5.75 mmol, 1.2 equiv) at room temperature under an argon atmosphere. After being stirred at the same temperature for 15 h, the reaction mixture was concentrated in vacuo to remove acetone. The resulting residue was diluted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1) to afford the MOM ester 5 (3.06 g, 4.73 mmol, 99%) as a white amorphous solid. [α]23D −16 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.74 (d, 2H, J = 7.5 Hz), 7.57–7.59 (m, 2H), 7.38 (t, 2H, J = 7.5 Hz), 7.29 (d, 2H, J = 7.5 Hz), 5.66 (d, 1H, J = 6.8 Hz), 5.41 (d, 1H, J = 8.5 Hz), 5.31 (d, 1H, J = 5.6 Hz), 5.27 (d, 1H, J = 5.6 Hz), 4.58–4.61 (m, 1H), 4.51 (d, 1H, J = 7.5 Hz), 4.36–4.44 (m, 2H), 4.22 (t, 1H, J = 6.9 Hz), 4.11–4.16 (m, 2H), 3.72 (s, 3H), 3.44–3.47 (m, 4H), 3.03 (dd, 1H, J = 13.4, 3.7 Hz), 2.91 (dd, 1H, J = 13.4, 5.4 Hz), 1.31 (d, 3H, J = 7.0 Hz), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 170.0, 156.7, 155.7, 143.7, 141.3, 127.7, 127.0, 125.0, 120.0, 91.8, 67.2, 63.7, 58.1, 58.0, 53.8, 52.5, 47.1, 43.7, 33.5, 19.4, 17.6, −1.6; IR (neat) 3335, 2953, 1722, 1511, 1450, 1339, 1249, 1210, 1157, 1081, 1047, 994, 928, 859, 837, 759, 741 cm−1; HRMS[ESI] m/z calcd for C31H42N2O9NaSSi [M+Na]+ 669.2272, found 669.2280.
3.3. Synthesis of the Tripeptide 1 by Solution-Phase Peptide Synthesis
3.3.1. The Tripeptide 1a
To a solution of the N-Fmoc-amine 5 (3.06 g, 4.73 mmol, 1.0 equiv) in dry MeCN (40 mL) was added Et2NH (10 mL) at room temperature under an argon atmosphere. After being stirred at the same temperature for 40 min, the reaction mixture was concentrated in vacuo. The resulting residue was azeotroped three times with MeCN to remove Et2NH, and the resulting crude amine was used for the next reaction without further purification.
To a solution of the crude amine in dry CH2Cl2 (50 mL) were added DIEA (1.7 mL, 9.46 mmol, 2.0 equiv), Cbz-Phe-OH (1.70 g, 5.68 mmol, 1.2 equiv), HOBt (773 mg, 5.68 mmol, 1.2 equiv) and EDCI·HCl (1.09 g, 5.68 mmol, 1.2 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 13 h, the reaction mixture was diluted with CH2Cl2. The organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1) to afford the tripeptide 6a (2.88 g, 4.08 mmol, 86% in 2 steps) as a white amorphous solid. [α]23D −25 (c 0.90, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.20–7.27 (m, 10H), 6.73 (d, 1H, J = 6.5 Hz), 5.45 (d, 1H, J = 9.2 Hz), 5.38 (d, 1H, J = 5.8 Hz), 5.29 (d, 1H, J = 5.8 Hz), 5.26 (d, 1H, J = 5.8 Hz), 5.08 (s, 2H), 4.72–4.74 (m, 1H), 4.48–4.50 (m, 2H), 4.15–4.17 (m, 2H), 3.75 (s, 3H), 3.48 (s, 3H), 3.37–3.41 (m, 1H), 3.00–3.18 (m, 3H), 2.84 (dd, 1H, J =13.9, 5.9 Hz), 1.27 (d, 3H, J = 7.0 Hz), 0.96–1.01 (m, 2H), 0.02 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 171.0, 169.5, 156.6, 155.9, 136.2, 136.1, 129.3, 128.6, 128.4, 128.1, 127.9, 127.0, 91.7, 67.0, 63.7, 58.1, 57.9, 56.0, 52.5, 52.2, 43.3, 38.1, 32.8, 19.2, 17.6, −1.6; IR (neat) 3314, 3030, 2953, 1721, 1519, 1454, 1338, 1249, 1215, 1155, 1083, 1048, 931, 860, 837, 750, 699 cm−1; HRMS [ESI] m/z calcd for C33H47N3O10NaSSi [M+Na]+ 728.2644, found 728.2650.
To a solution of the MOM ester 6a (2.88 g, 4.08 mmol, 1.0 equiv) in 1,4-dioxane (30 mL) was added 4 M HCl/1,4-dioxane (10 mL) at 0 °C under argon atmosphere. After being stirred at room temperature for 1 h, the reaction mixture was concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 50:1) to afford the carboxylic acid 1a (2.35 g, 3.55 mmol, 87%) as a white amorphous solid. [α]23D −13 (c 1.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 12.9 (brs, 1H), 8.38 (d, 1H, J = 7.7 Hz), 7.46 (d, 1H, J = 8.7 Hz), 7.19–7.31 (m, 11 H), 4.93 (s, 2H), 4,25–4.43 (m, 3H), 4.03–4.05 (m, 2H), 3.64 (s, 3H), 3.25–3.31 (m, 1H), 2.96–3.03 (m, 2H), 2.70–2.80 (m, 2H), 1.21 (d, 3H, J = 6.8 Hz), 0.90–0.92 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 171.6, 170.8, 156.2, 155.7, 138.0, 136.9, 129.2, 128.2, 128.0, 127.6, 127.3, 126.2, 65.1, 62.2, 58.5, 55.9, 52.2, 51.9, 41.8, 37.4, 32.1, 18.7, 17.3, −1.5; IR (neat) 3315, 3064,3030, 2953, 1721, 1518, 1454, 1439, 1340, 1287, 1250, 1215, 1180, 1081, 1050, 860, 837, 753, 698 cm−1; HRMS [ESI] m/z calcd for C31H43N3O9NaSSi [M+Na]+ 684.2381, found 684.2391.
3.3.2. The Tripeptide 1b
Compound 6b was prepared from the N-Fmoc-amine 5 (248 mg, 383 µmol) according to the procedure above described for 6a, and obtained in 69% yield (178 mg, 265 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1). [α]22D −17 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.18–7.28 (m, 5H), 6.75 (d, 1H, J = 7.2 Hz), 5.40 (d, 1H, J = 8.5 Hz), 5.26 (d, 1H, J = 5.8 Hz), 5.22 (d, 1H, J = 5.8 Hz), 5.01–5.03 (m, 1H), 4.69–4.71 (m, 1H), 4.39–4.46 (m, 2H), 4.13–4.15 (m, 2H), 3.74 (s, 3H), 3.44 (s, 3H), 3.35–3.37 (m, 1H), 3.11 (dd, 1H, J = 14.2, 5.8 Hz), 2.99–3.02 (m, 2H), 2.82 (dd, 1H, J = 13.8, 6.3 Hz), 1.36 (s, 9H), 1.26 (d, 3H, J = 7.2 Hz), 0.95–0.97 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.4, 171.1, 169.6, 156.7, 155.3, 136.5, 129.3, 128.7, 127.0, 91.7, 80.3, 63.7, 58.1, 58.0, 55.8, 52.5, 52.3, 43.5, 38.1, 33.1, 28.2, 19.3, 17.7, −1.5; IR (neat) 3317, 2953, 1719, 1510, 1454, 1366, 1339, 1249, 1210, 1168, 1086, 1048, 933, 860, 837, 776, 699 cm−1; HRMS[ESI] m/z calcd for C30H49N3O10NaSSi [M+Na]+ 650.2800, found 650.2819.
To a solution of the MOM ester 6b (158 mg, 235 μmol, 1.0 equiv) in 1,4-dioxane (4.20 mL) was added 4 M HCl/1,4-dioxane (0.6 mL) at 0 °C under argon atmosphere. After being stirred at room temperature for 2.5 h, the reaction mixture was concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 10:1) to afford the carboxylic acid 1b (109 mg, 173 μmol, 74%) as a white amorphous solid. [α]21D −12 (c 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, 1H, J = 7.2 Hz), 7.20–7.36 (m, 5H), 7.14–7.20 (m, 1H), 6.84 (d, 1H, J = 8.5 Hz), 4.36–4.45 (m, 1H), 4.16–4.35 (m, 2H), 3.99–4.09 (m, 2H), 3.64 (s, 3H), 3.19–3.28 (m, 1H), 2.90–3.06 (m, 2H), 2.66–2.83 (m, 2H), 1.27 (s, 9H), 1.21 (d, 3H, J = 6.5 Hz), 0.89–0.94 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 170.8, 156.2, 155.1, 138.0, 129.2, 127.9, 126.1, 78.0, 66.2, 58.4, 55.6, 52.0, 51.9, 41.8, 37.4, 32.2, 28.1, 18.7, 17.3, –1.5; IR (neat) 3320, 2954, 1721, 1512, 1453, 1367, 1339, 1249, 1170, 1080, 1049, 859, 837, 699 cm−1; HRMS[ESI] m/z calcd for C28H45N3O9NaSSi [M+Na]+ 650.2538, found 650.2546.
3.3.3. The Tripeptide 1c
Compound 6c was prepared from the N-Fmoc-amine 5 (367 mg, 568 μmol) according to the procedure above described for 6a, and obtained in 86% yield (388 mg, 488 μmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1). [α]20D −28 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.73 (d, 2H, J = 7.5 Hz), 7.50 (t, 2H, J = 7.5 Hz), 7.37 (t, 2H, J = 7.5 Hz), 7.21–7.29 (m, 7H), 6.70–6.82 (m, 1H), 5.44 (d, 2H, J = 8.5 Hz), 5.23–5.26 (m, 2H), 4.71–4.72 (m, 1H), 4.46–4.48 (m, 2H), 4.39–4.42 Hz (m, 1H), 4.27–4.30 (m, 1H), 4.13–4.17 (m, 3H), 3.71 (s, 3H), 3.43 (s, 3H), 3.34–3.40 (m, 1H), 2.98–3.10 Hz (m, 3H), 2.81–2.85 (m, 1H), 1.24 (d, 3H, J = 7.1 Hz), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 171.0, 169.5, 156.6, 155.9, 143.71, 143.66, 141.2, 136.2, 129.3, 128.7, 127.7, 127.0, 125.0, 119.9, 91.7, 77.2, 67.1, 63.7, 58.0, 52.5, 52.2, 47.0, 43.3, 38.2, 32.8, 29.2, 19.2, 17.6, −1.6; IR (neat) 3310, 3064, 3025, 2953, 1719, 1670, 1517, 1450, 1412, 1381, 1338, 1287, 1249, 1217, 1154, 1084, 1047, 932, 860, 837, 757, 742, 700 cm−1; HRMS[ESI] m/z calcd for C40H51N3O10NaSSi [M+Na]+ 816.2957, found 816.2960.
Compound 1c was prepared from the MOM ester 6c (340 mg, 428 µmol) according to the procedure above described for 1a, and obtained in 85% yield (272 mg, 362 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 50:1). [α]24D −14 (c 0.96, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, 1H, J = 7.5 Hz), 7.87 (d, 2H, J = 7.5 Hz), 7.60–7.64 (m, 3H), 7.16–7.42 (m, 10H), 4.25–4.40 (m, 3H), 4.09–4.16 (m, 3H), 4.03–4.05 (m, 2H), 3.64 (s, 3H), 3.25–3.26 (m, 1H), 2.95–3.03 (m, 2H), 2.78–2.80 (m, 2H), 1.19 (d, 3H, J = 7.0 Hz), 0.90–0.92 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.8, 171.5, 170.8, 156.2, 155.6, 143.7, 143.6, 140.6, 138.0, 129.2, 127.9, 127.5, 127.0, 126.1, 125.23, 125.17, 120.0, 65.6, 62.2, 58.4, 56.0, 52.3, 51.8, 46.5, 41.7, 37.4, 32.2, 18.7, 17.3, −1.6; IR (neat) 3313, 3064, 3028, 2953, 1721, 1516, 1450, 1338, 1249, 1216, 1180, 1081, 1048, 859, 837, 757, 742, 699 cm−1; HRMS [ESI] m/z calcd for C38H47N3O9NaSSi [M+Na]+ 772.2694, found 772.2715.
3.3.4. The Tripeptide 1d
Compound 6d was prepared from the N-Fmoc-amine 5 (480 mg, 742 μmol) according to the procedure above described for 6a, and obtained in 75% yield (344 mg, 558 μmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1 to hexane/acetone = 3:1). [α]23D −16 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25–7.32 (m, 5H), 7.01 (s, 1H), 5.67 (s, 1H), 5.48 (d, 1H, J = 9.2 Hz), 5.28 (d, 1H, J = 5.8 Hz), 5.23 (d, 1H, J = 5.8 Hz), 5.10 (s, 2H), 4.77 (dt, 1H, J = 6.3, 5.6 Hz), 4.48 (dd, 1H, J = 8.8, 3.0 Hz), 4.12–4.15 (m, 2H), 3.92 (d, 2H, J = 5.6 Hz), 3.72 (s, 3H), 3.36–3.44 (m, 4H), 3.03 (dd, 1H, J = 13.6, 4.0 Hz), 2.86 (dd, 1H, J = 13.9, 6.2 Hz), 1.27 (d, 3H, J = 7.2 Hz), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 169.8, 169.2, 156.6, 156.5, 136.1, 128.5, 128.1, 128.0, 91.7, 77.2, 67.1, 63.7, 57.9, 52.5, 52.1, 44.4, 43.3, 32.7, 19.1, 17.6, −1.6; IR (neat) 3325, 2953, 1723, 1515, 1453, 1381, 1339, 1249, 1156, 1088, 1048, 927, 860, 837, 754, 698 cm−1; HRMS[ESI] m/z calcd for C26H41N3O10NaSSi [M+Na]+ 638.2174, found 638.2180.
Compound 1d was prepared from the MOM ester 6d (215 mg, 349 µmol) according to the procedure above described for 1a, and obtained in 45% yield (89.6 mg, 157 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 50:1 to 10:1). [α]24D +2.7 (c 0.95, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 12.9 (brs, 1H), 8.18 (d, 1H, J = 8.0 H), 7.30–7.44 (m, 7H), 5.02 (s, 2H), 4.37–4.40 (m, 1H), 4.23 (dd, 1H, J = 8.0, 6.0 Hz), 4.03–4.05 (m, 2H), 3.66 (d, 2H, J = 6.3 Hz), 3.63 (s, 3H), 3.18–3.20 (m, 1H), 2.91 (dd, 1H, J = 13.5, 5.1 Hz), 2.72 (dd, 1H, J = 13.5, 8.1 Hz), 1.18 (d, 3H, J = 6.8 Hz), 0.90–0.93 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 170.7, 169.0, 156.4, 156.2, 137.0, 128.2, 127.7, 127.6, 65.4, 62.2, 58.4, 51.9, 51.8, 43.2, 41.8, 32.3, 18.6, 17.3, −1.6; IR (neat) 3327, 2953, 1722, 1523, 1453, 1438, 1340, 1249, 1081, 1050, 860, 837, 697 cm−1; HRMS [ESI] m/z calcd for C24H37N3O9NaSSi [M+Na]+ 594.1912, found 594.1924.
3.3.5. The Tripeptide 1e
To a solution of N-Fmoc-amine 5 (359 mg, 554 μmol, 1.0 equiv) in dry MeCN (4.4 mL) was added Et2NH (1.1 mL) at room temperature under an argon atmosphere. After being stirred at the same temperature for 1.5 h, the reaction mixture was concentrated in vacuo. The resulting residue was azeotroped three times with MeCN, and the resulting crude amine was used for the next reaction without further purification.
To a solution of the crude amine in dry CH2Cl2 (5.5 mL) were added DIEA (193 μL, 1.11 mmol, 2.0 equiv), Cbz-Ile-OH (177 mg, 665 μmol, 1.2 equiv) and HATU (253 mg, 665 μmol, 1.2 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 7 h, the reaction mixture was diluted with CH2Cl2. The organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1) to afford the tripeptide 6e (373 mg, 539 mmol, 97% in 2 steps) as a yellowish amorphous solid. [α]23D −19 (c 0.98, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.29–7.34 (m, 5H), 6.82 (d, 1H, J = 6.5 Hz), 5.51 (d, 1H, J = 9.2 Hz), 5.43 (d, 1H, J = 8.7 Hz), 5.32 (d, 1H, J = 5.8 Hz), 5.27 (d, 1H, J = 5.8 Hz), 5.11 (s, 2H), 4.78–4.80 (m, 1H), 4.52 (dd, 1H, J = 9.3, 3.0 Hz), 4.14–4.17 (m, 3H), 3.75 (s, 3H), 3.48 (s, 3H), 3.37–3.48 (m, 1H), 3.04 (dd, 1H, J = 13.8, 4.3 Hz), 2.89 (dd, 1H, J = 13.8, 6.0 Hz), 1.89–1.91 (m, 1H), 1.60–1.70 (m, 1H) 1.30 (d, 3H, J = 7.0 Hz), 1.07–1.26 (m, 1H), 0.90–1.01 (m, 8H), 0.03 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.4, 171.2, 169.7, 156.7, 156.2, 136.2, 128.5, 128.1, 128.0, 91.7, 67.1, 63.7, 59.6, 58.1, 58.0, 52.5, 52.0, 43.5, 37.4, 32.9, 24.6, 19.3, 17.6, 15.4, 11.3, −1.6; IR (neat) 3311, 2959, 1723, 1666, 1524, 1454, 1382, 1339, 1284, 1248, 1156, 1087, 1045, 932, 860, 837, 697 cm−1; HRMS[ESI] m/z calcd for C30H49N3O10NaSSi [M+Na]+ 694.2800, found 694.2814.
Compound 1e was prepared from the MOM ester 6e (373 mg, 539 µmol) according to the procedure above described for 1a, and obtained in 67% yield (228 mg, 362 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeCN = 3:1 to CH2Cl2/MeOH = 20:1). [α]21D −6.5 (c 0.99, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 12.8 (brs, 1H), 8.21 (d, 1H, J = 7.7 Hz), 7.24–7.33 (m, 7H), 5.02 (s, 2H), 4.35–4.38 (m, 1H), 4.25 (dd, 1H, J = 7.4, 5.7 Hz), 4.03–4.05 (m, 2H), 3.91–3.99 (m, 1H), 3.62–3.64 (m, 3H), 3.21 (s, 1H), 2.92 (dd, 1H, J = 13.0, 5.1 Hz), 2.72 (dd, 1H, J = 13.0, 8.2 Hz), 1.63–1.82 (m, 1H), 1.31–1.49 (m, 1H), 1.03–1.23 (m, 4H), 0.72–0.96 (m, 8H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 171.2, 170.8, 156.2, 155. 9, 137.0, 128.2, 127.6, 127.5, 65.3, 62.2, 59.0, 58.4, 51.90, 51.85, 41.6, 36.6, 32.1, 24.2, 18.6, 17.3, 15.2, 10.8, −1.5; IR (neat) 3316, 2959, 1721, 1666, 1517, 1454, 1340, 1286, 1249, 1216, 1179, 1082, 1045, 859, 837 cm−1; HRMS [ESI] m/z calcd for C28H45N3O9NaSSi [M+Na]+ 650.2538, found 650.2551.
3.3.6. The Tripeptide 1f
Compound 6f was prepared from the N-Fmoc-amine 5 (268 mg, 414 μmol) according to the procedure above described for 6a, and obtained in 74% yield (242 mg, 307 μmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1). [α]22D −21 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25–7.32 (m, 5H), 6.92–6.94 (m, 1H), 5.47–5.49 (m, 2H), 5.30 (d, 1H, J = 5.8 Hz), 5.23 (d, 1H, J = 5.8 Hz), 5.08 (s, 2H), 4.74 (dt, 1H, J = 6.8, 5.4 Hz), 4.65 (brs, 1H), 4.48 (dd, 1H, J = 8.7, 2.4 Hz), 4.21–4.22 (m, 1H), 4.13–4.15 (m, 2H), 3.73 (s, 3H), 3.37–3.43 (m, 4H), 3.02–3.04 (m, 3H), 2.86 (dd, 1H, J = 13.2, 5.9 Hz), 1.25–1.85 (m, 18H), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ171.8, 171.2, 169.7, 156.7, 156.2, 156.1, 136.2, 128.4, 128.1, 128.0, 91.7, 79.0, 67.0, 63.7, 58.1, 57.9, 54.7, 52.5, 52.1, 43.3, 39.8, 32.7, 31.9, 29.6, 28.4, 22.3, 19.2, 17.6, −1.6; IR (neat) 3319. 2952, 1714, 1511, 1454, 1365, 1339, 1249, 1169, 1086, 1046, 860, 837 cm−1; HRMS[ESI] m/z calcd for C35H58N4O12NaSSi [M+Na]+ 809.3433, found 809.3434.
Compound 1f was prepared from the MOM ester 6f (215 mg, 279 µmol) according to the procedure above described for 1b, and obtained in 87% yield (180 mg, 242 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeCN = 1:1 to CH2Cl2/MeOH = 10:1). [α]24D −13 (c 1.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 7.91 (d, 1H, J = 7.2 Hz), 7.28–7.32 (m, 5H), 7.11–7.12 (m, 1H), 6.90–6.91 (m, 1H), 6.47–6.48 (m, 1H), 5.02 (s, 2H), 4.32–4.37 (m, 1H), 4.23 (dd, 1H, J = 8.1, 5.9 Hz), 3.99–4.06 (m, 3H), 3.63 (s, 3H), 3.19–3.25 (m, 1H), 2.93–3.02 (m, 3H), 2,74 (dd, 1H, J = 13.4, 7.6 Hz), 1.61–1.64 (m, 1H), 1.51–1.53 (m, 1H), 1.13–1.37 (m, 16H), 0.90–0.93 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.8, 171.7, 170.7, 156.1, 155.8, 155.4, 136.9, 128.2, 127.6, 127.5, 66.2, 65.3, 62.1, 58.4, 54.5, 52.0, 51.7, 41.6, 32.2, 31.6, 29.1, 28.1, 27.8, 22.6, 18.6, 17.2, −1.6; IR (neat) 3325, 2953, 1713, 1515, 1453, 1411, 1391, 1366, 1340, 1249, 1214, 1172, 1081, 1047, 860, 837, 754, 697 cm−1; HRMS [ESI] m/z calcd for C33H54N4O11NaSSi [M+Na]+ 765.3171, found 765.3179.
3.4. The Photocatalytic AviMeCys Formation Using 1
3.4.1. The β-Thioenamides (Z)-9a and (E)-9a
To a solution of the carboxylic acid 6a (66.6 mg, 100 µmol, 1.0 equiv) and N-hydroxyphthalimide (18.1 mg, 110 µmol, 1.1 equiv) in dry CH2Cl2 (1.0 mL) was added DIC (17.3 µL, 110 µmol, 1.1 equiv) at room temperature under an argon atmosphere, and the mixture was stirred at the same temperature for 30 min. After complete consumption of 6a (monitored by TLC analysis), the reaction mixture was cooled to −40 °C. A solution of eosin Y-Na2 (0.7 mg, 1.00 μmol, 1 mol%), Hantzsch ester (25.5 mg, 100 µmol, 1.0 equiv) and diphenyl diselenide (62.8 mg, 200 µmol, 2.0 equiv) in dry DMF (1.5 mL, used immediately after freeze-pump-thaw cycling) was then added to the above mixture at −40 °C. After being stirred at the same temperature for 30 min under irradiated Blue LEDs, Et3N (250 μL, 1.79 mmol, 18 equiv) was added to the solution at −40 °C. After being stirred at room temperature for 3 h, the reaction mixture was quenched with saturated aqueous NaHCO3, and stirred for 12 h. The aqueous layer was extracted three times with Et2O. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by preparative TLC (eluted with hexane/EtOAc = 7:2) to afford the β-thioenamide (Z)-9a (26.2 mg, 42.5 µmol, 42%) as a white amorphous solid and the β-thioenamide (E)-9a (19.6 mg, 31.8 µmol, 32%) as a white amorphous solid. (Z)-9a: [α]24D −41 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.42 (d, 1H, J = 10.6 Hz), 7.19–7.34 (m, 10H), 7.15 (dd, 1H, J = 10.6, 7.2 Hz), 5.49–5.50 (m, 1H), 5.40–5.41 (m, 1H), 5.32 (d, 1H, J = 7.2 Hz), 5.07–5.09 (m, 2H), 4.56–4.58 (m, 1H), 4.51 (dd, 1H, J = 9.2, 3.9 Hz), 4.14–4.15 (m, 2H), 3.65 (s, 3H), 3.31–3.32 (m, 1H), 3.19 (dd, 1H, J = 13.9, 6.2 Hz), 3.09–3.11 (m, 1H), 1.32 (d, 3H, J = 7.2 Hz), 0.95–0.97 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 168.9, 156.8, 156.2, 136.2, 136.1, 129.7, 129.3, 128.9, 128.6, 128.3, 128.2, 127.2, 99.7, 67.3, 63.9, 58.9, 56.4, 52.6, 45.8, 38.1, 18.3, 17.8, −1.4; IR (neat) 3310, 3064, 3031, 2952, 1697, 1628, 1498, 1455, 1380, 1337, 1248, 1178, 1080, 1046, 860, 837, 742, 698 cm−1; HRMS[ESI] m/z calcd for C30H41N3O7NaSSi [M+Na]+ 638.2327, found 638.2330. (E)-9a: [α]23D −3.5 (c 0.89, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.01 (m, 1H), 7.14–7.32 (m, 10H), 6.99 (dd, 1H, J = 13.7, 6.6 Hz), 5.59 (d, 1H, J = 13.7 Hz), 5.42–5.44 (m, 2H), 5.05 (s, 2H), 4.48 (dd, 1H, J = 5.5, 2.7 Hz), 4.40–4.42 (m, 1H), 4.13–4.17 (m, 2H), 3.64 (s, 3H), 3.34–3.38 (m, 1H), 3.06 (d, 2H, J = 6.8 Hz), 1.29 (d, 3H, J = 7.5 Hz), 1.00–1.01 (m, 2H), 0.03 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.2, 168.3, 156.8, 156.3, 136.0, 135.9, 129.4, 129.3, 129.0, 128.7, 128.5, 128.2, 127.4, 104.1, 67.5, 63.9, 57.9, 56.4, 52.5, 45.3, 38.2, 18.6, 17.8, −1.4; IR (neat) 3303, 2953, 1725, 1688, 1669, 1629, 1505, 1454, 1341, 1288, 1261, 1246, 1209, 1176, 1079, 1046, 938, 862, 835, 750, 697 cm−1; HRMS[ESI] m/z calcd for C30H41N3O7NaSSi [M+Na]+ 638.2327, found 638.2334.
3.4.2. The β-Thioenamides (Z)-9b and (E)-9b
Compounds (Z)-9b and (E)-9b were prepared from the carboxylic acid 6b (63.7 mg, 101 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 7:2, hexane/IPA = 20:1) to be obtained in 32% yield (18.6 mg, 32.0 μmol) as a white amorphous solid and 32% yield (18.9 mg, 32.5 μmol) as a white amorphous solid, respectively. (Z)-9b: [α]24D −49 (c 1.2, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.42 (d, 1H, J = 10.9 Hz), 7.28–7.30 (m, 2H), 7.22–7.24 (m, 3H), 7.12–7.15 (m, 1H), 5.45–5.47 (m, 1H), 5.28–5.29 (m, 1H), 5.14–5.16 (m, 1H), 4.52 (d, 2H, J = 10.3 Hz), 4.14–4.17 (m, 2H), 3.70 (s, 3H), 3.34–3.36 (m, 1H), 3.18 (dd, 1H, J = 14.0, 5.8 Hz), 3.04–3.06 (m, 1H), 1.39 (s, 9H), 1.32 (d, 3H, J = 6.8 Hz), 0.98–0.99 (m, 2H), 0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 169.3, 156.8, 155.6, 136.5, 129.5, 129.3, 128.8, 127.1, 99.6, 80.6, 63.9, 58.7, 55.8, 52.6, 45.8, 37.9, 28.3, 18.4, 17.8, −1.4; IR (neat) 3324, 2953, 1690, 1627, 1499, 1366, 1338, 1285, 1249, 1171, 1080, 1047, 860, 837, 754, 699 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2490. (E)-9b: [α]24D −6.8 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.04–8.05 (m, 1H), 7.28 (t, 2H, J = 7.5 Hz), 7.22 (t, 1H, J = 7.5 Hz), 7.15 (d, 2H, J = 7.5 Hz), 7.00 (dd, 1H, J = 13.7, 10.9 Hz), 5.59 (d, 1H, J = 13.7 Hz), 5.42 (d, 1H, J = 6.0 Hz), 5.06 (d, 1H, J = 8.2 Hz), 4.47 (dd, 1H, J = 9.2, 3.1 Hz), 4.33 (s, 1H), 4.15–4.16 (m, 2H), 3.65 (s, 3H), 3.37–3.38 (m, 1H), 3.05–3.06 (m, 1H), 2.99–3.00 (m, 1H), 1.37 (s, 9H), 1.28 (d, 3H, J = 6.8 Hz), 0.98–1.00 (m, 2H), 0.04 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.2, 168.7, 156.8, 155.8, 136.3, 129.6, 129.3, 128.8, 127.2, 103.6, 80.8, 63.5, 57.9, 56.0, 52.5, 45.3, 38.0, 28.3, 18.5, 17.8, −1.4; IR (neat) 3310, 2954, 1723, 1681, 1628, 1513, 1454, 1391, 1367, 1339, 1289, 1248, 1211, 1169, 1079, 1047, 941, 860, 837, 754, 698 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2487.
3.4.3. The β-Thioenamides (Z)-9c and (E)-9c
To a solution of the carboxylic acid 6c (75.2 mg, 100 µmol, 1.0 equiv) and N-hydroxyphthalimide (18.0 mg, 110 µmol, 1.1 equiv) in dry CH2Cl2 (1.0 mL) was added DIC (17.3 μL, 110 µmol, 1.1 equiv) at room temperature under an argon atmosphere, and the mixture was stirred at the same temperature for 5 h. After complete consumption of 5c (monitored by TLC analysis), the reaction mixture was cooled to −40 °C. A solution of Eosin Y (696 μg, 1.00 μmol, 1 mol%), Hantzsch ester (25.4 mg, 100 µmol, 1.0 equiv) and diphenyl diselenide (62.6 mg, 200 µmol, 2.0 equiv) in dry DMF (1.5 mL, used immediately after Freeze-Pump-Thaw cycling) was then added to the above mixture at −40 °C. After being stirred at the same temperature for 30 min under irradiated Blue LEDs, Et3N (41.9 μL, 300 μmol, 3.0 equiv) was added to the solution at −40 °C. After being stirred at room temperature for 3 h, the reaction mixture was quenched with saturated aqueous NaHCO3, diluted with Et2O, and stirred for 12 h. The organic layer was separated, and the aqueous layer was extracted three times with Et2O. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by preparative TLC (eluted with hexane/EtOAc = 7:2, hexane/IPA = 10:1) to afford the β-thioenamide (Z)-9c (14.6 mg, 20.7 µmol, 21%) as a white amorphous solid and the β-thioenamide (E)-9c (11.1 mg, 15.8 µmol, 16%) as a white amorphous solid. (Z)-9c: [α]24D −25 (c 0.83, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.41–8.42 (m, 1H), 7.75 (d, 2H, J = 7.5 Hz), 7.49–7.52 (m, 2H), 7.39 (t, 2H, J = 7.5 Hz), 7.25–7.28 (m, 7H), 7.17 (dd, 1H, J = 10.8, 7.6 Hz) 5.53–5.55 (m, 1H), 5.38–5.40 (m, 1H), 5.33 (d, 1H, J = 4.6 Hz), 4.59–4.61 (m, 1H), 4.50–4.52 (m, 1H), 4.42 (dd, 1H, J = 10.8, 7.5 Hz), 4.34–4.35 (m, 1H), 4.12–4.18 (m, 3H), 3.68 (s, 3H), 3.31–3.33 (m, 1H), 3.12–3.19 (m, 2H), 1.32 (d, 3H, J = 6.8 Hz), 0.93–0.95 (m, 2H), 0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.4, 168.9, 156.8, 156.2, 143.83, 143.80, 141.4, 136.2, 129.8, 129.4, 129.0, 127.9, 127.4, 127.2, 125.2, 120.1, 99.8, 67.4, 64.0, 59.0, 56.4, 52.7, 47.2, 45.9, 38.3, 18.3, 17.8, −1.4; IR (neat) 3308, 3064, 3028, 2952, 1696, 1628, 1499, 1451, 1336, 1248, 1178, 1080, 1045, 859, 837, 757, 740, 699 cm−1; HRMS [ESI] m/z calcd for C37H45N3O7NaSSi [M+Na]+ 726.2640, found 726.2649. (E)-9c: [α]24D −16 (c 0.59, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.76 (d, 2H, J = 7.5 Hz), 7.50 (t, 2H, J = 7.5 Hz), 7.40 (t, 2H, J = 7.5 Hz), 7.25–7.29 (m, 8H), 6.95–6.99 (m, 1H), 5.58 (d, 1H, J = 13.7 Hz), 5.39 (d, 1H, J = 9.6 Hz), 5.26–5.28 (m, 1H), 4.45–4.47 (m, 2H), 4.34–4.36 (m, 2H), 4.15–4.17 (m, 3H), 3.63 (s, 3H), 3.36–3.37 (m, 1H), 3.06–3.07 (m, 2H), 1.28 (d, 3H, J = 6.8 Hz), 0.99–1.00 (m, 2H), 0.04 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.1, 168.0, 156.7, 156.3, 143.6, 141.4, 136.0, 129.3, 129.0, 127.9, 127.4, 127.2, 125.0, 120.1, 104.2, 67.3, 63.8, 57.9, 56.3, 52.5, 47.1, 45.2, 38.0, 18.5, 17.8, −1.4; IR (neat) 3316, 3064, 3027, 2950, 1691, 1626, 1509, 1450, 1338, 1289, 1247, 1216, 1079, 1045, 937, 859, 837, 757, 739, 700 cm−1; HRMS [ESI] m/z calcd for C37H45N3O7NaSSi [M+Na]+ 726.2640, found 726.2643.
3.4.4. The β-Thioenamides (Z)-9d and (E)-9d
Compounds (Z)-9d and (E)-9d were prepared from the carboxylic acid 6d (57.1 mg, 99.9 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 7:2, toluene/acetone = 8:1, hexane/IPA = 10:1) to be obtained in 39% yield (20.2 mg, 38.4 μmol) as a white amorphous solid and 19% yield (9.9 mg, 19 μmol) as a white amorphous solid, respectively. (Z)-9d: [α]23D +3.0 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.57–8.59 (m, 1H), 7.25–7.34 (m, 5H), 7.14 (dd, 1H, J = 10.9, 7.5 Hz), 5.75–5.77 (m, 1H), 5.59–5.61 (m, 1H), 5.32 (d, 1H, J = 7.5 Hz), 5.18 (d, 1H, J = 12.3 Hz), 5.14 (d, 1H, J = 12.3 Hz), 4.54 (dd, 1H, J = 8.9, 2.7 Hz), 4.12–4.14 (m, 2H), 3.98 (d, 2H, J = 5.5 Hz), 3.64 (s, 3H), 3.35–3.37 (m, 1H), 1.34 (d, 3H, J = 6.8 Hz), 0.93–0.95 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 167.1, 156.9, 156.8, 136.1, 129.3, 128.6, 128.4, 128.2, 100.1, 67.5, 63.9, 58.7, 52.5, 46.4, 44.9, 18.5, 17.8, −1.4; IR (neat) 3320, 2952, 1700, 1629, 1517, 1338, 1248, 1176, 1081, 1046, 860, 837, 738, 697 cm−1; HRMS [ESI] m/z calcd for C23H35N3O7NaSSi [M+Na]+ 548.1857, found 548.1864. (E)-9d: [α]23D +5.0 (c 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.31–8.32 (m, 1H), 7.31–7.36 (m, 5H), 7.05 (dd, 1H, J = 13.7, 10.9 Hz), 5.72 (d, 1H, J = 13.7 Hz), 5.55–5.56 (m, 1H), 5.45 (d, 1H, J = 8.9 Hz), 5.12 (s, 2H), 4.48 (dd, 1H, J = 8.9, 3.4 Hz), 4.15–4.16 (m, 2H), 3.87 (d, 2H, J = 5.5 Hz), 3.70 (s, 3H), 3.37–3.39 (m, 1H), 1.29 (d, 3H, J = 6.8 Hz), 0.98–1.00 (m, 2H), 0.03, (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 166.4, 156.9, 156.7, 135.9, 129.5, 128.7, 128.5, 128.2, 103.7, 67.6, 63.8, 57.9, 52.6, 45.3, 44.8, 18.4, 17.8, −1.4; IR (neat) 3311, 2953, 1696, 1627, 1512, 1454, 1338, 1248, 1174, 1080, 1047, 860, 837, 697 cm−1; HRMS [ESI] m/z calcd for C23H35N3O7NaSSi [M+Na]+ 548.1857, found 548.1866.
3.4.5. The β-Thioenamides (Z)-9e and (E)-9e
Compounds (Z)-9e and (E)-9e were prepared from the carboxylic acid 6e (62.2 mg, 99.1 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 7:2) to be obtained in 36% yield (20.5 mg, 35.2 μmol) as a white amorphous solid and 32% yield (18.3 mg, 31.5 μmol) as a white amorphous solid, respectively. (Z)-9e: [α]24D −45 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.52 (d, 1H, J = 10.6 Hz), 7.30–7.33 (m, 5H), 7.19 (dd, 1H, J = 10.6, 7.5 Hz), 5.66 (d, 1H, J = 8.2 Hz), 5.41 (d, 1H, J = 10.0 Hz), 5.34 (d, 1H, J = 7.5 Hz), 5.15 (d, 1H, J = 12.3 Hz), 5.10 (d, 1H, J = 12.3 Hz), 4.55 (dd, 1H, J = 10.0, 5.0 Hz), 4.26–4.27 (m, 1H), 4.11–4.15 (m, 2H), 3.61 (s, 3H), 3.33–3.34 (m, 1H), 2.03–2.04 (m, 1H), 1.48–1.49 (m, 1H), 1.37 (d, 3H, J = 6.8 Hz), 1.18–1.24 (m, 1H), 0.97 (d, 3H, J = 7.6 Hz), 0.94–0.97 (m, 2H), 0.91 (t, 3H, J = 7.6 Hz), 0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.5, 169.3, 156.8, 156.5, 136.2, 130.0, 128.6, 128.34, 128.26, 99.1, 67.4, 63.9, 60.1, 59.2, 52.6, 45.6, 37.3, 24.6, 18.2, 17.8, 15.7, 11.6, −1.4; IR (neat) 3314, 3065, 3033, 2959, 1720, 1695, 1628, 1512, 1381, 1337, 1283, 1248, 1178, 1127, 1080, 1043, 938, 860, 837, 773, 738, 696 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2487. (E)-9e: [α]24D +8.8 (c 0.78, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.35–8.37 (m, 1H), 7.30–7.35 (m, 5H), 7.04 (dd, 1H, J = 13.7, 10.3 Hz), 5.71 (d, 1H, J = 13.7 Hz), 5.46–5.47 (m, 2H), 5.10 (d, 1H, J = 12.0 Hz), 5.05 (d, 1H, J = 12.0 Hz), 4.48 (dd, 1H, J = 8.9, 3.4 Hz), 4.14–4.16 (m, 2H), 4.01 (t, 1H, J = 7.9 Hz), 3.65 (s, 3H), 3.40–3.41 (m, 1H), 1.85–1.93 (m, 1H), 1.48–1.50 (m, 1H), 1.29 (d, 3H, J = 6.8 Hz), 1.08–1.11 (m, 1H), 0.98–1.00 (m, 2H), 0.91 (d, 3H, J = 6.8 Hz), 0.87 (t, 3H, J = 7.5 Hz), 0.02 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 168.8, 156.8, 156.7, 136.0, 129.5, 128.7, 128.4, 128.1, 103.9, 67.4, 63.8, 59.9, 57.9, 52.5, 45.4, 37.1, 24.8, 18.6, 17.8, 15.6, 11.3, −1.4; IR (neat) 3297, 2960, 2929, 1747, 1714, 1692, 1664, 1630, 1515, 1455, 1380, 1335, 1283, 1242, 1211, 1176, 1081, 1041, 862, 836, 697 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2485.
3.4.6. The β-Thioenamides (Z)-9f and (E)-9f
Compounds (Z)-9f and (E)-9f were prepared from the carboxylic acid 6f (74.9 mg, 101 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 3.5:1 to 2:1, hexane/IPA = 20:1, toluene/acetone = 20:1, hexane/tBuOH = 9:1) to be obtained in 28% yield (19.9 mg, 28.6 μmol) as a white amorphous solid and 16% yield (11.5 mg, 16.5 μmol) as a white amorphous solid, respectively. (Z)-9f: [α]23D −7.6 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.58 (d, 1H, J = 10.3 Hz), 7.30–7.34 (m, 5H), 7.14 (dd, 1H, J = 10.3, 6.8 Hz), 5.63–5.65 (m, 2H), 5.31 (d, 1H, J = 6.8 Hz), 5.15 (d, 1H, J = 12.3 Hz), 5.10 (d, 1H, J = 12.3 Hz), 4.62–4.61 (m, 1H), 4.55 (dd, 1H, J = 9.6, 3.4 Hz), 4.24–4.25 (m, 1H), 4.12–4.14 (m, 2H), 3.63 (s, 3H), 3.38–3.40 (m, 1H), 3.16–3.18 (m, 1H), 3.07–3.08 (m, 1H), 1.96–1.97 (m, 1H), 1.82–1.83 (m, 1H), 1.69–1.70 (m, 1H), 1.41–1.48 (m, 12H), 1.35 (d, 3H, J = 7.5 Hz), 0.95–0.96 (m, 2H), 0.03 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.4, 169.7, 156.9, 156.6, 156.4, 136.2, 129.6, 128.6, 128.3, 99.9, 79.3, 67.4, 63.8, 58.8, 55.3, 52.5, 46.1, 39.7, 31.4, 29.7, 28.5, 22.5, 18.5, 17.8, −1.4; IR (neat) 3325, 2952, 1698, 1628, 1522, 1365, 1337, 1249, 1172, 1080, 1045, 860, 837, 754, 697 cm−1; HRMS [ESI] m/z calcd for C32H52N4O9NaSSi [M+Na]+ 719.3116, found 719.3137. (E)-9f: [α]23D +0.1 (c 0.58, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.57–8.58 (m, 1H), 7.30–7.35 (m, 5H), 7.04 (dd, 1H, J = 13.3, 10.6 Hz), 5.71 (d, 1H, J = 13.3 Hz), 5.60–5.62 (m, 1H), 5.44 (d, 1H, J = 10.6 Hz), 5.08 (s, 2H), 4.68–4.70 (m, 1H), 4.47 (dd, 1H, J = 8.9, 3.4 Hz), 4.14–4.16 (m, 3H), 3.66 (s, 3H), 3.38–3.40 (m, 1H), 3.06–3.08 (m, 2H), 1.96–1.98 (m, 1H), 1.83–1.85 (m, 1H), 1.63–1.65 (m, 1H), 1.28–1.48 (m, 3H), 1.40 (s, 9H), 1.29 (d, 3H, J = 6.8 Hz), 0.98–1.00 (m, 2H), 0.02 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 169.3, 156.8, 156.7, 156.5, 136.0, 129.9, 128.6, 128.4, 128.2, 103.5, 79.5, 67.4, 63.8, 57.9, 54.8, 52.5, 45.4, 39.4, 31.3, 29.5, 28.5, 22.3, 18.6, 17.8, −1.4; IR (neat) 3313, 2952, 1694, 1628, 1513, 1454, 1365, 1337, 1248, 1172, 1081, 1045, 860, 837, 754 cm−1; HRMS [ESI] m/z calcd for C32H52N4O9NaSSi [M+Na]+ 719.3116, found 719.3125.
4. Conclusions
In summary, we have demonstrated the formation of AviMeCys using lanthionine-bearing peptides 1. The decarboxylative selenoetherification of the NHPI esters 7 smoothly proceeded at a low temperature (−40 °C) in the presence of 1 mol% of eosin Y-Na2 as a photocatalyst, and subsequent β-elimination in a one-pot operation resulted in the corresponding β-thioenamides 9 in moderate to good yields without losing the sulfide-bridged motif. The carbamate-type protecting groups, such as Cbz, Teoc, Boc and Fmoc groups, were tolerant under the reaction conditions, suggesting that the reaction should be useful in synthesizing structurally complicated RiPPs. The synthesis of neothioviridamide [49,50] and its related natural products [51,52,53,54] is underway.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12121615/s1, Figure S1: Experimental setup for photocatalyzed oxidative decarboxylation; Figure S2. Emission spectrum for Kessil A160WE Tuna Blue; 1H and 13C{1H} NMR spectra of synthetic compounds.
Author Contributions
Conceptualization, M.K., K.O. and T.D.; methodology, M.K., K.O. and T.D.; formal analysis, M.K. and K.O.; investigation, M.K. and K.O.; data curation, M.K. and K.O.; writing—original draft preparation, M.K., K.O. and T.D.; writing—review and editing, M.K., K.O. and T.D.; supervision, T.D.; project administration, T.D.; funding acquisition, M.K., K.O. and T.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI Grant Number JP21K15216 and 22H02740, and JST SPRING Grant Number JPMJSP2114 (Tohoku University Advanced Graduate School Pioneering Research Support Project for PhD Students). This research was partially supported by Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP22ama121038.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Montalbán-López, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; Van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Do, T.; Link, A.J. Mechanisms of action of ribosomally synthesized and posttranslationally modified peptides (RiPPs). J. Ind. Microbiol. Biotechnol. 2021, 1, 1524–1540. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, K.J.; van der Donk, W.A. Ribosomally synthesized and post-translationally modified peptide natural product discovery in the genomic era. Curr. Opin. Chem. Biol. 2017, 38, 36–44. [Google Scholar] [CrossRef]
- Katz, B.A.; Johnson, C.; Cass, R.T. Structure-based design of high affinity streptavidin binding cyclic peptide ligands containing thioether crosslinks. J. Am. Chem. Soc. 1995, 117, 8541–8547. [Google Scholar] [CrossRef]
- Aimetti, A.A.; Shoemaker, R.K.; Lin, C.-C.; Anseth, K.S. On-resin peptide macrocyclization using thiol–ene click chemistry. Chem. Commun. 2010, 46, 4061–4063. [Google Scholar] [CrossRef] [Green Version]
- Galande, A.K.; Bramlett, K.S.; Burris, T.P.; Wittliff, J.L.; Spatola, A.F. Thioether side chain cyclization for helical peptide formation: Inhibitors of estrogen receptor-coactivator interactions. J. Pept. Res. 2004, 63, 297–302. [Google Scholar] [CrossRef]
- Rathman, B.M.; Del Valle, J.R. Late-Stage Sidechain-to-Backbone Macrocyclization of N-Amino Peptides. Org. Lett. 2022, 24, 1536–1540. [Google Scholar] [CrossRef]
- Chatterjee, C.; Paul, M.; Xie, L.; van der Donk, W.A. Biosynthesis and Mode of Action of Lantibiotics. Chem. Rev. 2005, 105, 633–684. [Google Scholar] [CrossRef]
- Smith, L.; Hillman, J.D. Therapeutic potential of type A (I) lantibiotics, a group of cationic peptide antibiotics. Curr. Opin. Macrobiol. 2008, 11, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Sit, C.S.; Yoganathan, S.; Vederas, J.C. Biosynthesis of aminovinyl-cysteine-containing peptides and its application in the production of potential drug candidates. Acc. Chem. Res. 2011, 44, 261–268. [Google Scholar] [CrossRef]
- Grant-Mackie, E.S.; Williams, E.T.; Harris, P.W.R.; Brimble, M.A. Aminovinyl Cysteine Containing Peptides: A Unique Motif That Imparts Key Biological Activity. JACS Au 2021, 1, 1527–1540. [Google Scholar] [CrossRef]
- De Leon Rodriguez, L.M.; Williams, E.T.; Brimble, M.A. Chemical Synthesis of Bioactive Naturally Derived Cyclic Peptides Containing Ene-Like Rigidifying Motifs. Chem. Eur. J. 2018, 24, 17869–17880. [Google Scholar] [CrossRef] [PubMed]
- Aydilllo, C.; Avenoza, A.; Busto, J.H.; Jiménez-Osés, G.; Pergrina, J.M.; Zurbano, M.M. A biomimetic approach to lanthionines. Org. Lett. 2012, 14, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Jiménez, M.I.; Aydillo, C.; Navo, C.D.; Avenoza, A.; Corzana, F.; Jiménez-Osés, G.; Zurbano, M.M.; Busto, J.H.; Peregrina, J.M. Bifunctional Chiral Dehydroalanines for Peptide Coupling and Stereoselective S-Michael Addition. Org. Lett. 2016, 18, 2796–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; van der Donk, W.A. Biomimetic Stereoselective Formation of Methyllanthionine. Org. Lett. 2002, 4, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, A.; Lopatniuk, M.; Luzhetskyy, A.; Müller, R.; Koehnke, J. Total In Vitro Biosynthesis of the Thioamitide Thioholgamide and Investigation of the Pathway. J. Am. Chem. Soc. 2022, 144, 5136–5144. [Google Scholar] [CrossRef]
- Lutz, J.; Don, V.S.; Kumar, R.; Taylor, C.M. Influence of sulfur on acid-mediated enamide formation. Org. Lett. 2017, 19, 5146–5149. [Google Scholar] [CrossRef]
- Lutz, J.; Taylor, C.M. Synthesis of the Aminovinylcysteine-Containing C-Terminal Macrocycle of the Linaridins. Org. Lett. 2020, 22, 1874–1877. [Google Scholar] [CrossRef]
- Banerjee, B.; Litvinov, D.N.; Kang, J.; Bettale, J.D.; Castle, S.L. Stereoselective additions of thiyl radicals to terminal ynamides. Org. Lett. 2010, 12, 2650–2652. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Tsuji, J. Palladium-Catalyzed Decarboxylation-Dehydrogenation of Allyl β-Keto Carboxylates and Allyl Enol Carbonates as a Novel Synthetic Method for α-Substituted α,β-Unsaturated Ketones. J. Am. Chem. Soc. 1982, 104, 5844–5846. [Google Scholar] [CrossRef]
- García-Reynaga, P.; Carrillo, K.A.; VanNieuwenhze, S.M. Decarbonylative Approach to the Synthesis of Enamides from Amino Acids: Stereoselective Synthesis of the (Z)-Aminovinyl-D-Cysteine Unit of Mersacidin. Org. Lett. 2012, 14, 1030–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, K.A.; Van Nieuwenhze, S.M. Synthesis of the AviMeCys-Containing D-Ring of Mersacidin. Org. Lett. 2012, 14, 1034–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.A.; Mandrup Kandemir, J.; Walsh, S.J.; Spring, D.R. Photocatalytic methods for amino acid modification. Chem. Soc. Rev. 2021, 50, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Bottecchia, C.; Noël, T. Photocatalytic Modification of Amino Acids, Peptides, and Proteins. Chem. Eur. J. 2019, 25, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malins, L.R. Decarboxylative couplings as versatile tools for late-stage peptide modifications. Pep. Sci. 2018, 110, e24049. [Google Scholar] [CrossRef]
- Murarka, S. N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal. 2018, 360, 1735–1753. [Google Scholar] [CrossRef]
- Li, Z.; Gentry, Z.; Murphy, B.; Vannieuwenhze, M.S. Scalable synthesis of orthogonally protected β-methyllanthionines by indium(III)-mediated ring opening of aziridines. Org. Lett. 2019, 21, 2200–2203. [Google Scholar] [CrossRef]
- Russell, G.A.; Tashtoush, H. Free-radical chain-substitution reactions of alkylmercury halides. J. Am. Chem. Soc. 1983, 105, 1398–1399. [Google Scholar] [CrossRef]
- Perkins, M.J.; Turner, E.S. SH2 reactions of diphenyl diselenide; preparation and reactions of bridgehead selenides. J. Chem. Soc. Chem. Commun. 1981, 3, 139–140. [Google Scholar] [CrossRef]
- Russell, G.A.; Ngoviwatchai, P.; Tashtoush, H.I.; Pla-Dalmau, A.; Khanna, R.K. Reactions of alkylmercurials with heteroatom-centered acceptor radicals. J. Am. Chem. Soc. 1988, 110, 3530–3538. [Google Scholar] [CrossRef]
- Jiang, M.; Yang, H.; Fu, H. Visible-Light Photoredox Synthesis of Chiral α-Selenoamino Acids. Org. Lett. 2016, 18, 1968–1971. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Lumb, J.P. Mimicking oxidative radical cyclizations of lignan biosynthesis using redox-neutral photocatalysis. Nat. Chem. 2021, 13, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Mautner, H.G.; Chu, S.-H.; Gunther, W.H.H. The Aminolysis of Thioacyl and Selenoacyl Analogs. J. Am. Chem. Soc. 1963, 85, 3458–3462. [Google Scholar] [CrossRef]
- Durek, T.; Alewood, P.F. Preformed selenoesters enable rapid native chemical ligation at intractable sites. Angew. Chem. Int. Ed. 2011, 50, 12042–12045. [Google Scholar] [CrossRef]
- Raj, M.; Wu, H.; Blosser, S.L.; Vittoria, M.A.; Arora, P.S. Aldehyde capture ligation for synthesis of native peptide bonds. J. Am. Chem. Soc. 2015, 137, 6932–6940. [Google Scholar] [CrossRef]
- Okada, K.; Okamoto, K.; Morita, N.; Okubo, K.; Oda, M. Photosensitized Decarboxylative Michael Addition through N-(Acyloxy)phthalimides via an Electron-Transfer Mechanism. J. Am. Chem. Soc. 1991, 113, 9401–9402. [Google Scholar] [CrossRef]
- Pratsch, G.; Lackner, G.L.; Overman, L.E. Constructing Quaternary Carbons from N -(Acyloxy)phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis. J. Org. Chem. 2015, 80, 6025–6036. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, V.; Singh, P.P. Eosin Y Catalysed Photoredox Synthesis: A Review. RSC Adv. 2017, 7, 31377–31392. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Xu, R.; Chen, Y. Donor–Acceptor Complex Enables Alkoxyl Radical Generation for Metal-Free C(sp3)–C(sp3) Cleavage and Allylation/Alkenylation. Angew. Chem. Int. Ed. 2017, 56, 12619–12623. [Google Scholar] [CrossRef]
- Crisenza, G.E.M.; Mazzarella, D.; Melchiorre, P. Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [Google Scholar] [CrossRef]
- Van Bergen, T.J.; Hedstrand, D.M.; Kruizinga, W.H.; Kellogg, R.M. Hydride Transfer from 1,4-Dihydropyridine to sp3-Hybridized Carbon in Sulfonium Salts and Activated Halides. Studies with NAD(P)H Models. J. Org. Chem. 1979, 44, 4953–4962. [Google Scholar] [CrossRef]
- Nakamura, K.; Fujii, M.; Mekata, H.; Oka, S.; Ohno, A. Desulfonylation of β-keto sulfones with the hantzsch ester, an nad(p)h model. Chem. Lett. 1986, 15, 87–88. [Google Scholar] [CrossRef]
- Ortiz-López, F.J.; Carretero-Molina, D.; Sánchez-Hidalgo, M.; Martín, J.; González, I.; Román-Hurtado, F.; de la Cruz, M.; García-Fernández, S.; Reyes, F.; Deisinger, J.P.; et al. Cacaoidin, First Member of the New Lanthidin RiPP Family. Angew. Chem. Int. Ed. 2020, 59, 12654–12658. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chatterjee, D.K.; Jani, R.H.; Blumbach, J.; Ganguli, B.N.; Klesel, N.; Limbert, M.; Seibert, G. Mersacidin, a New Antibiotic from Bacillus. J. Antibiot. 1992, 45, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Zhang, F.; Cheng, Z.; Bashiri, G.; Wang, J.; Hong, J.; Wang, Y.; Xu, L.; Chen, X.; Huang, S.-X.; et al. Functional Genome Mining Reveals a Class V Lanthipeptide Containing a D-Amino Acid Introduced by an F420H2-Dependent Reductase. Angew. Chem. Int. Ed. 2020, 59, 18029–18035. [Google Scholar] [CrossRef]
- Elkhalifa, M.; Elbaum, M.B.; Chenoweth, D.M.; Molander, G.A. Solid-Phase Photochemical Decarboxylative Hydroalkylation of Peptides. Org. Lett. 2021, 23, 8219–8223. [Google Scholar] [CrossRef]
- Milewska, K.D.; Malins, L.R. Synthesis of Amino Acid α-Thioethers and Late-Stage Incorporation into Peptides. Org. Lett. 2022, 24, 3680–3685. [Google Scholar] [CrossRef]
- Schwarz, M.K.; Tumelty, D.; Gallop, M.A. Solid-Phase Synthesis of 3,5-Disubstituted 2,3-Dihydro-1,5-benzothiazepin-4(5H)-ones. J. Org. Chem. 1999, 64, 2219–2231. [Google Scholar] [CrossRef]
- Kawahara, T.; Izumikawa, M.; Kozone, I.; Hashimoto, J.; Kagaya, N.; Koiwai, H.; Komatsu, M.; Fujie, M.; Sato, N.; Ikeda, H.; et al. Neothioviridamide, a Polythioamide Compound Produced by Heterologous Expression of a Streptomyces sp. Cryptic RiPP Biosynthetic Gene Cluster. J. Nat. Prod. 2018, 81, 264–269. [Google Scholar] [CrossRef]
- Kjaerulff, L.; Sikandar, A.; Zaburannyi, N.; Adam, S.; Herrmann, J.; Koehnke, J.; Müller, R. Thioholgamides: Thioamide-Containing Cytotoxic RiPP Natural Products. ACS Chem. Biol. 2017, 12, 2837–2841. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Sasaki, K.; Adachi, H.; Furihata, K.; Nagai, K.; Shin-ya, K. Thioviridamide, a Novel Apoptosis Inducer in Transformed Cells from Streptomyces olivoviridis. J. Antibiot. 2006, 59, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumikawa, M.; Kozone, I.; Hashimoto, J.; Kagaya, N.; Takagi, M.; Koiwai, H.; Komatsu, M.; Fujie, M.; Satoh, N.; Ikeda, H.; et al. Novel thioviridamide derivative—JBIR-140: Heterologous expression of the gene cluster for thioviridamide biosynthesis. J. Antibiot. 2015, 68, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Lacret, R.; Cappello, A.R.; Truman, A.W. A Genomics-Based Approach Identifies a Thioviridamide-Like Compound with Selective Anticancer Activity. ACS Chem. Biol. 2017, 12, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Fiorillo, M.; Brindisi, M.; Curcio, R.; Dolce, V.; Lacret, R.; Truman, A.W.; Sotgia, F.; Lisanti, M.P.; Cappello, A.R. Thioalbamide, A Thioamidated Peptide from Amycolatopsis alba, Affects Tumor Growth and Stemness by Inducing Metabolic Dysfunction and Oxidative Stress. Cells 2019, 8, 1408. [Google Scholar] [CrossRef]
Scheme 1.
AviMeCys formation via decarboxylation/decarbonylation of lanthionine derivatives.

Scheme 2.
Preparation of the lanthionine-bearing peptides 1.

Scheme 3.
Photocatalytic selenoetherification/β-elimination of the lanthionine-bearing peptide 1a.

Scheme 4.
Scope of lanthionine-bearing peptides in the AviMeCys formation. a 3.0 equiv of Et3N was used.
Scheme 4.
Scope of lanthionine-bearing peptides in the AviMeCys formation. a 3.0 equiv of Et3N was used.

Scheme 5.
Plausible reaction mechanism.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Preliminary screening of photocatalysts for selenoetherification/β-elimination a.
 | ||
|---|---|---|
| Entry | Photocatalyst | Yield b |
| 1 | [Ru(bpy)3](PF6)2 | 49% (Z: 28%, E: 21%) |
| 2 | [Ir(dtbbpy)(ppy)2]PF6 | 48% (Z: 28%, E: 20%) |
| 3 | Rose bengal-Na2 | 52% (Z: 30%, E: 22%) |
| 4 | Eosin Y-Na2 | 59% (Z: 34%, E: 25%) |
| 5 | none | 53% (Z: 31%, E: 22%) |
| 6 c | Eosin Y-Na2 | - |
a All reactions were conducted on a 0.1 mmol scale. b Isolated yield based on 1a. c No reaction was performed in the absence of the blue light.
Table 2.
Optimization of reaction conditions a.
 | ||||
|---|---|---|---|---|
| Entry | Reductant (Equiv) | Solvent | Temp. (°C) | Yield b |
| 1 | Hantzsch ester (2.0) | DMF | 0 | 59% (Z: 34%, E: 25%) |
| 2 | Hantzsch ester (2.0) | CH2Cl2 | 0 | 33% (Z: 20%, E: 13%) |
| 3 | Hantzsch ester (2.0) | MeCN | 0 | 52% (Z: 33%, E: 19%) |
| 4 | Hantzsch ester (2.0) | DMA | 0 | 53% (Z: 30%, E: 23%) |
| 5 | Hantzsch ester (2.0) | DMSO | 0 | 45% (Z: 26%, E: 19%) |
| 6 | 1-benzyl-1,4-dihydronicotinamide (2.0) | DMF | 0 | 38% (Z: 22%, E: 16%) |
| 7 | γ-terpinene (2.0) | DMF | 0 | 6% (Z: 3%, E: 3%) |
| 8 | DIEA (2.0) | DMF | 0 | 16% (Z: 9%, E: 7%) |
| 9 | Hantzsch ester (1.0) | DMF | 0 | 68% (Z: 36%, E: 32%) |
| 10 | Hantzsch ester (0.5) | DMF | 0 | 51% (Z: 28%, E: 23%) |
| 11 c | none | DMF | 0 | 22% (Z: 13%, E: 9%) |
| 12 | Hantzsch ester (1.0) | DMF | −40 | 74% (Z: 42%, E: 32%) |
| 13 | Hantzsch ester (1.0) | DMF | −78 | 34% (Z: 19%, E: 15%) |
| 14 d | Hantzsch ester (1.0) | DMF | −40 | 51% (Z/E = 57:43) e |
a All reactions were conducted on a 0.1 mmol scale. b Isolated yield based on 1a. c Selenoetherification was conducted for 2 h. d 1.0 mmol scale. e The product was obtained as a Z/E mixture. The ratio was determined by 1H NMR.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kumashiro, M.; Ohsawa, K.; Doi, T. Photocatalyzed Oxidative Decarboxylation Forming Aminovinylcysteine Containing Peptides. Catalysts 2022, 12, 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12121615
AMA Style
Kumashiro M, Ohsawa K, Doi T. Photocatalyzed Oxidative Decarboxylation Forming Aminovinylcysteine Containing Peptides. Catalysts. 2022; 12(12):1615. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12121615
Chicago/Turabian StyleKumashiro, Masaya, Kosuke Ohsawa, and Takayuki Doi. 2022. "Photocatalyzed Oxidative Decarboxylation Forming Aminovinylcysteine Containing Peptides" Catalysts 12, no. 12: 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12121615
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.