
Density Functional Theory Study on the Influence of Cation and Anion Elements Doping on the Surface of Ti3C2 on the Adsorption Performance of Formaldehyde

Abstract
:1. Introduction
2. Calculation Method and Structural Model
3. Results and Discussion
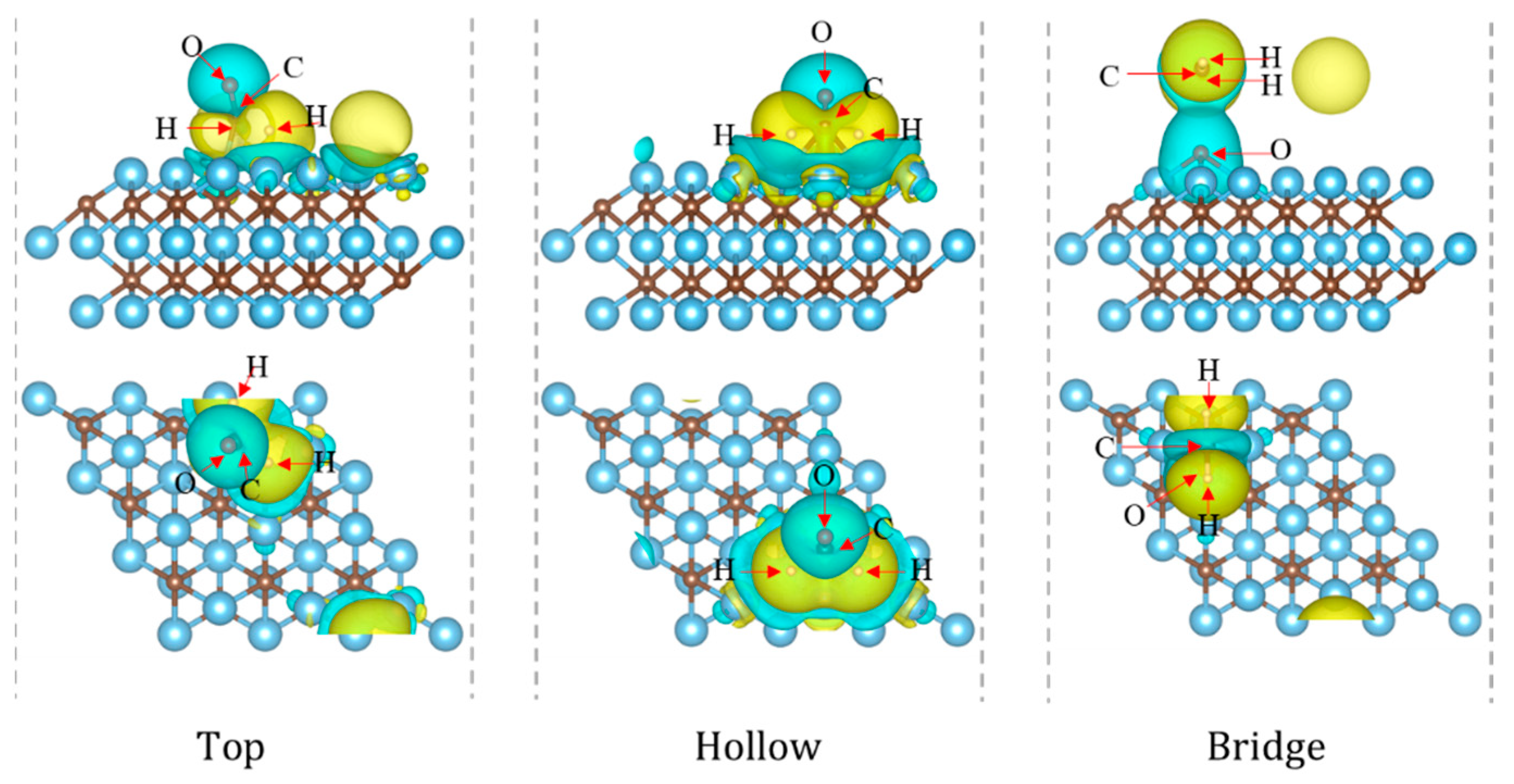
3.1. The Adsorption Characteristics of HCHO on the Surface of Ti3C2
3.2. The Adsorption of Formaldehyde by Element-Doped Ti3C2
3.2.1. Doping System Structure Optimization
3.2.2. Adsorption Characteristics of Doped System to HCHO
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Le, V.T.; Vasseghian, Y.; Doan, V.D.; Nguyen, T.T.T.; Vo, T.-T.T.; Do, H.H.; Vu, K.B.; Vu, Q.H.; Lam, T.D.; Tran, V.A. Flexible and high-sensitivity sensor based on Ti3C2–MoS2 MXene composite for the detection of toxic gases. Chemosphere 2022, 291, 133025. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, W.; Li, G.; Ao, Z.; An, T. Al-modified C2N adsorption enhancement mechanism for for-maldehyde degradation and a density functional theory study of potential catalytic activity. Chin. J. Catal. 2019, 40, 664–673. [Google Scholar]
- Zhang, H.P.; Luo, X.G.; Lin, X.Y.; Lu, X.; Leng, Y.; Song, H.-T. Density functional theory calculations on the adsorption of formaldehyde and other harmful gases on pure, Ti-doped, or N-doped graphene sheets. Appl. Surf. Sci. 2013, 283, 559–565. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, D.; Wei, T.; Wang, L.; Li, X.; Liu, B. Adsorption characteristics of formaldehyde on nitrogen doped graphene-based single atom ad-sorbents: A DFT study. Appl. Surf. Sci. 2019, 493, 1260–1267. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, W.; Hamreh, S. A density functional theory study on the formaldehyde detection mechanism by Pd-decorated ZnO nanotube. J. Phys. Chem. Solids 2020, 144, 109511. [Google Scholar]
- Zhou, X.; Zhao, C.; Chen, C.; Chen, J.; Li, Y. DFT study on adsorption of formaldehyde on pure, Pd-doped, Si-doped single-walled carbon nanotube. Appl. Surf. Sci. 2020, 525, 146595. [Google Scholar]
- Kuganathan, N.; Anurakavan, S.; Abiman, P.; Iyngaran, P.; Gkanas, E.I.; Chroneos, A. Adsorption of lead on the surfaces of pristine and B, Si and N-doped gra-phene. Phys. B Condens. Matter 2021, 600, 412639. [Google Scholar] [CrossRef]
- Luo , H.; Cao, Y.; Zhou, J.; Feng, J.; Cao, J.; Guo, H. Adsorption of NO2, NH3 on monolayer MoS2 doped with Al, Si, and P: A first-principles study. Chem. Phys. Lett. 2016, 643, 27–33. [Google Scholar]
- Zhao, B.; Shang, C.; Zhou, B.; Zhang, R.Q.; Wang, J.J.; Chen, Z.Q.; Jiang, M. Adsorption and dissociation of H2O molecule on the doped monolayer MoS2 with B/Si. Appl. Surf. Sci. 2019, 481, 994–1000. [Google Scholar] [CrossRef]
- Jiang, X.; Yue, X.; Li, Y.; Wei, X.; Zheng, Q.; Xie, F.; Lin, D.; Qu, G. Anion-Cation-dual doped tremella-like nickel phosphides for electrocatalytic water oxidation. Chem. Eng. J. 2021, 426, 130718. [Google Scholar] [CrossRef]
- Kang, W.; Wang, Y.; Wan, Y.; Tuo, Y.; Wang, X.; Sun, D. Embedding anion-doped Fe7S8 in N-doped carbon matrix and shell for fast and stable sodium storage. Mater. Chem. Phys. 2021, 264, 124456. [Google Scholar] [CrossRef]
- Zhu, Y.; Lin, Q.; Wang, Z.; Qi, D.; Yin, Y.; Liu, Y.; Zhang, X.; Shao, Z.; Wang, H. Chlorine-anion doping induced multi-factor optimization in perovskties for boosting intrinsic oxygen evolution. J. Energy Chem. 2020, 52, 115–120. [Google Scholar] [CrossRef]
- Ujjan, Z.A.; Bhatti, M.A.; Shah, A.A.; Tahira, A.; Shaikh, N.M.; Kumar, S.; Mugheri, A.Q.; Medany, S.S.; Nafady, A.; Alnjiman, F.; et al. Simultaneous doping of sulfur and chloride ions into ZnO nanorods for improved photocatalytic properties towards degradation of methylene blue. Ceram. Int. 2021, 48, 5535–5545. [Google Scholar] [CrossRef]
- Thupsuri, S.; Tabtimsai, C.; Ruangpornvisuti, V.; Wanno, B. A study of the transition metal doped boron nitride nanosheets as promising candidates for hydrogen and formaldehyde adsorptions. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 134, 114859. [Google Scholar] [CrossRef]
- Fang, Y.; Yang, D.D.; Xiang, C.Y.; Shi, M.; Zhao, H.; Asadi, H. A density functional study on the formaldehyde recognition by Al-doped ZnO nanosheet. J. Mol. Graph. Model. 2020, 99, 107630. [Google Scholar] [CrossRef]
- Serinay, N.; Fellah, M.F. Acetaldehyde adsorption and detection: A Density Functional Theory Study on Al-doped Graphene. Vacuum 2020, 175, 109279. [Google Scholar] [CrossRef]
- Zhou, Q.; Yuan, L.; Yang, X.; Fu, Z.; Tang, Y.; Wang, C.; Zhang, H. DFT study of formaldehyde adsorption on vacancy defected graphene doped with B, N, and S. Chem. Phys. 2014, 440, 80–86. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, Y.; Li, L.; Song, K.; Wang, X.; Wang, C.; Jian, X.; Ji, C.; Qian, P. Formaldehyde oxidation on Co-doped reduced CeO2 (111): First-principles calculations. Surf. Sci. 2020, 701, 121693. [Google Scholar] [CrossRef]
- Liu, H.; Xia, Q.; Zhang, A.; Luo, W. First-principles study of formaldehyde adsorption on TiO2 surface. Mater. Rev. 2015, 29, 149–154. [Google Scholar]
- Zhou, J.; Liu, G.; Jiang, Q.; Zhao, W.; Ao, Z.; An, T. Density functional theory study on single-atom catalyst Ti/Ti3C2O2 for the catalytic oxidation of formaldehyde. Chin. J. Catal. 2020, 41, 1633–1644. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, S.; Ji, J.; Wei, S. The adsorption activity of OH, O, F and Au on two-dimensional Ti2C and Ti3C2 surfaces. Acta Phys. Chim. Sin. 2015, 31, 369–376. [Google Scholar]
- Zhang, Y.; Zhou, Z.; Lan, J.; Zhang, P. Prediction of Ti3C2O2 MXene as an effective capturer of for-maldehyde. Appl. Surf. Sci. 2019, 469, 770–774. [Google Scholar] [CrossRef]
- Ta, Q.T.H.; Tran, N.M.; Tri, N.N.; Sreedhar, A.; Noh, J.-S. Highly surface-active Si-doped TiO2/Ti3C2TX heterostructure for gas sensing and photodegradation of toxic matters. Chem. Eng. J. 2021, 425, 13143. [Google Scholar] [CrossRef]
- Obodo, K.O.; Ouma, C.; Modisha, P.M.; Bessarabov, D. Density functional theory calculation of Ti3C2 MXene monolayer as catalytic support for platinum towards the dehydrogenation of methylcyclohexane. Appl. Surf. Sci. 2020, 529, 147186. [Google Scholar] [CrossRef]
- Long, R.; Yu, Z.; Tan, Q.; Feng, X.; Zhu, X.; Li, X.; Wang, P. Ti3C2 MXene/NH2-MIL-88B(Fe): Research on the adsorption kinetics and photocatalytic performance of an efficient integrated photocatalytic adsorbent. Appl. Surf. Sci. 2021, 570, 151244. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Wan, X.; Jiang, J.; Ai, L. Edge-rich MoS2 nanosheets anchored on layered Ti3C2 MXene for highly efficient and rapid catalytic reduction of 4-nitrophenol and methylene blue. J. Alloy Compd. 2022, 570, 161900. [Google Scholar] [CrossRef]
- Yu, M.; Liang, H.; Zhan, R.; Xu, L.; Niu, J. Sm-doped g-C3N4/Ti3C2 MXene heterojunction for visible-light photocatalytic degradation of ciprofloxacin. Chin. Chem. Lett. 2020, 32, 2155–2158. [Google Scholar] [CrossRef]
- Liu, N.; Lu, N.; Yu, H.T.; Chen, S.; Quan, X. Efficient day-night photocatalysis performance of 2D/2D Ti3C2/Porous g-C3N4 nanolayers composite and its application in the degradation of organic pollutants. Chemosphere 2019, 246, 125760. [Google Scholar] [CrossRef]
- Bao, X.; Li, H.; Wang, Z.; Tong, F.; Liu, M.; Zheng, Z.; Wang, P.; Cheng, H.; Liu, Y.; Dai, Y.; et al. TiO2/Ti3C2 as an efficient photocatalyst for selective oxidation of benzyl alcohol to benzaldehyde. Appl. Catal. B Environ. 2021, 286, 119885. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Hermawan, A.; Zhu, J.; Yin, S. Surface engineering of Ti3C2Tx MXene by oxygen plasma irradiation as room temperature ethanol sensor. Funct. Mater. Lett. 2022, 15, 2251007. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Hermawan, A.; Zhu, J.; Yin, S. A facile method for preparation of porous nitro-gen-doped Ti3C2Tx MXene for highly responsive acetone detection at high temperature. Funct. Mater. Lett. 2021, 14, 2151043. [Google Scholar] [CrossRef]
- Sun, G.; Kürti, J.; Rajczy, P.; Kertesz, M.; Hafner, J.; Kresse, G. Performance of the Vienna ab initio simulation package (VASP) in chemical applications. J. Mol. Struct. 2015, 624, 37–45. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Shein, I.R.; Ivanovskii, A.L. Graphene-like titanium carbides and nitrides Tin+1Cn, Tin+1Nn (n = 1, 2, and 3) from de-intercalated MAX phases: First-principles probing of their structural, electronic properties and relative stability. Comput. Mater. Sci. 2012, 65, 104–114. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, D.; Wu, Q.; Wang, H.; Wang, L.; Liu, B.; Zhou, A.; He, J. MXene: A New Family of Promising Hydrogen Storage Medium. J. Phys. Chem. A Mol. Spectrosc. Kinet. Environ. Gen. Theory 2013, 117, 14253–14260. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Teng, B.-T.; Zhao, Y.; Wu, F.-M.; Wen, X.-D.; Chen, Q.-P.; Huang, W.-X. A density functional theory study of CF3CH2I adsorption and reaction on Ag(111). Surf. Sci. 2012, 606, 1227–1232. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron Localization Function. Angew. Chem. Int. Ed. Engl. 1992, 36, 1808–1832. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Method | rC–O/Å | /Å | /Å | Etotal/eV | Eads/eV |
|---|---|---|---|---|---|
| Top | 1.09 | 1.41 | 1.41 | −421.32 | −7.58 |
| Hollow | 1.10 | 1.42 | 1.41 | −421.29 | −7.55 |
| Bridge | - | 1.18 | 1.18 | −419.16 | −5.42 |
| Adsorption Site | Adsorption of Basal | Etotal/eV | Esubstrate/eV | rC–O/Å | /Å | /Å | Eads/eV |
|---|---|---|---|---|---|---|---|
| Top | Ti3C2 | −421.32 | −391.61 | 1.09 | 1.41 | 1.41 | −7.58 |
| Cu-Ti3C2 | −416.26 | −386.09 | 1.10 | 1.42 | 1.43 | −8.04 | |
| Pt-Ti3C2 | −416.23 | −389.03 | - | 1.13 | 1.11 | −5.07 | |
| Co-Ti3C2 | −412.73 | −389.96 | - | 1.13 | 1.11 | −0.64 | |
| Si-Ti3C2 | −418.21 | −388.34 | 1.09 | 1.38 | 1.43 | −7.74 | |
| F-Ti3C2 | −415.40 | −385.52 | - | 1.10 | 1.13 | −7.75 | |
| Cl-Ti3C2 | −413.81 | −385.11 | - | 1.10 | 1.12 | −6.57 | |
| Br-Ti3C2 | −413.23 | −384.92 | - | 1.10 | 1.12 | −6.18 | |
| Hollow | Ti3C2 | −421.28 | −391.61 | 1.10 | 1.42 | 1.41 | −7.54 |
| Cu-Ti3C2 | −414.88 | −386.09 | 1.10 | 1.30 | 1.28 | −6.66 | |
| Pt-Ti3C2 | −414.03 | −389.04 | - | 1.11 | 1.13 | −2.86 | |
| Co-Ti3C2 | −411.83 | −389.95 | - | 1.11 | 1.11 | 0.25 | |
| Si-Ti3C2 | −418.56 | −388.34 | 1.10 | 1.43 | 1.43 | −8.09 | |
| F-Ti3C2 | −412.09 | −385.53 | 1.55 | 1.10 | 1.10 | −4.43 | |
| Cl-Ti3C2 | −414.62 | −385.12 | - | 1.11 | 1.12 | −7.37 | |
| Br-Ti3C2 | −413.23 | −384.92 | - | 1.13 | 1.10 | −6.18 | |
| Bridge | Ti3C2 | −419.16 | −391.61 | - | 1.18 | 1.18 | −5.42 |
| Cu-Ti3C2 | −413.67 | −386.09 | - | 1.18 | 1.18 | −5.45 | |
| Pt-Ti3C2 | −413.84 | −389.03 | 1.90 | 1.10 | 1.09 | −2.68 | |
| Co-Ti3C2 | −412.33 | −389.95 | - | 1.13 | 1.11 | −0.25 | |
| Si-Ti3C2 | −415.92 | −388.34 | - | 1.18 | 1.18 | −5.45 | |
| F-Ti3C2 | −415.57 | −385.52 | - | 1.11 | 1.13 | −7.92 | |
| Cl-Ti3C2 | −414.16 | −385.11 | - | 1.10 | 1.13 | −6.92 | |
| Br-Ti3C2 | −408.43 | −384.92 | - | 1.09 | 1.09 | −1.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Q.; Zhu, B.; Zhu, Z.; Chen, M.; Guo, J. Density Functional Theory Study on the Influence of Cation and Anion Elements Doping on the Surface of Ti3C2 on the Adsorption Performance of Formaldehyde. Catalysts 2022, 12, 387. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12040387
Guo Q, Zhu B, Zhu Z, Chen M, Guo J. Density Functional Theory Study on the Influence of Cation and Anion Elements Doping on the Surface of Ti3C2 on the Adsorption Performance of Formaldehyde. Catalysts. 2022; 12(4):387. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12040387
Chicago/Turabian StyleGuo, Qianyu, Baikang Zhu, Zhouhao Zhu, Mengshan Chen, and Jian Guo. 2022. "Density Functional Theory Study on the Influence of Cation and Anion Elements Doping on the Surface of Ti3C2 on the Adsorption Performance of Formaldehyde" Catalysts 12, no. 4: 387. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12040387