
Systematic Functional and Computational Analysis of Glucose-Binding Residues in Glycoside Hydrolase Family GH116

 ,
,  ,
,
Abstract
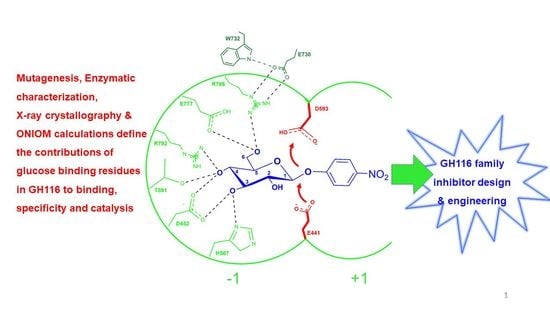
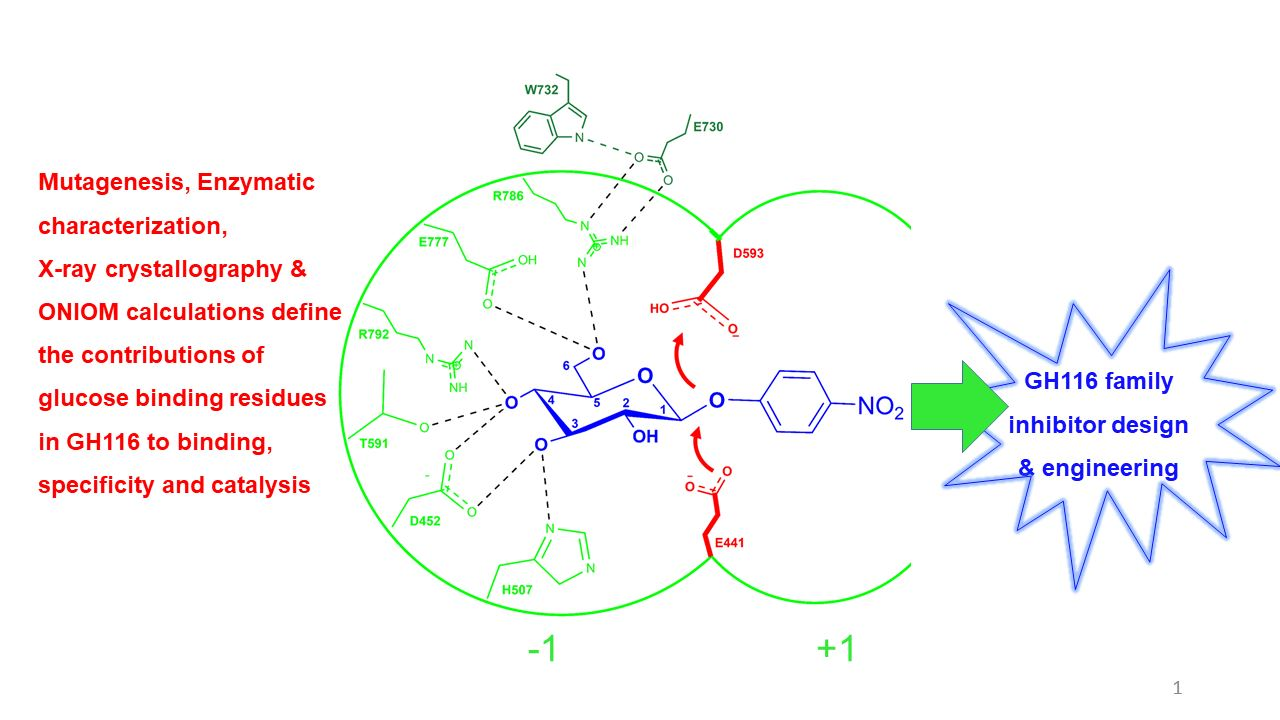
:
1. Introduction
2. Results
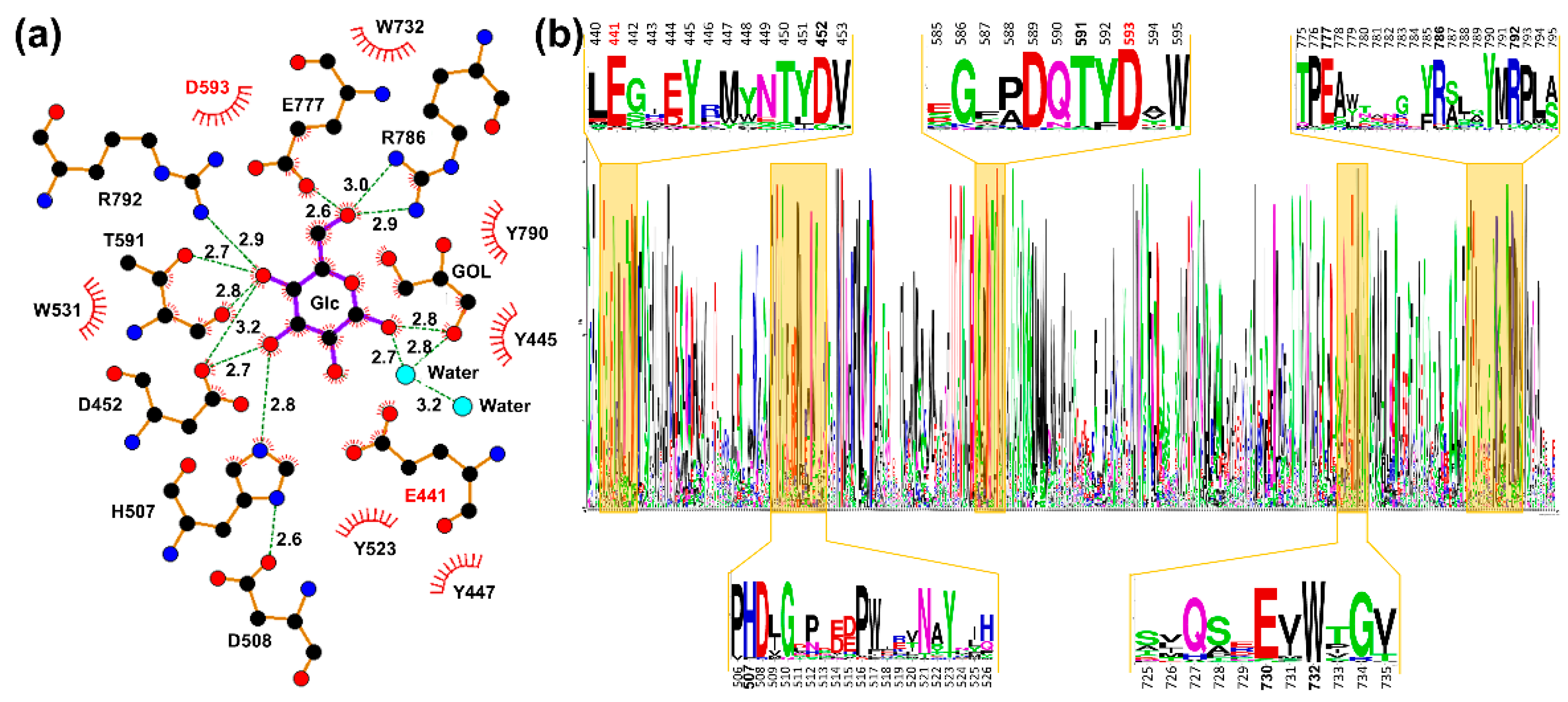
2.1. Analysis of Glucose-Binding Residues
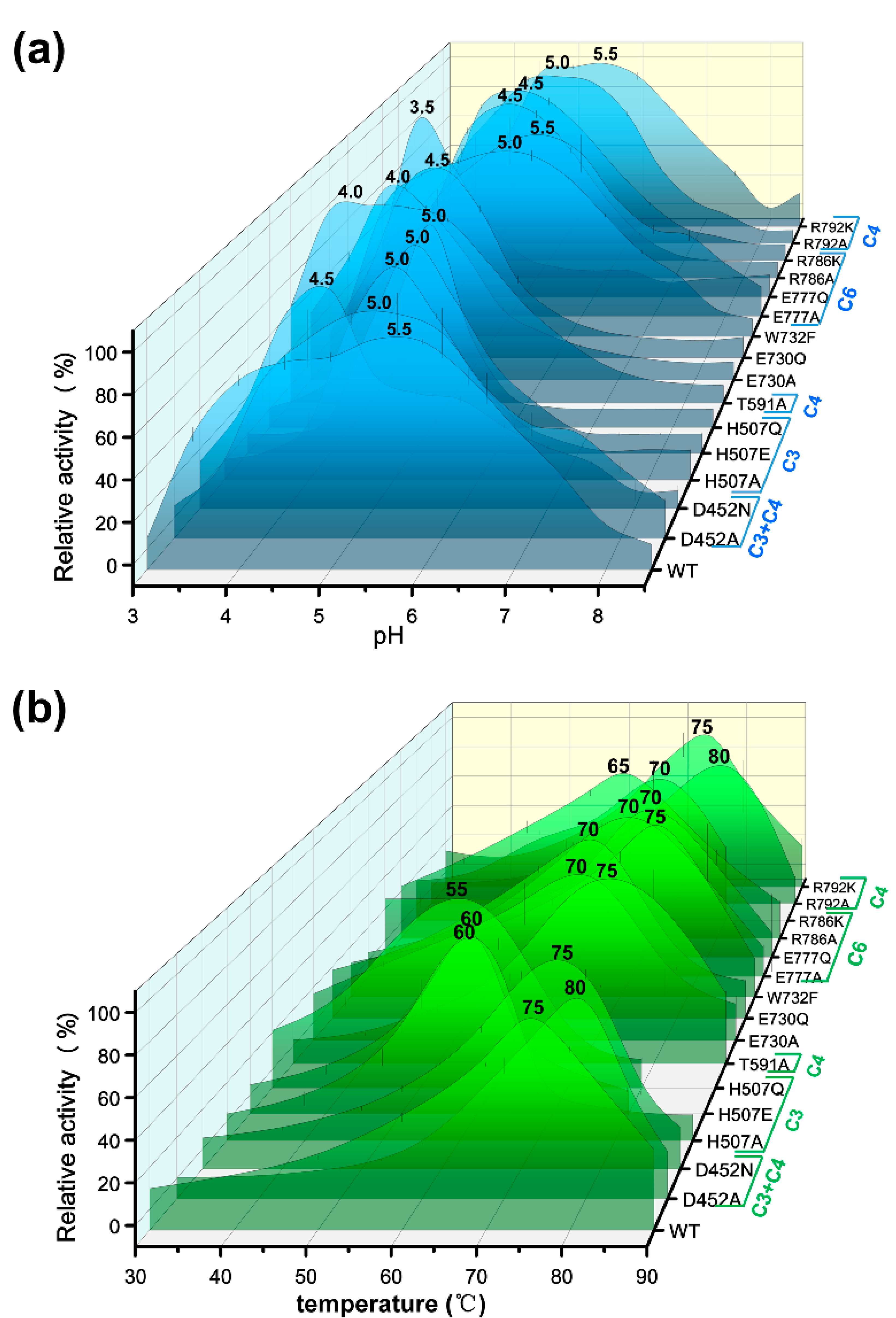
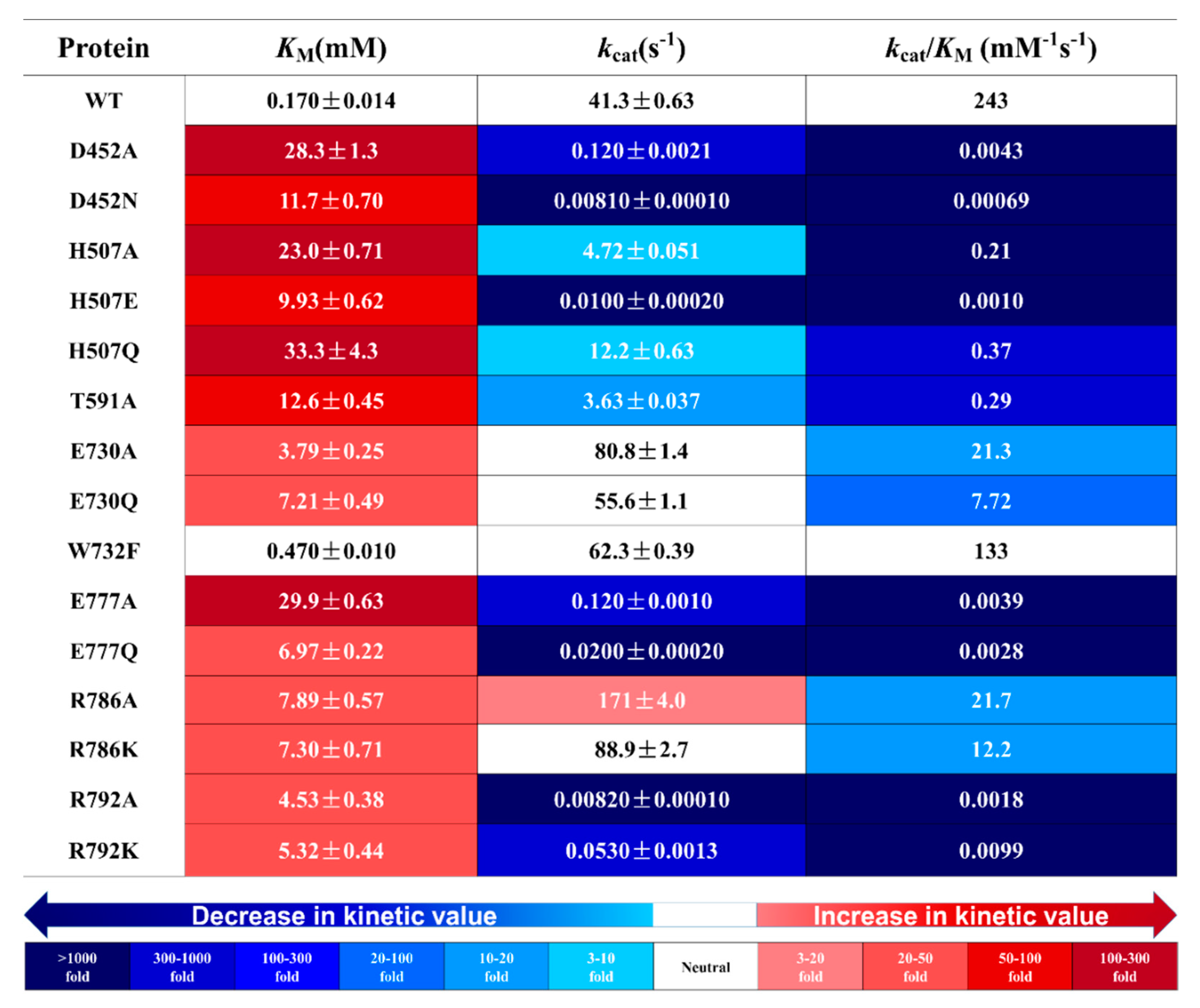
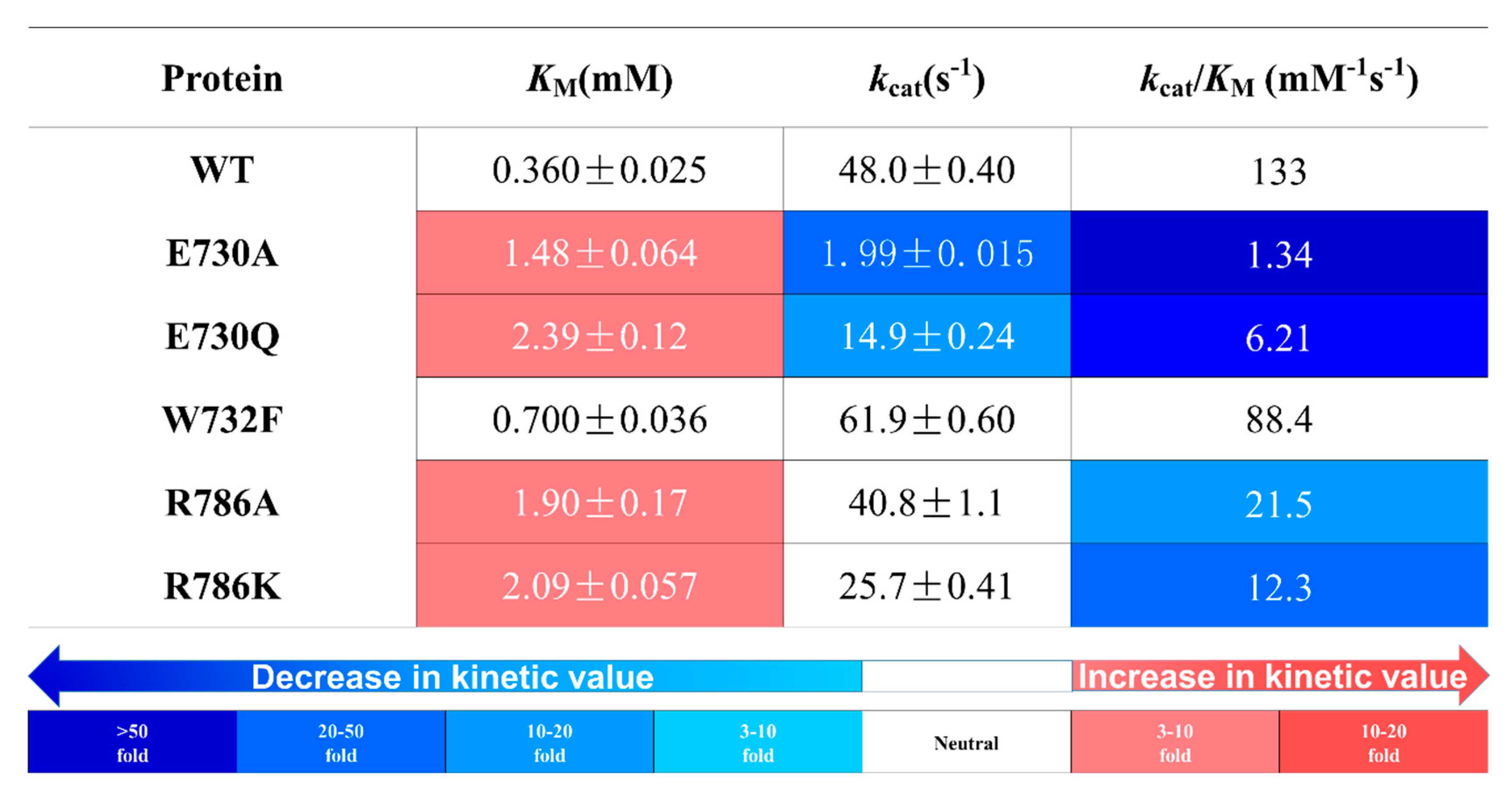
2.2. Effects of Glucose-Binding Residue Mutations on the Activity of TxGH116
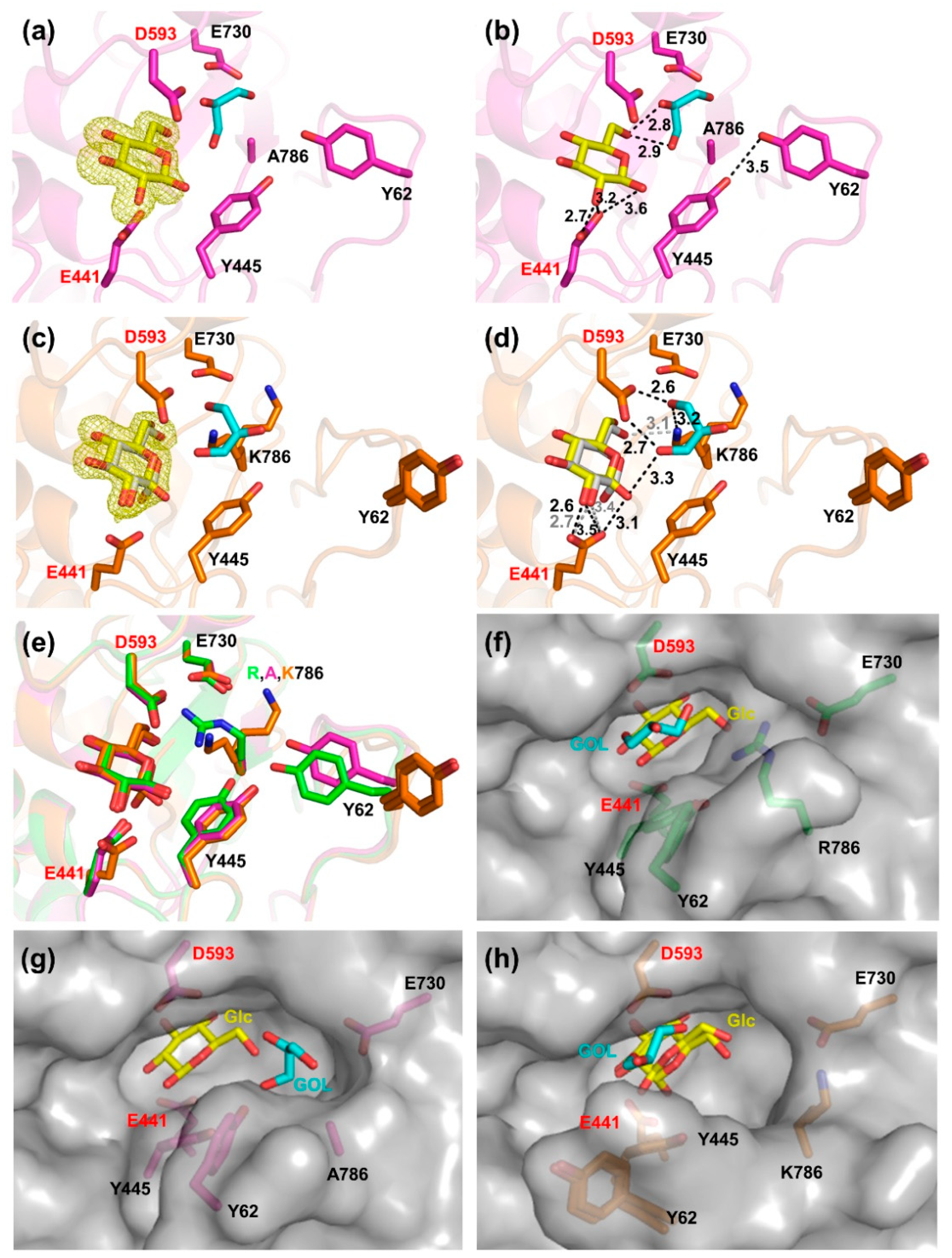
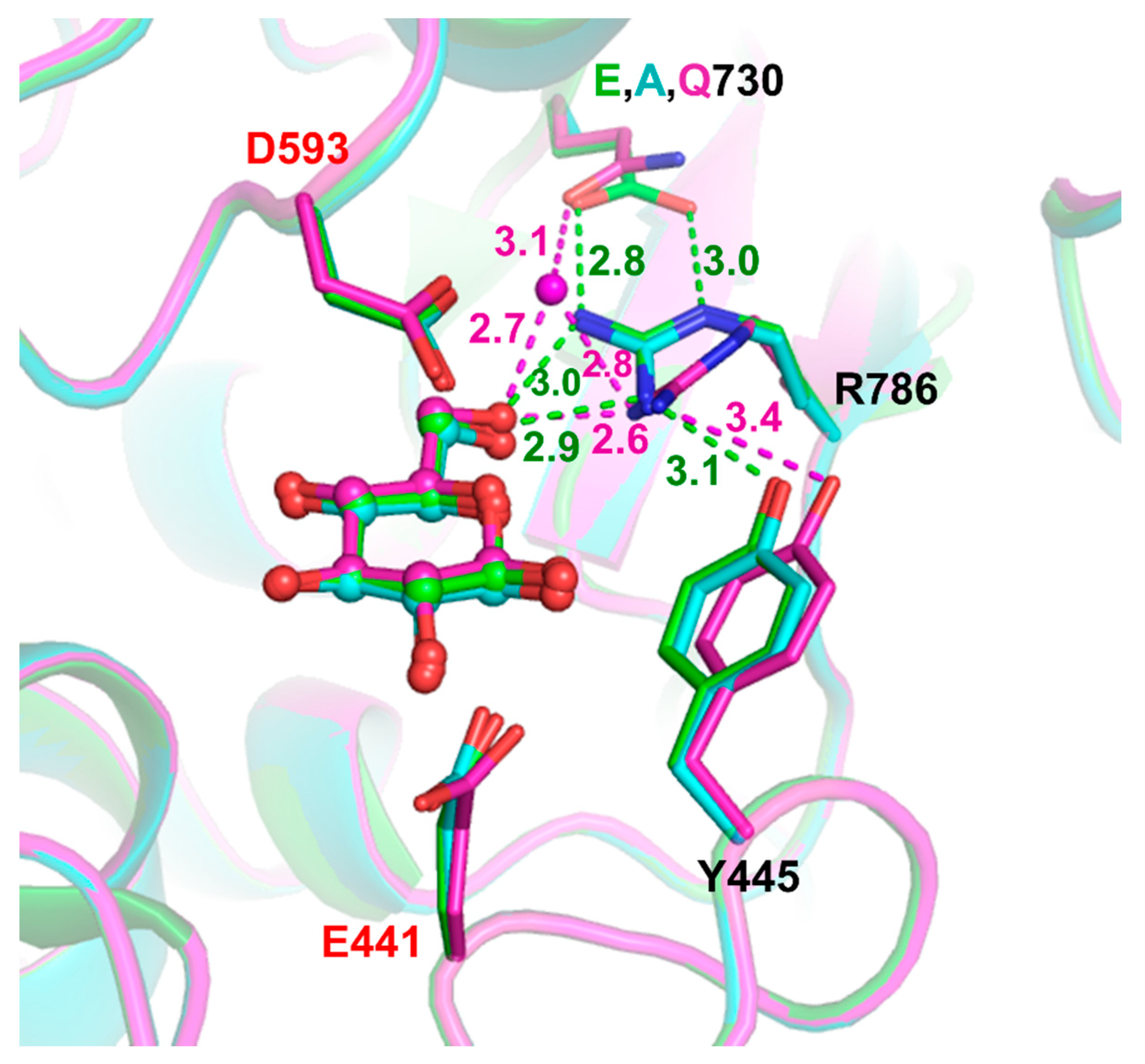
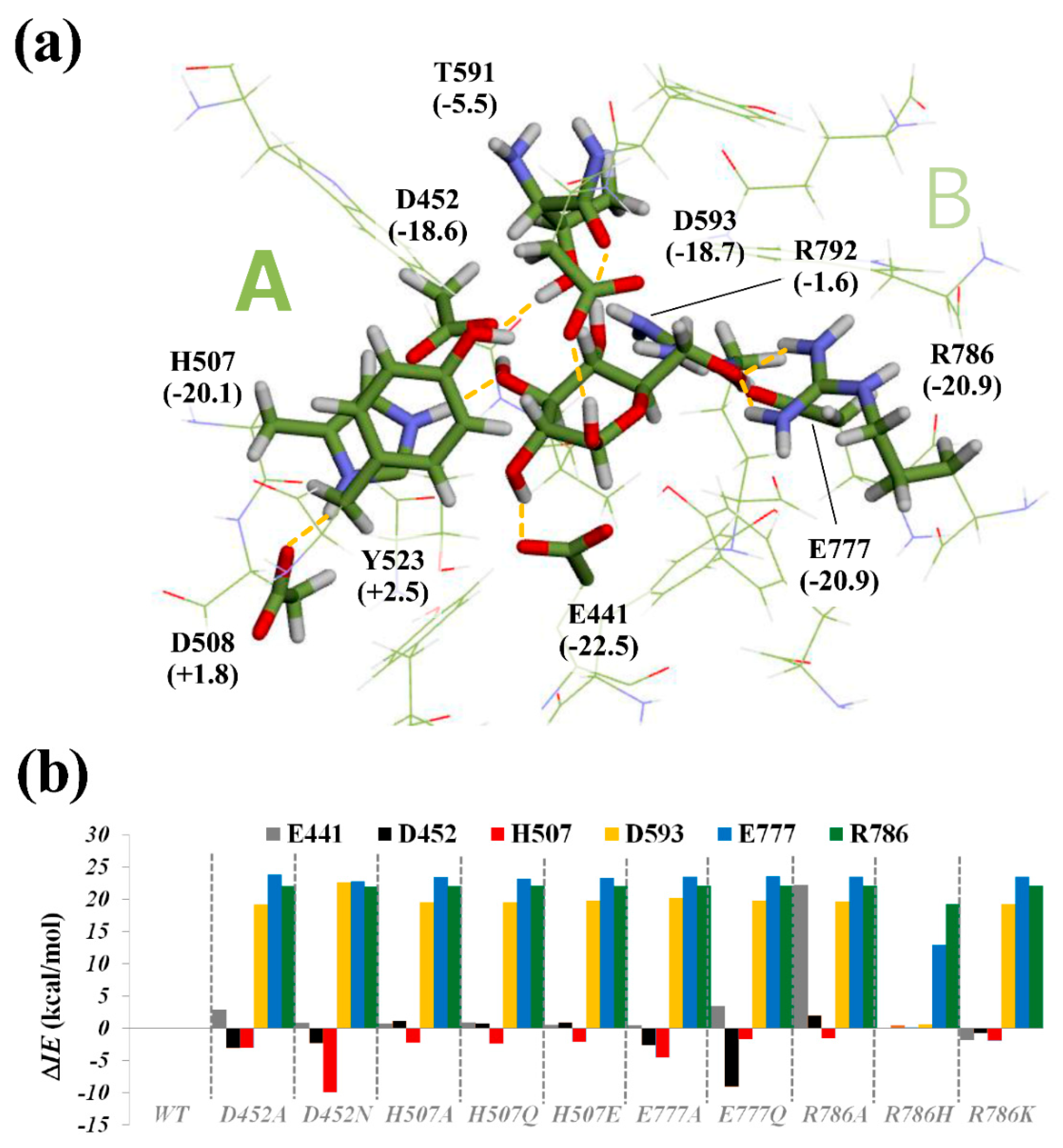
2.3. Structural Investigation of R786 and E730 Mutants
2.4. Inhibition of pNPGlc Hydrolysis by Glucose and Cellobiose
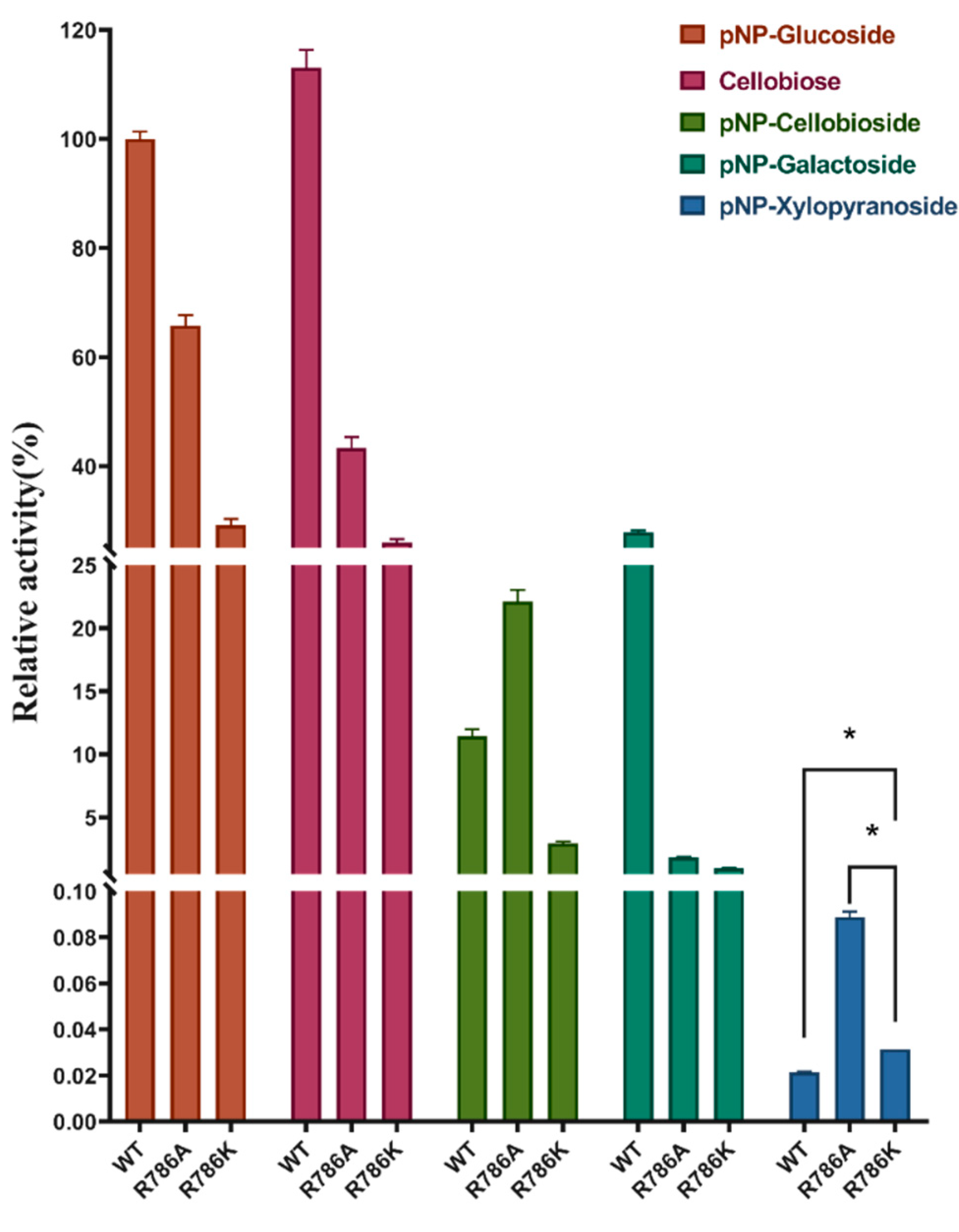
2.5. Effect of R786 Mutations on Sugar Specificity
2.6. Computational Analysis of Glucose-Binding Residues
3. Discussion
4. Materials and Methods
4.1. Conserved Residue Alignment and Site-Directed Mutagenesis of TxGH116 Amino Acid Residues Acting in Glucose Binding
4.2. Protein Expression and Purification and Enzymatic Characterization
4.3. Peroxidase/Glucose Oxidase (PGO) Assay of Oligosaccharide Substrates
4.4. pH and Temperature Dependence
4.5. Glucose and Cellobiose Inhibition Kinetics of Different Mutants
4.6. Protein Crystallization
4.7. Synchrotron X-ray Diffraction and Structure Solution
4.8. Computational Procedures: Binding Energy and Particular Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holliday, G.L.; Bartlett, G.J.; Almonacid, D.E.; O’Boyle, N.M.; Murray-Rust, P.; Thornton, J.M.; Mitchell, J.B. MACiE: A database of enzyme reaction mechanisms. Bioinformatics 2005, 21, 4315–4316. [Google Scholar] [CrossRef] [Green Version]
- Holliday, G.L.; Mitchell, J.B.; Thornton, J.M. Understanding the functional roles of amino acid residues in enzyme catalysis. J. Mol. Biol. 2009, 390, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Ketudat Cairns, J.R.; Mahong, B.; Baiya, S.; Jeon, J.S. Beta-Glucosidases: Multitasking, moonlighting or simply misunderstood? Plant Sci. 2015, 241, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarter, J.D.; Withers, S.G. Mechanisms of enzymatic glycoside hydrolysis. Curr. Opin. Struct. Biol. 1994, 4, 885–892. [Google Scholar] [CrossRef]
- Zechel, D.L.; Withers, S.G. Glycosidase mechanisms: Anatomy of a finely tuned catalyst. Acc. Chem. Res. 2000, 33, 11–18. [Google Scholar] [PubMed]
- McCarthy, J.K.; Uzelac, A.; Davis, D.F.; Eveleigh, D.E. Improved catalytic efficiency and active site modification of 1,4-beta-D-glucan glucohydrolase A from Thermotoga neapolitana by directed evolution. J. Biol. Chem. 2004, 279, 11495–11502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmeister, W.P.; Cottaz, S.; Driguez, H.; Iori, R.; Palmieri, S.; Henrissat, B. The crystal structures of Sinapis alba myrosinase and a covalent glycosyl-enzyme intermediate provide insights into the substrate recognition and active-site machinery of an S-glycosidase. Structure 1997, 5, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Kurakata, Y.; Uechi, A.; Yoshida, H.; Kamitori, S.; Sakano, Y.; Nishikawa, A.; Tonozuka, T. Structural insights into the substrate specificity and function of Escherichia coli K12 YgjK, a glucosidase belonging to the glycoside hydrolase family 63. J. Mol. Biol. 2008, 381, 116–128. [Google Scholar] [CrossRef]
- Vocadlo, D.J.; Davies, G.J. Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 2008, 12, 539–555. [Google Scholar] [CrossRef]
- Chuenchor, W.; Pengthaisong, S.; Robinson, R.C.; Yuvaniyama, J.; Svasti, J.; Cairns, J.R. The structural basis of oligosaccharide binding by rice BGlu1 beta-glucosidase. J. Struct. Biol. 2011, 173, 169–179. [Google Scholar] [CrossRef]
- Geronimo, I.; Payne, C.M.; Sandgren, M. The role of catalytic residue pKa on the hydrolysis/transglycosylation partition in family 3 beta-glucosidases. Org. Biomol. Chem. 2018, 16, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dopitova, R.; Mazura, P.; Janda, L.; Chaloupkova, R.; Jerabek, P.; Damborsky, J.; Filipi, T.; Kiran, N.S.; Brzobohaty, B. Functional analysis of the aglycone-binding site of the maize beta-glucosidase Zm-p60.1. FEBS J. 2008, 275, 6123–6135. [Google Scholar] [CrossRef] [PubMed]
- Pengthaisong, S.; Chen, C.F.; Withers, S.G.; Kuaprasert, B.; Ketudat Cairns, J.R. Rice BGlu1 glycosynthase and wild type transglycosylation activities distinguished by cyclophellitol inhibition. Carbohydr. Res. 2012, 352, 51–59. [Google Scholar] [CrossRef]
- Lucas, J.E.; Siegel, J.B. Quantitative functional characterization of conserved molecular interactions in the active site of mannitol 2-dehydrogenase. Protein Sci. 2015, 24, 936–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, S.; Wu, X.; Liu, S.; Li, D.; Xu, H.; Gao, P.; Chen, G.; Wang, L. Subsite-specific contributions of different aromatic residues in the active site architecture of glycoside hydrolase family 12. Sci. Rep. 2015, 5, 18357. [Google Scholar] [CrossRef] [PubMed]
- Charoenwattanasatien, R.; Pengthaisong, S.; Breen, I.; Mutoh, R.; Sansenya, S.; Hua, Y.; Tankrathok, A.; Wu, L.; Songsiriritthigul, C.; Tanaka, H.; et al. Bacterial beta-Glucosidase Reveals the Structural and Functional Basis of Genetic Defects in Human Glucocerebrosidase 2 (GBA2). ACS Chem. Biol. 2016, 11, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Jeng, W.-Y.; Wang, N.-C.; Lin, C.-T.; Chang, W.-J.; Liu, C.-I.; Wang, A.-J. High-resolution structures of Neotermes koshunensis β-glucosidase mutants provide insights into the catalytic mechanism and the synthesis of glucoconjugates. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Woeste, M.A.; Wachten, D. The enigmatic role of GBA2 in controlling locomotor function. Front. Mol. Neurosci. 2017, 10, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, S.; Stewart, J.; van der Spoel, A.C. Truncated mutants of beta-glucosidase 2 (GBA2) are localized in the mitochondrial matrix and cause mitochondrial fragmentation. PLoS ONE 2020, 15, e0233856. [Google Scholar] [CrossRef] [PubMed]
- van Weely, S.; Brandsma, M.; Strijland, A.; Tager, J.M.; Aerts, J.M. Demonstration of the existence of a second, non-lysosomal glucocerebrosidase that is not deficient in Gaucher disease. Biochim. Biophys. Acta 1993, 1181, 55–62. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Jatooratthawichot, P.; Talabnin, C.; Ngiwsara, L.; Rustam, Y.H.; Svasti, J.; Reid, G.E.; Ketudat Cairns, J.R. Effect of expression of human glucosylceramidase 2 isoforms on lipid profiles in COS-7 cells. Metabolites 2020, 10, 488. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.-L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [Green Version]
- Bouscary, A.; Quessada, C.; René, F.; Spedding, M.; Henriques, A.; Ngo, S.; Loeffler, J.-P. Drug repositioning in neurodegeneration: An overview of the use of ambroxol in neurodegenerative diseases. Eur. J. Pharmacol. 2020, 884, 173446. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Kallemeijn, W.W.; Lelieveld, L.T.; Mirzaian, M.; Zoutendijk, I.; Vardi, A.; Futerman, A.H.; Meijer, A.H.; Spaink, H.P.; Overkleeft, H.S. In vivo inactivation of glycosidases by conduritol B epoxide and cyclophellitol as revealed by activity-based protein profiling. FEBS J. 2019, 286, 584–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artola, M.; Wu, L.; Ferraz, M.J.; Kuo, C.L.; Raich, L.; Breen, I.Z.; Offen, W.A.; Codee, J.D.C.; van der Marel, G.A.; Rovira, C.; et al. 1,6-Cyclophellitol cyclosulfates: A new class of irreversible glycosidase inhibitor. ACS Cent. Sci. 2017, 3, 784–793. [Google Scholar] [CrossRef]
- Lahav, D.l.; Liu, B.; van den Berg, R.J.; van den Nieuwendijk, A.M.; Wennekes, T.; Ghisaidoobe, A.T.; Breen, I.; Ferraz, M.J.; Kuo, C.-L.; Wu, L. A fluorescence polarization activity-based protein profiling assay in the discovery of potent, selective inhibitors for human nonlysosomal glucosylceramidase. J. Am. Chem. Soc. 2017, 139, 14192–14197. [Google Scholar] [CrossRef]
- Artola, M.; Kuo, C.-L.; Lelieveld, L.T.; Rowland, R.J.; van der Marel, G.A.; Codée, J.D.; Boot, R.G.; Davies, G.J.; Aerts, J.M.; Overkleeft, H.S. Functionalized cyclophellitols are selective glucocerebrosidase inhibitors and induce a bona fide neuropathic Gaucher model in zebrafish. J. Am. Chem. Soc. 2019, 141, 4214–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorantla, J.N.; Pengthaisong, S.; Choknud, S.; Kaewpuang, T.; Manyum, T.; Promarak, V.; Ketudat Cairns, J.R. Gram scale production of 1-azido-β-d-glucose via enzyme catalysis for the synthesis of 1,2,3-triazole-glucosides. RSC Adv. 2019, 9, 6211–6220. [Google Scholar] [CrossRef] [Green Version]
- Gorantla, J.N.; Maniganda, S.; Pengthaisong, S.; Ngiwsara, L.; Sawangareetrakul, P.; Chokchaisiri, S.; Kittakoop, P.; Svasti, J.; Ketudat Cairns, J.R. Chemoenzymatic and Protecting-Group-Free Synthesis of 1, 4-Substituted 1, 2, 3-Triazole-α-d-glucosides with Potent Inhibitory Activity toward Lysosomal α-Glucosidase. ACS Omega 2021, 6, 25710–25719. [Google Scholar] [CrossRef] [PubMed]
- Tankrathok, A.; Iglesias-Fernández, J.; Williams, R.J.; Pengthaisong, S.; Baiya, S.; Hakki, Z.; Robinson, R.C.; Hrmova, M.; Rovira, C.; Williams, S.J. A single glycosidase harnesses different pyranoside ring transition state conformations for hydrolysis of mannosides and glucosides. ACS Catal. 2015, 5, 6041–6051. [Google Scholar] [CrossRef]
- Pengthaisong, S.; Hua, Y.; Ketudat Cairns, J.R. Structural basis for transglycosylation in glycoside hydrolase family GH116 glycosynthases. Arch. Biochem. Biophys. 2021, 706, 108924. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Quirke, J.C.K.; Crich, D. Glycoside hydrolases restrict the side chain conformation of their substrates to gain additional transition state stabilization. J. Am. Chem. Soc. 2020, 142, 16965–16973. [Google Scholar] [CrossRef] [PubMed]
- Saen-oon, S.; Kuno, M.; Hannongbua, S. Binding energy analysis for wild-type and Y181C mutant HIV-1 RT/8-Cl TIBO complex structures: Quantum chemical calculations based on the ONIOM method. Proteins 2005, 61, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Nunrium, P.; Kuno, M.; Saen-oon, S.; Hannongbua, S. Particular interaction between efavirenz and the HIV-1 reverse transcriptase binding site as explained by the ONIOM2 method. Chem. Phys. Lett. 2005, 405, 198–202. [Google Scholar] [CrossRef]
- Kuno, M.; Hannongbua, S.; Morokuma, K. Theoretical investigation on nevirapine and HIV-1 reverse transcriptase binding site interaction, based on ONIOM method. Chem. Phys. Lett. 2003, 380, 456–463. [Google Scholar] [CrossRef]
- Romaniouk, A.; Vijay, I.K. Structure-function relationships in glucosidase I: Amino acids involved in binding the substrate to the enzyme. Glycobiology 1997, 7, 399–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namchuk, M.N.; Withers, S.G. Mechanism of Agrobacterium beta-glucosidase: Kinetic analysis of the role of noncovalent enzyme/substrate interactions. Biochemistry 1995, 34, 16194–16202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, Q.; Zhang, W.; Carmichael, I.; Serianni, A.S. DFT and NMR studies of 2JCOH, 3JHCOH, and 3JCCOH spin-couplings in saccharides: C-O torsional bias and H-bonding in aqueous solution. J. Org. Chem. 2007, 72, 7071–7082. [Google Scholar] [CrossRef] [PubMed]
- Quirke, J.C.K.; Crich, D. Side chain conformation restriction in the catalysis of glycosidic bond formation by Leloir glycosyltransferases, glycoside phosphorylases, and transglycosidases. ACS Catal. 2021, 11, 5069–5078. [Google Scholar] [CrossRef] [PubMed]
- Sultana, S.; Reichbauer, J.; Schule, R.; Mochel, F.; Synofzik, M.; van der Spoel, A.C. Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Espina, G.; Eley, K.; Pompidor, G.; Schneider, T.R.; Crennell, S.J.; Danson, M.J. A novel β-xylosidase structure from Geobacillus thermoglucosidasius: The first crystal structure of a glycoside hydrolase family GH52 enzyme reveals unpredicted similarity to other glycoside hydrolase folds. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobucci-Ponzano, B.; Aurilia, V.; Riccio, G.; Henrissat, B.; Coutinho, P.M.; Strazzulli, A.; Padula, A.; Corsaro, M.M.; Pieretti, G.; Pocsfalvi, G.; et al. A new archaeal beta-glycosidase from Sulfolobus solfataricus: Seeding a novel retaining beta-glycan-specific glycoside hydrolase family along with the human non-lysosomal glucosylceramidase GBA2. J. Biol. Chem. 2010, 285, 20691–20703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Mistry, J.; Schuster-Bockler, B.; Griffiths-Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R.; et al. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIlvaine, T. A buffer solution for colorimetric comparison. J. Biol. Chem. 1921, 49, 183–186. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Sansenya, S.; Mutoh, R.; Charoenwattanasatien, R.; Kurisu, G.; Ketudat Cairns, J.R. Expression and crystallization of a bacterial glycoside hydrolase family 116 beta-glucosidase from Thermoanaerobacterium xylanolyticum. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 41–44. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzym. 1997, 276, 307–326. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM method and its applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonsri, P.; Kuno, M.; Hannongbua, S. Key interactions of the mutant HIV-1 reverse transcriptase/efavirenz: An evidence obtained from ONIOM method. Med. Chem. Commun. 2011, 2, 1181–1187. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Glucose Ki (mM) | Cellobiose Ki (mM) |
|---|---|---|
| WT | 4.5 1 | 0.94 |
| R786A | 6.5 | 16.9 |
| R786K | 3.4 | 8.3 |
| E730A | 19.0 | N.D. 2 |
| E730Q | 4.4 | N.D. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, M.; Pengthaisong, S.; Charoenwattanasatien, R.; Thinkumrob, N.; Jitonnom, J.; Ketudat Cairns, J.R. Systematic Functional and Computational Analysis of Glucose-Binding Residues in Glycoside Hydrolase Family GH116. Catalysts 2022, 12, 343. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12030343
Huang M, Pengthaisong S, Charoenwattanasatien R, Thinkumrob N, Jitonnom J, Ketudat Cairns JR. Systematic Functional and Computational Analysis of Glucose-Binding Residues in Glycoside Hydrolase Family GH116. Catalysts. 2022; 12(3):343. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12030343
Chicago/Turabian StyleHuang, Meng, Salila Pengthaisong, Ratana Charoenwattanasatien, Natechanok Thinkumrob, Jitrayut Jitonnom, and James R. Ketudat Cairns. 2022. "Systematic Functional and Computational Analysis of Glucose-Binding Residues in Glycoside Hydrolase Family GH116" Catalysts 12, no. 3: 343. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12030343