
High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites


National Energy Technology Laboratory, U.S. Department of Energy, Pittsburgh, PA 15236, USA
Catalysts 2022, 12(5), 505; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050505
Submission received: 7 March 2022
/
Revised: 26 April 2022
/
Accepted: 28 April 2022
/
Published: 30 April 2022
(This article belongs to the Special Issue 10th Anniversary of Catalysts: Achievements in Computational Catalysis Techniques and Applications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Previous experimental breakthroughs reveal the potential to create novel heterogeneous catalysts for the electroreduction of CO2 to a high-value product CO using ligand-protected Au-based nanoclusters. Since the chemical composition and geometric structures have been precisely defined, it is possible to adopt robust design guidelines for the development of practical catalysts and to fundamentally elucidate the underlying reaction mechanism. In this short review, the computational progress made to understand the experimentally observed reduction process on the following subset of materials—Au25(SR)18−, Au24Pd(SR)18, Au23(SR)16− and Au21Cd2(SR)16−—is described. A significant finding from our first-principles mechanistic studies is that CO2 conversion on the fully ligand protected nanoclusters is thermodynamically unfavorable due to the very weak binding of intermediates on the surface region. However, the reaction becomes feasible when either Au or S sites are exposed through the removal of a ligand. The results particularly point to the role of undercoordinated S sites in the creation of highly functional heterogeneous catalysts that are both active and selective for the CO2 conversion process. The incorporation of dopants could significantly influence the catalytic reactivity of the nanoclusters. As demonstrated in the case of the monopalladium substitution in Au25(SR)18−, the presence of the foreign atom leads to an enhancement of CO production selectivity due to the greater stabilization of the intermediates. With the Cd substitution doping of Au23(SR)16−, the improvement in performance is also attributed to the enhanced binding strength of the intermediates on the geometrically modified surface of the nanocluster.
1. Introduction
The relentless warming trend caused by human greenhouse CO2 emission, primarily from burning fossil fuels, is regarded as an alarming measure of the Earth’s altered climate. The consequence is visible in the local and regional extreme weather events that are disruptive to modern life. Despite a global agreement to strengthen climate commitments, the overall CO2 emission is on track to keep rising, unless a range of effective technologies is developed and deployed. The electro-reduction of captured greenhouse gas CO2 is a promising class of electrochemical transformation in the broad scheme of carbon capture and utilization [1,2,3,4,5,6,7,8,9,10,11,12]. Powered by renewable electricity, it can potentially mitigate the increasing level of CO2 in the atmosphere, while providing a promising pathway towards sustainable and clean CO2 upgrade into a high value product at mild conditions. Among the economically viable products from this process is CO due to its intrinsic properties and its role as a starting reagent in other synthetic pathways.
The electrochemical reduction of CO2 (CO2RR) to CO involves two major steps occurring at the interface between the cathode and electrolyte:
CO2 + 2H+ + 2e− = CO + H2O
Each step involves the transfer of an electron, as the reactant is transformed into a product:
CO2 + H+ + e− = intermediate
intermediate + H+ + e− = CO + H2O
The first proton–electron transfer step involves the activation of C–O bonds to form the related intermediate. In the second step, the intermediate rearranges to products followed by desorption from the surface of the catalyst into the electrolyte. The proton–electron pair originates from the water splitting in the anode:
H2O = 1/2O2 + 2H+ + 2e−
The above process could be sustainable if driven electrochemically using electrical energy from renewable sources, such as wind and solar [13,14,15].
CO2RR, however, presents major performance challenges in terms of efficiency as a particular intractable issue is the CO2 molecule being fully oxidized and thermodynamically stable. Although the equilibrium redox potential of CO2 to CO is merely −0.11 V vs. the reversible hydrogen electrode (RHE), a larger overpotential (defined as the difference between the equilibrium potential and the onset or practical potential) of about −1 V is required due to the significant cost of disrupting the CO2 bonds [2]. When a far larger overpotential is applied, the undesirable hydrogen evolution reaction (HER) competes with CO2RR in the aqueous system generating H2 as a side product. Thus, the acceleration of the kinetics of CO2RR and the concomitant suppression of the competing HER are of great significance for large scale applications.
Facilitating efficiency and selectivity can be promoted via the rational design of effective electrocatalysts. Transition metal nanoparticles (NPs) of transition metals are explored for CO2RR as these materials have a quite solid track record of being used as catalysts for other related reactions, such as CO oxidation [16,17], the oxygen reduction reaction [18,19,20,21] and purely HER [22,23]. Among transition metals, the noble metal Au and Ag stand out in exclusively reducing CO2 to CO. Their excellent mechanical property permits the fabrication of materials with a different size, shape and component to improve CO2RR performance. A cyclic voltammetry experiment on a 200 nm Ag NP revealed a current density peak at a moderate overpotential of about −0.75 V associated with CO production [24]. The feature grows as the NP shrinks from 200 to 1 nm, indicating that the rate of CO2 conversion is sensitive to the particle size. Varying the shape also plays a role as evidenced by an experimentally reported Ag nanoporous catalyst that is very selective for CO products [25]. The current density shows a 300-fold increase over its polycrystalline counterpart at an overpotential of ≤−0.5 V. Based on the powder X-ray diffraction pattern, the observed enhancement is linked to an increase in the number of under-coordinated facets. This is attributed to the favorable binding of the intermediates, resulting in smaller overpotentials needed to overcome the thermodynamic barrier. Experiments on triangular Ag nanoplates with predominant Ag(100) yield a 96.8% selectivity under −0.86 V [26]. Size effect is also utilized to tune the catalytic activity and selectivity of Au NPs [27]. Differently sized NPs ranging from 4 to 10 nm show a prominent size-dependent activity/selectivity in the CO2RR with the 8 nm NP reaching a Faradaic efficiency (FE) of 90% at −0.67 V. Based on density functional theory (DFT) calculations, CO evolution is more favored on the edge sites, while the competitive HER is more active on the corner sites. A significant conclusion is the activity/selectivity of Au NPs can be tuned by varying the size to optimize the ratio of corner and edge sites. A similar conclusion was reached for the size-dependent catalytic activity studies of Au NPs in the size range of ~1–8 nm [28]. A dramatic increase in current density, albeit with a decrease in selectivity for CO, was observed, with decreasing Au particle size. The DFT calculations revealed that changes in the population of low-coordinated edge sites with a stronger chemisorption property are responsible for the upsurge in HER activity.
An emerging strategy is to explore the use of a new class of nanomaterials, the ligand-protected Au-based nanoclusters (NCs). Unlike conventional NPs, these materials exhibit well-defined atomic structures with 10–100 metal atoms in the underlying inorganic core that form selective bonds with ligand stabilizers [29,30,31,32]. As in a few atom clusters, these exhibit molecular-like electronic structures owing to strong quantum confinements effects [33,34]. They are reported to promote the thermal oxidation of styrene-, CO- and sulfur-containing compounds [35,36,37,38]. Thiolate-protected NCs consisting of 25 gold atoms in the core region (Au25(SR)18−, where SR is the thiolate ligand) are the first NCs to be tested for CO2RR [39]. Under aqueous conditions, CO FE reaches nearly 100% at −1.0 V, which is a marked improvement over conventional Au NPs and bulk Au electrodes. It was found that the charged state and the electrochemical catalytic activity are strongly correlated, with the negatively charged species showing the best CO2RR activity, followed by its neutral and the positively charged counterparts [40,41]. The monopalladium-doped NC Au24Pd(SR)18 was reported to achieve nearly 100% CO FE at potential ranging from −0.6 V to −1.2 V, a marked improvement over the pristine species whose FE declines beginning at −0.9 V. Au19Cd2(SR)16−, a doped counterpart of Au23(SR)16−, made by introducing Cd atoms in the exterior portion, was reported to further promote CO2RR with CO selectivity of 90–95% at −0.5 to −0.9 V. This material exhibits the highest CO2RR activity (2200 mA mg−1 at −1.0 V) among the reported NCs.
Obtaining a full understanding of the reaction mechanisms responsible for CO2RR on these NCs is of great interest from a fundamental standpoint, providing complementary data points for the rational design of materials. Since it is not yet plausible to observe the reaction pathways experimentally, it is necessary to explore theoretical avenues. The precise atomic structures available from crystallographic data have made it possible to gain important mechanistic insights into the CO2 conversion on these NCs using theoretical simulation. It also permits further investigation into the synergistic effects on the reaction mechanism brought about by alloying. In the next section, we review our previous complementary theoretical simulation, showing how the outcome helps to shape a mechanistic understanding of CO2RR on these materials.
2. Theoretical Studies of Au-Based Nanoclusters
2.1. Aspects of Modeling
A particular aspect of the modelling of CO2RR is the quantification of binding strength between a given catalyst and the intermediates. The binding states of the adsorbates are influenced not only by the constituent elements of the catalyst, but also the nature of the active sites and any chemical additives. Hence, calculation is an effective strategy that could potentially unveil structure-activity relationships. In what follows, we provide a description of the modeling approaches used for CO2RR to CO on pertinent Au-based NCs under a electrochemical environment.
The thermodynamics for electrochemical reactions were obtained using the computational hydrogen electrode (CHE) approach [42]. At the heart of this technique is the assumption that the free energy of the proton–electron pair () is equal to that of the hydrogen gas () at potential U = 0 V at 101,325 Pa of H2, 298 K and all pH values. When U ≠ 0 V, a potential linear shift of –eU is applied on the free energy. For a proton–electron transfer to adsorbate *A, *A + H+(aq) + e−→*AH, the free energy change becomes , where are the free energy of the product, free energy of the reactant, free energy of gas phase H2 and the elementary positive charge, respectively. For a given reaction step, the limiting potential is obtained, which is defined as the value that makes it exothermic, ΔG 0 eV. For a reaction pathway, the corresponding limiting potential is given by the step with the highest reaction free energy. A key assumption is that the proton–electron transfer barrier in each step is assumed to be surmountable at room temperature as the appropriate potential is applied.
The pathway towards CO production may take the following 2e− CO2RR mechanism [43,44]:
CO2 + H+(aq) + e− + *→*COOH
*COOH + H+(aq) + e−→*CO + H2O
*CO→CO + *
In these formulae, the adsorption of CO2 is accompanied by coupling with an electron/proton pair to initiate the catalysis reaction. The *CO intermediate stems from *COOH after electron/proton transfer and the removal of water. In the last step, the adsorbed CO formed in the second reduction step undergoes molecular desorption. For the competing HER reaction, the 2e− pathway producing H2 may be represented by [28,45]:
H+(aq) + e− + *→*H
*H + H+(aq) + e−→* + H2
In this mechanism, a hydronium ion initially adsorbs on the surface and forms a hydrogen radical, followed by combination of two hydrogen radicals to form a hydrogen molecule. The limiting potentials for H2 evolution and CO2 reduction were determined numerically according to and , respectively. In order not to be severely contaminated by HER, the limiting potential of this process should be as negative as possible compared to CO2RR. The descriptor that represents the difference in the limiting potentials, , can then be used to ascertain the trend in selectivity. That is, a positive value corresponds to preference for CO2RR and the more positive the magnitude becomes, the higher its selectivity over HER.
The relaxation of the geometry was performed using the DFT approach. The employed DFT method was implemented using the Vienna Ab Initio Simulation Package (VASP) [46]. The exchange-correlation functional used in this study was based in the framework of Perdew, Burke and Ernzerhof [47], while the core-electrons were represented by the projector-augmented wave (PAW) method [48]. The Kohn-Sham one electron valence eigenstates were expanded in terms of plane-wave basis sets with a cutoff energy of 500–600 eV. The ionic and electronic convergence limit were set to 0.03 eV/Å and 1 × 10−5 eV, respectively, while the Methfessel-Paxton scheme [49] was utilized with a smearing width of 0.1 eV. A three-dimensional periodic cubic box with 30 Å side was inserted in the simulation models to minimize unphysical interactions. The sampling of the Brillouin zone was conducted with a Γ-point k-point mesh.
The Poisson-Boltzmann implicit solvent model implemented in VASP by Matthew and Hennig was employed [50]. The background dielectric constant of water was set to ɛb ~80 with a cutoff charge density of 0.0025 Å−3. The cavitation energies were evaluated employing a surface tension parameter of 0.525 meV/Å2. Free energy corrections for adsorbed species were applied assuming harmonic degrees of freedom and those for gaseous species were quantified in the ideal gas limit. The free energy for the aqeous species was evaluated by applying free energy corrections to their room temperature state.
2.2. Theoretical Insights
2.2.1. Carbon Dioxide Reduction on Au25(SR)18−
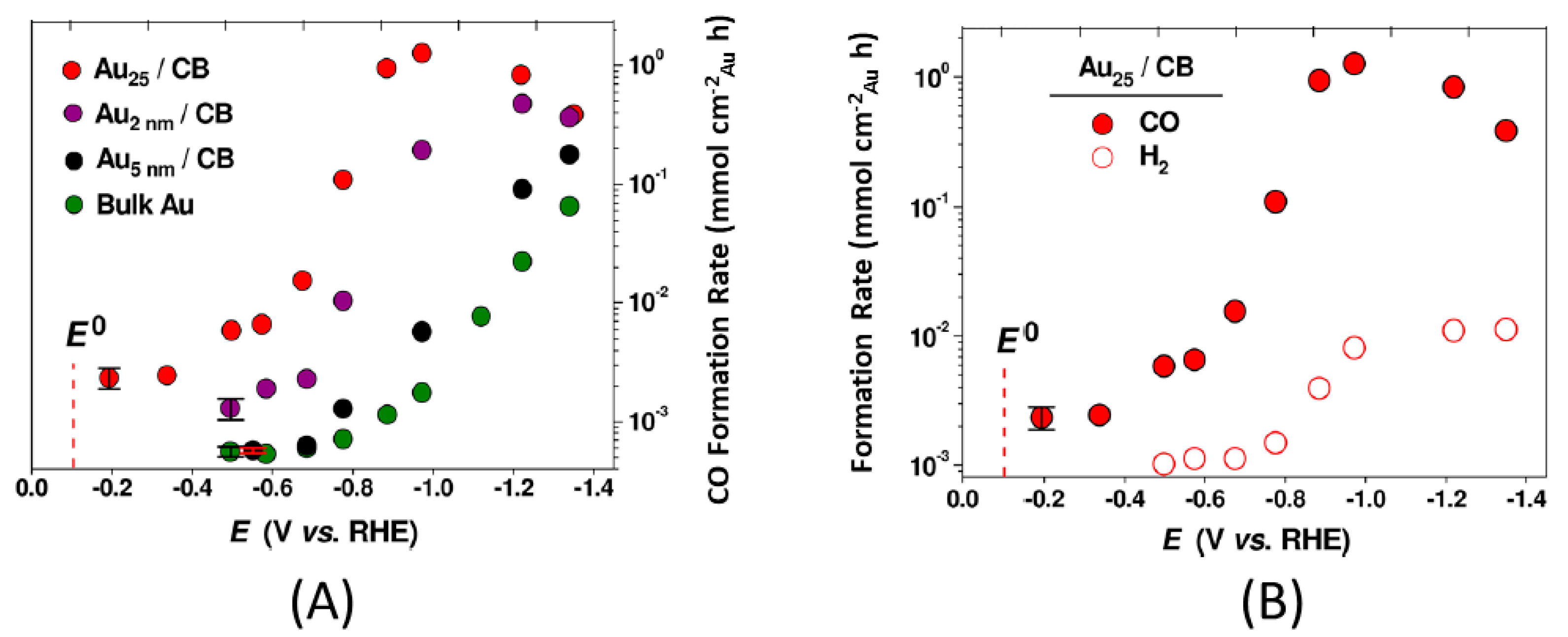
In this section, our group’s effort to understand the mechanistic pathways of CO2RR to CO on Au25(SR)18− is presented. The experimental work of Kauffman et al. brought Au25(SR)18− to the spotlight where it was demonstrated that the NC is active in CO2RR. It can successfully reduce CO2 to CO beginning with an overpotential of −0.19 V (Figure 1A) [39,40], indicating that Au25(SR)18− is superior to Au nanoparticles and bulk Au by 0.2–0.3 V. The FE for selective CO production reaches approximately 100% at −1.0 V (Figure 1B), which shows that almost every electron injected is harnessed in the conversion process. Additionally, the CO formation rate is 7−700 times higher than the conventional Au NPs and bulk Au electrodes at this industrially relevant potential. The corresponding neutral and positively charged counterparts, Au25(SR)18 and Au25(SR)18+, yield comparable products and onset potentials. However, the current density, turnover frequency and FE are higher in Au25(SR)18−.
Though the experimental structures of the NCs are available experimentally, simulation models that perfectly match the data can become computationally prohibitive. For example, Au25(SR)18− would require a 349-atom representation when each of the 18-atom ligand (–SR = –SCH2CH2C6H5) is included. Moreover, for plane-wave basis sets with a typical energy cutoff of 500–600 eV, a sizeable periodic box to blot out any unphysical effect due to image interaction needs to be employed.
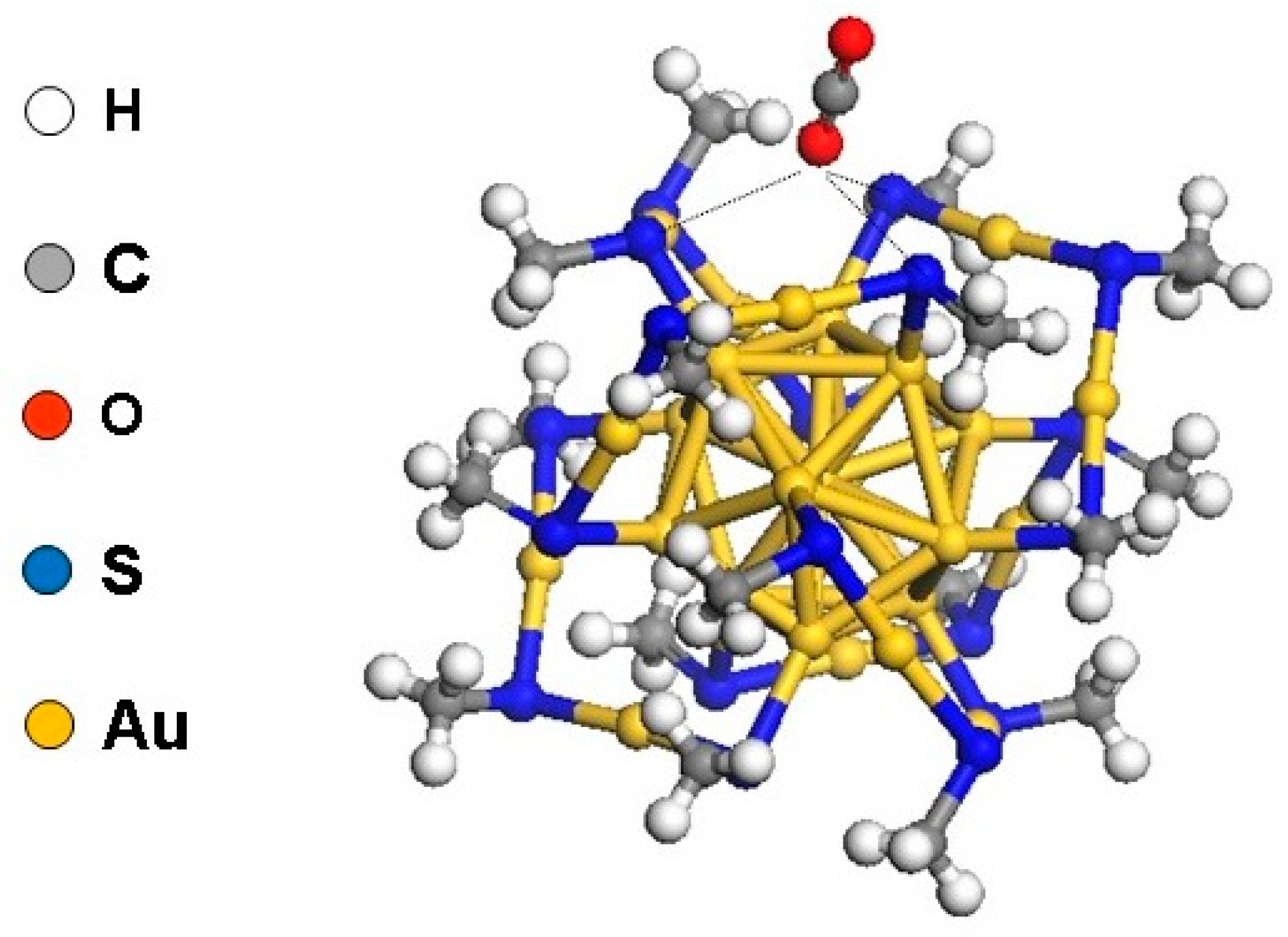
To remedy this, we utilized a methyl thiolate (–SCH3) to represent the real ligand, thereby reducing the model to a relatively manageable 115 atom representation [39]. The use of a truncated version of the ligands can be traced back to a previous DFT work in which a smaller –SH ligand representation is used [51]. Our model is an upgrade in that it mimics better the overall electronegativity of –SCH2CH2C6H5. With a viable and realistic atomic representation at hand, we explored the active sites in the intact nanocluster. With CO2 as the adsorbate, our calculations identified several configurations with binding energies of up to −0.2 eV. The most energetically favorable structure consisted of the oxygen end of CO2 interacting with three adjacent sulfur atoms (Figure 2). However, the theoretical picture of a weak CO2 binding was problematic since the adsorbed molecule could desorb at ambient temperature. At this juncture, we were guided by the prevailing conjecture that the active sites were located on the intact NC [35,36,37,38].
With our initial foray providing a practical Au25(SCH3)18− model, baseline data and further intuition, we then embarked on more comprehensive studies to refine our understanding. Merely looking at the initial CO2 adsorption offers a very limited picture of the process. Other aspects, such as the subsequent chemical transformation, including the reaction thermodynamics, need to be addressed. In the follow up studies, we looked at the reaction profile using the widely used CHE method [52]. We also took into account previous efforts showing both the inactivity of the intact nanocluster in the context of thermal CO oxidation [35], and the critical role of undercoordinated sites as the driving force for aldehyde hydrogenation and other oxidation reactions [53,54].
The free energy profile for the reaction pathway based on CO2RR steps outlined in Section 2.1 and the optimized intermediate structures on the intact NC are displayed in Figure 3. For an external potential of U = 0 V, the protonation of CO2 to form COOH is endothermic, requiring 2.04 eV, while the dissociation of the protonated COOH is exothermic. After surface CO is formed, it can detach with a small barrier to form the gas phase molecule. The COOH formation is the potential limiting step with an overpotential of −2.04 V. However, this is about one order of magnitude larger than the experimental value at which the initiation of CO production is observed (~−0.1 V) [39]. A significant finding is that CO2RR is unlikely to occur in intact form at room temperature and low overpotentials.
The suggestion that undercoordinated atoms in the negatively charged nanocluster promoted thermal aldehyde hydrogenation and other oxidation reactions opened new vistas for further exploration [35,53,54]. For example, in situ IR and complementary DFT calculations showed the intact NC as inert for thermal CO oxidation [35]. Oxidation took place when a defect was introduced on the surface region through the partial removal of ligands. This was a vivid indicator of property-structure relationships in that the improved performance was attributed to the presence of a defect termination with low coordination numbers.
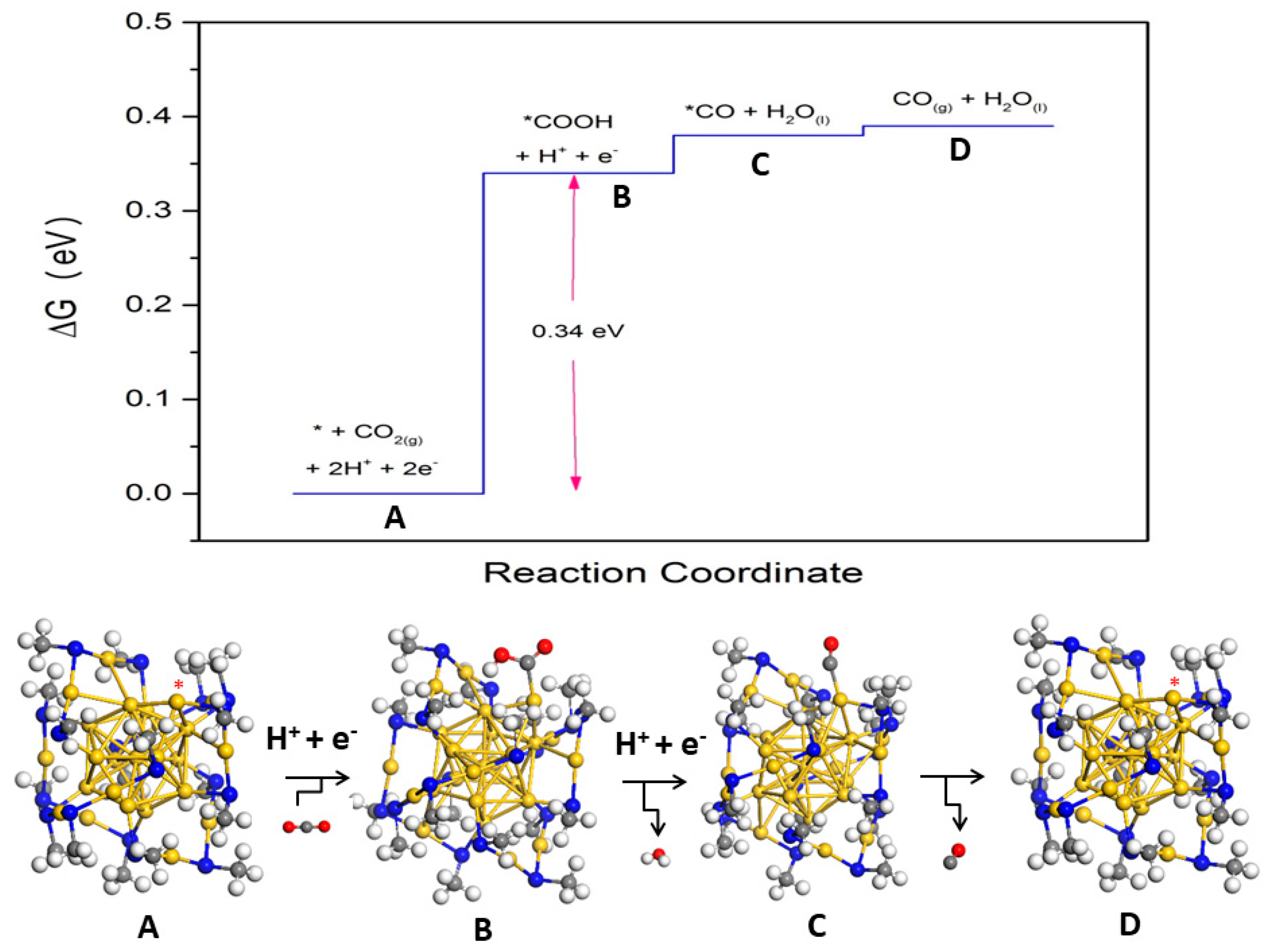
The CO2RR process was reexamined by generating via a single dethiolation yielding undercoordinated Au as the active site. The energies required for the formation of the adsorbed intermediates (*COOH and *CO) were predicted to be dramatically lower than the intact case (Figure 4). The lower limit of the overpotential associated with the first reduction step was predicted to be −0.34 V, which was in better agreement with the measured value. The possibility of a non-metal S site as the reaction center provides an alternative scenario [55]. Instead of the whole ligand, only the hydrocarbon portion was removed, producing a defect consisting of an exposed S atom. Undercoordinated S atoms were also proposed as active sites for cubane-shaped Ni–Fe–S [56], and MoS2 catalysts [57]. An endothermic and exothermic ΔG for the electrochemical formation of Au and S sites, respectively, were predicted indicating that the stripping away of CH3 is more thermodynamically favorable. A significant finding is that the S site for CO2 reduction was also active for H2 evolution. To gain further insight into the selectivity trend, Austin et al. used the descriptor , where the first and second term refer to limiting potentials for CO2RR and HER. Unlike the Au site, CO2RR was predicted to be selective on the S site as evidenced by the positive value [55]. Thus, an improved coupling between theory and experiments was attained when the S site was considered as the reaction center.
2.2.2. Promotion of Carbon Dioxide Reduction through Doping
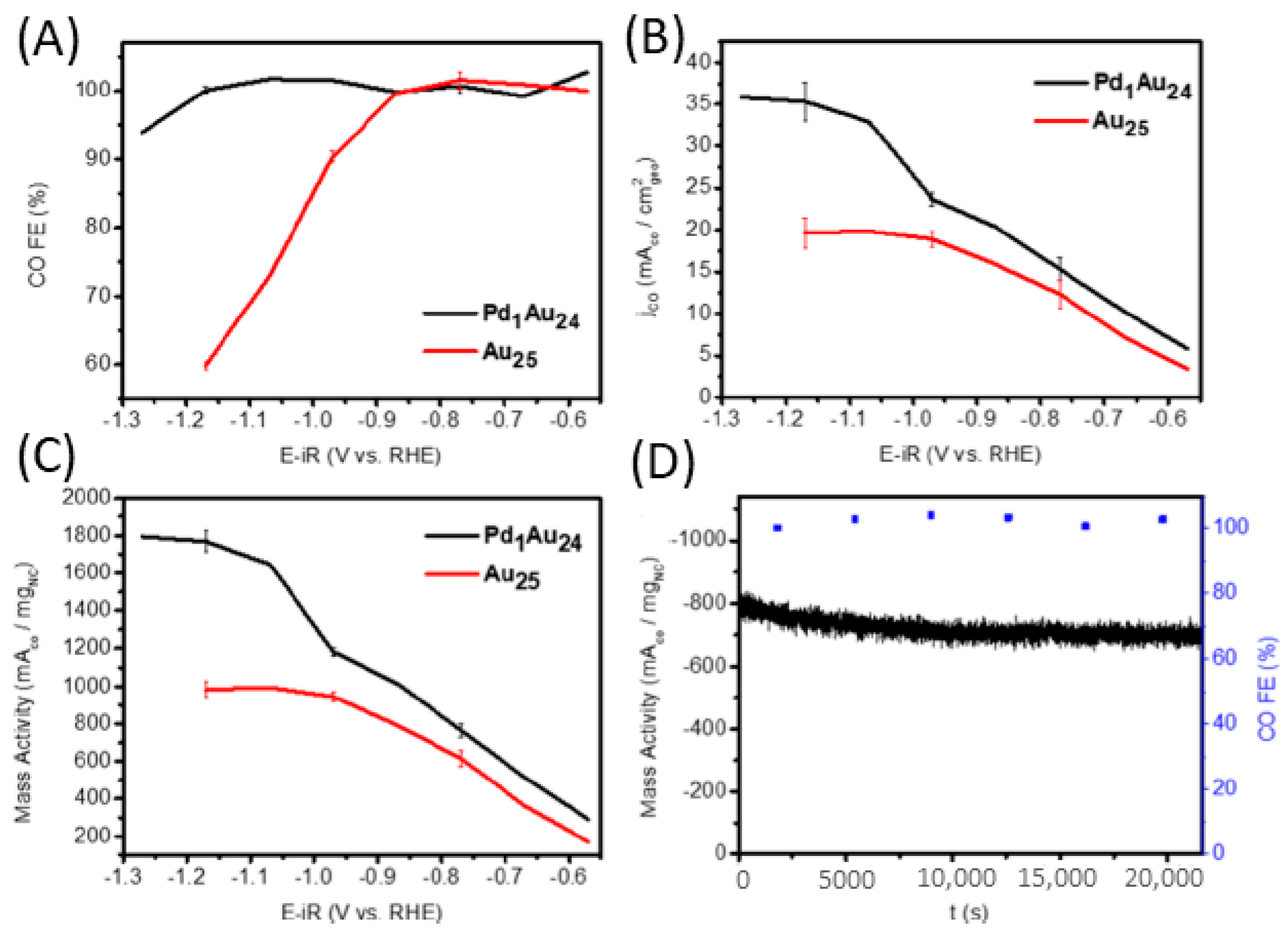
We explored the incorporation of dopant as an approach to facilitate CO2RR. From the scanning tunneling electron microscopy (STEM) images, both size and significant morphology can be ruled out upon monopalladium substitution in Au25(SR)18−, namely, the monodispersed ~0.8 nm size was essentially preserved (Figure 5A,B). In situ spectroscopy analysis combined with CHE calculations revealed that the substitution promoted electroreduction activity and selectivity [58]. Figure 6A–C show the measured electrochemical CO2RR activity of Au24Pd(SR)18 versus Au25(SR)18, indicating CO and H2 as the primary products of both materials. The potential-dependent CO FE showed a significant amount of the molecule was generated, reaching approximately 100% at a potential range of −0.6 to −0.9 V. Beyond −0.9 V, the catalytic performance of Au25(SR)18 decreased, whereas the doped counterpart was not significantly compromised. The CO partial current density also revealed that Au24Pd(SR)18 exhibited higher CO partial current density and mass activity at the same conditions. Unlike conventional Au NPs and bulk Au electrodes, Au24Pd(SR)18 is capable of attaining 100% CO FE over a much wider potential range and a near-complete HER suppression up to −1.2 V. Moreover, it possesses a high stability even under the negative potential for CO2RR (Figure 6D). Thus, doping the parent NC with a Pd atom can elevate both activity and selectivity.
A significant conclusion drawn in Section 2.2.1 is that the intact cluster can be ruled out as an active catalyst, highlighting the role of undercoordinated sites in facilitating CO2RR. The removal of one of the thiol ligands unveils a reactive metal site that lowers the formation of the crucial COOH intermediate for CO2 reduction. Another feasible scenario is also predicted, namely, the electrochemical displacement of the hydrocarbon portion of the thiol ligand to form an exposed S as the reaction center. We used Au25(SCH3)18 and Au24Pd(SCH3)18 simulation models to represent the intact parent and the doped NCs and removed the SCH3 and CH3 portions to expose Au and S sites. The calculations predicted that the Pd atom prefers to reside in the core site of the activated NCs, and the spatial preference did not change in the presence of the CO, COOH and H intermediates.
Figure 7A,B summarize the predicted free energy diagram of CO2RR and HER on the exposed metal site of Au25(SCH3)17 and Au24Pd(SCH3)17. In both cases, the potential limiting step for CO2RR was predicted to be the first electron/proton transfer step. The binding of the COOH intermediate in the doped NC was slightly stronger than the parent NC, resulting in the lowering of the potential requirement ( = −0.58 V compared to = −0.67 for the parent NC). For HER, the protonation of adsorbed H to form gaseous H2 is predicted to be the step that dictates the potential requirements since it occurs at positive free energy. The limiting potential in the pure case ( = −0.08 V) is lower than the doped one ( = −0.17 V). The predicted for the parent and the doped version are −0.59 V and −0.41 V, suggesting that selective CO2 reduction is not favorable on the exposed metal site.
Since the above calculations ruled out the possibility of selective CO2 reduction on the metal site, the alternate scenario involving exposed sulfur as the active site was considered. The calculated CO2RR and HER free energy diagrams in Figure 7C,D show the first electrochemical step as the limiting step of CO2RR in both NCs. While the formation of *COOH species still required an increase in free energy, the key difference was the enhanced stability of this intermediate on sulfur. The adsorption of both COOH and H intermediates were found to be more favorable than on the Au site. On both NCs, the electrochemical H2 desorption remained to be the potential limiting step since it was the most endothermic step. The calculated yielded values of −0.45 and 0.24 V for Au25S(SCH3)17 and Au24PdS(SCH3)17. Although our results showed that the Au25S(SCH3)17 cluster can be active for CO2RR, it is only the Au24PdS(SCH3)17 that is selective to CO2RR due to the positive value of . Thus, we find that the Pd dopant can play a critical role in stabilizing the key intermediates that better facilitates CO2RR. Moreover, the presence of the dopant leads to a more selective CO production when the reaction center consists of exposed S sites.
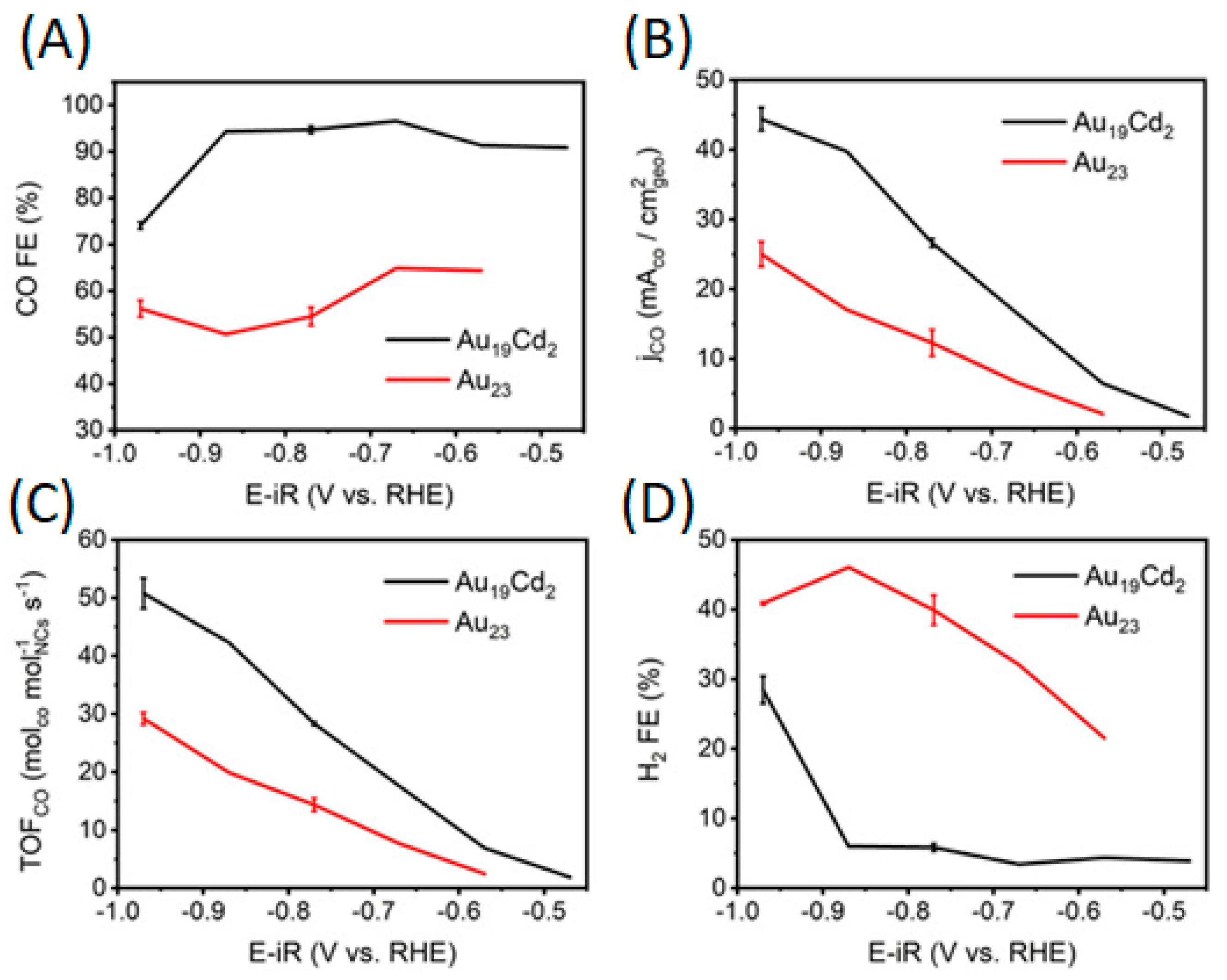
Another approach explored was to exclusively dope the NC surface without compromising the inner kernel metal structure of the parent material. Our work on the surface level modification of Au23(SR)16 through doubly Cd substitution revealed that this was a viable scheme [59]. The CO2RR activities of the Au23(SR)16− and Au19Cd2(SR)16− NCs were compared by conducting constant-potential electrolysis measurements at various applied potentials. It was found that the surface-doped version exhibited higher current densities for CO production (Figure 8B) and turnover frequencies with reduced onset potential (Figure 8C) than the undoped case. The doped version yielded CO as the major product between −0.5 and −0.9 V with HER becoming more competitive for potentials larger than −0.9 V (Figure 8A,D). In contrast, H2 production was more dominant in the undoped version at all potentials. Notably, Au19Cd2(SR)16− demonstrates a larger CO partial current density than the previously reported Au NC and Au NP [59]. The ~40 mA/cm2 value at −0.9 V is superior to the 2–18 mA/cm2 values reported for Au44(TBBT)28, Au47Cd2 (TBBT)31 (TBBT = tert-butylbenzenethiolate) and ultrathin Au–Pd nanocrystals at the same potential [60,61]. Additionally, the partial current density of ~45 mA/cm2 at −1.0 V is about 33–90% higher than Au NP, Mo-doped Au NP and Au NP containing a CeOx interface [62,63,64].
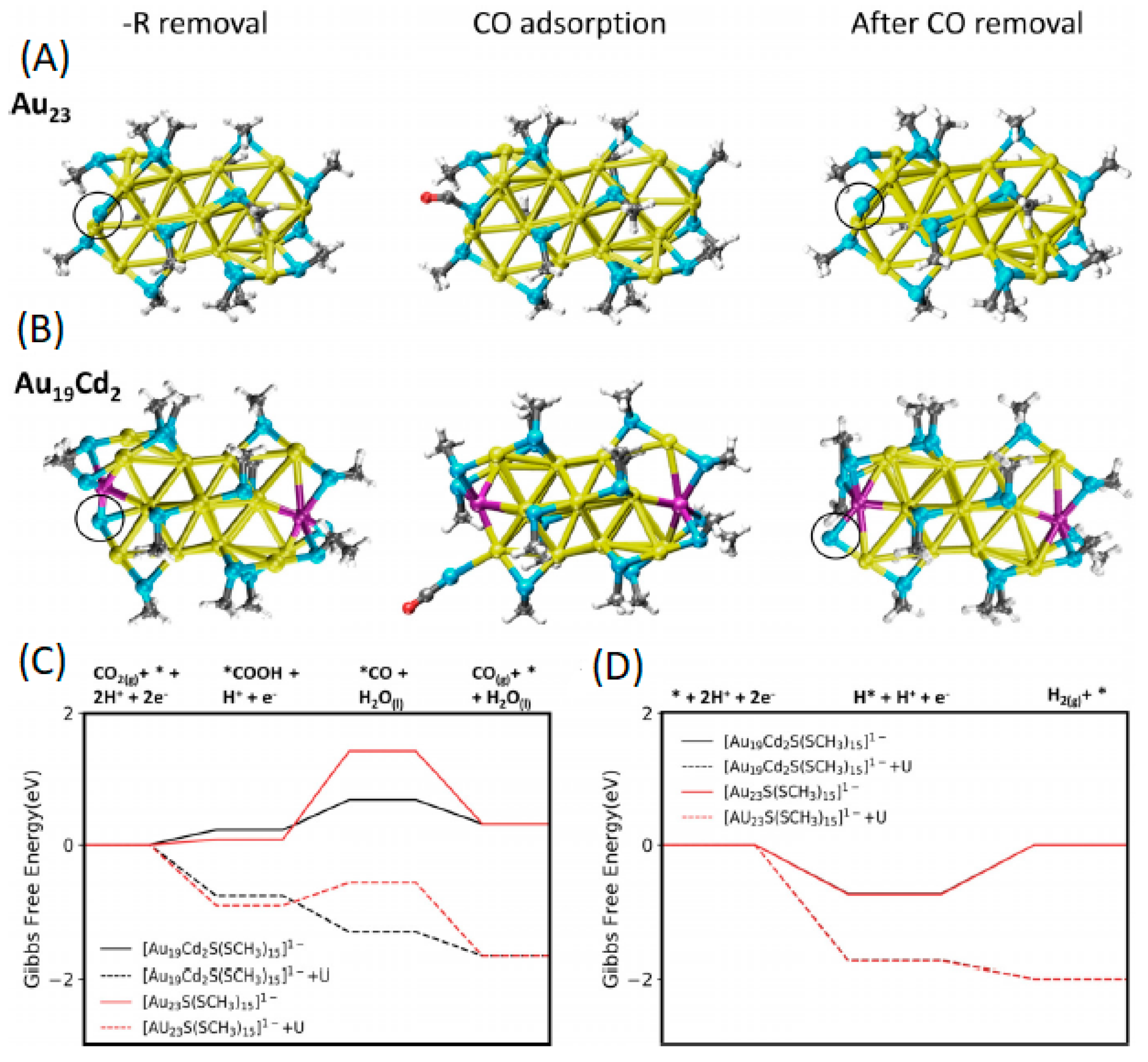
Figure 9A,B show the structure of the model Au23S(SCH3)15− and Au19Cd2S(SCH3)15− NCs containing exposed S as the active site, generated through the removal of a –CH3 from Au23(SCH3)16− and Au19Cd2(SCH3)16−. The predicted CO2RR and HER energy profiles are depicted in Figure 9C,D. It is noted that the HER energy profiles for both NCs are comparable. While the CO2RR step was predicted to be the potential limiting for both NCs, the process is less thermodynamically challenging in the doped case due to the stronger binding of CO. The calculated for the doped version was predicted to be more positive by ~0.9 V, in agreement with the experimental selectivity trend.
3. Summary and Outlook
In this short review, we described the complementary theoretical efforts to understand the experimentally observed CO2RR activity and selectivity in Au25(SR)18−, Au24Pd(SR)18, Au23(SR)16− and Au21Cd2(SR)16−. We focused on a particular structural variation—–ligand removal—and how it alters the CO2RR activity and selectivity on the nanoclusters. The first-principles studies indicated that exposed S sites, possibly created during preparation or under reaction conditions, may be the active reaction center in all the NCs investigated. In contrast to the Au site, it stabilizes the pertinent CO2RR intermediates while destabilizing H, yielding a non-negative . These results provide insights into the structure–property relationship in that the thermodynamics of the electrochemistry can be correlated to the undercoordinated S surface sites as the reaction center.
Further work must still be performed to elevate our fundamental understanding of the CO2RR mechanism on these NCs. We note that much of our focus is on the determination of reaction energetics and thermodynamics. The availability of algorithms to simulate the hydrogenation by H+ + e− species presents an opportunity to evaluate the reaction barriers under reaction conditions [65,66]. The obtained kinetic picture will lead to an increased coupling with experimental data, such as the measured current densities, which otherwise cannot be achieved by merely looking at the thermodynamics. Such effort may be challenging because of the huge computational expenses required to predict the reaction barriers within the DFT framework. The widely used Climbing Image Nudged Elastic Band (CI-NEB) method, for example, entails the discretization of the reaction pathway into a finite number of images to locate the transition state [67]. For the NCs considered in this paper, the calculation using plane wave basis sets can become more expensive as each image would contain the NC model enclosed in a periodic box large, enough to cancel out spurious interaction. Additionally, explicit water molecules may have to be included in the images to account for the H+ + e− transfer to the target intermediates, potentially requiring many hundreds of energies and gradients evaluations. However, continuing advances in high performance computing technology may expand the scope of first-principles simulations towards such type of explorations.
From a practical standpoint, more research remains to be pursued for Au-based NCs to realize its full potential. At present, the electrocatalysts considered in this review have not been integrated to practical devices to further evaluate their potential for the low-cost and scalable deployment for CO2 conversion to CO. Such setup would permit the evaluation of the integrity of the materials at realistic conditions. Aside from the Au NCs, membrane materials and electrocatalyst’s support are integral to the device. To comprehend the potential role of these components in determining the outcome of the CO2 conversion, it is necessary to invest effort into this aspect. The introduction of non-aqueous electrolytes plus the optimization of other conditions, such as flow rate and pressure, may significantly enhance the performance metrics as well. Thus, while there is always a need for fundamental research, a future focus that would pave the way for a practical CO2 conversion to CO application is highly desirable.
Funding
This researcher received no external funding.
Acknowledgments
This project was funded by the United States Department of Energy, National Energy Technology Laboratory. This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of author(s) expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Gamboa-Aldeco, M.E., Eds.; Springer: New York, NY, USA, 2008. [Google Scholar]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef]
- Dry, M.E. The fischer—Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Schulz, H. Short history and present trends of Fischer—Tropsch synthesis. Appl. Catal. A 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Lan, G.; Yang, J.; Ye, R.P.; Boyjoo, Y.; Liang, J.; Liu, X.; Li, Y.; Liu, J.; Qian, K. Sustainable carbon materials toward emerging applications. Small Methods 2021, 5, 2001250. [Google Scholar] [CrossRef]
- Liu, J.; Cai, C.; Wang, Y.; Liu, Y.; Huang, L.; Tian, T.; Yao, Y.; Wei, J.; Chen, R.; Zhang, K. A biomimetic plasmonic nanoreactor for reliable metabolite detection. Adv. Sci. 2020, 7, 1903730. [Google Scholar] [CrossRef]
- Zu, Y.; Yao, H.; Wang, Y.; Yan, L.; Gu, Z.; Chen, C.; Gao, L.; Yin, W. The age of bioinspired molybdenum-involved nanozymes: Synthesis, catalytic mechanisms, and biomedical applications. View 2021, 2, 20200188. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Singh, M.R.; Clark, E.L.; Bell, A.T. Thermodynamic and achievable efficiencies for solar-driven electrochemical reduction of carbon dioxide to transportation fuels. Proc. Natl. Acad. Sci. USA 2015, 112, E6111–E6118. [Google Scholar] [CrossRef] [Green Version]
- Lopez, N.; Nørskov, J.K. Catalytic CO oxidation by a gold nanoparticle: A density functional study. J. Am. Chem. Soc. 2002, 124, 11262–11263. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.; Clausen, B.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Vogel, W.; Lamy, C.; Alonso-Vante, N. Structure and electrocatalytic activity of carbon-supported Pt− Ni alloy nanoparticles toward the oxygen reduction reaction. J. Phys. Chem. B 2004, 108, 11024–11034. [Google Scholar] [CrossRef]
- Chen, S.; Ferreira, P.J.; Sheng, W.; Yabuuchi, N.; Allard, L.F.; Shao-Horn, Y. Enhanced activity for oxygen reduction reaction on “Pt3Co” nanoparticles: Direct evidence of percolated and sandwich-segregation structures. J. Am. Chem. Soc. 2008, 130, 13818–13819. [Google Scholar] [CrossRef]
- Mazumder, V.; Chi, M.; More, K.L.; Sun, S. Core/shell Pd/FePt nanoparticles as an active and durable catalyst for the oxygen reduction reaction. J. Am. Chem. Soc. 2010, 132, 7848–7849. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Xie, L.; Liang, Y.; Hong, G.; Dai, H. MoS2 nanoparticles grown on graphene: An advanced catalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2011, 133, 7296–7299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Khojin, A.; Jhong, H.-R.M.; Rosen, B.A.; Zhu, W.; Ma, S.; Kenis, P.J.; Masel, R.I. Nanoparticle silver catalysts that show enhanced activity for carbon dioxide electrolysis. J. Phys. Chem. C 2013, 117, 1627–1632. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tao, H.; Zeng, L.; Liu, Q.; Xu, Z.; Liu, Q.; Luo, J.-L. Shape-dependent electrocatalytic reduction of CO2 to CO on triangular silver nanoplates. J. Am. Chem. Soc. 2017, 139, 2160–2163. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Michalski, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Roldan Cuenya, B. Exceptional Size—Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Gates, B.C. Supported gold catalysts: New properties offered by nanometer and sub-nanometer structures. Chem. Commun. 2013, 49, 7876–7877. [Google Scholar] [CrossRef]
- Herzing, A.A.; Kiely, C.J.; Carley, A.F.; Landon, P.; Hutchings, G.J. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. [Google Scholar] [CrossRef]
- Corma, A.; Concepción, P.; Boronat, M.; Sabater, M.J.; Navas, J.; Yacaman, M.J.; Larios, E.; Posadas, A.; López-Quintela, M.A.; Buceta, D. Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity. Nat. Chem. 2013, 5, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Oliver-Meseguer, J.; Cabrero-Antonino, J.R.; Domínguez, I.; Leyva-Pérez, A.; Corma, A. Small gold clusters formed in solution give reaction turnover numbers of 107 at room temperature. Science 2012, 338, 1452–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.M.; Khoury, J.T.; Schaaff, T.G.; Shafigullin, M.N.; Vezmar, I.; Whetten, R.L. Optical absorption spectra of nanocrystal gold molecules. J. Phys. Chem. B 1997, 101, 3706–3712. [Google Scholar] [CrossRef] [Green Version]
- Jin, R. Quantum sized, thiolate-protected gold nanoclusters. Nanoscale 2010, 2, 343–362. [Google Scholar] [PubMed]
- Wu, Z.; Jiang, D.-E.; Mann, A.K.; Mullins, D.R.; Qiao, Z.-A.; Allard, L.F.; Zeng, C.; Jin, R.; Overbury, S.H. Thiolate ligands as a double-edged sword for CO oxidation on CeO2 supported Au25 (SCH2CH2Ph) 18 nanoclusters. J. Am. Chem. Soc. 2014, 136, 6111–6122. [Google Scholar] [CrossRef]
- Gaur, S.; Wu, H.; Stanley, G.G.; More, K.; Kumar, C.S.; Spivey, J.J. CO oxidation studies over cluster-derived Au/TiO2 and AUROlite™ Au/TiO2 catalysts using DRIFTS. Catal. Today 2013, 208, 72–81. [Google Scholar] [CrossRef]
- Zhu, Y.; Qian, H.; Zhu, M.; Jin, R. Thiolate-Protected Aun Nanoclusters as Catalysts for Selective Oxidation and Hydrogenation Processes. Adv. Mater. 2010, 22, 1915–1920. [Google Scholar] [CrossRef]
- Nie, X.; Qian, H.; Ge, Q.; Xu, H.; Jin, R. CO oxidation catalyzed by oxide-supported Au25(SR)18 nanoclusters and identification of perimeter sites as active centers. ACS Nano 2012, 6, 6014–6022. [Google Scholar] [CrossRef]
- Kauffman, D.; Alfonso, D.; Matranga, C.; Qian, H.; Jin, R. Experimental and Computational Investigation of Au25 Clusters and CO2: A Unique Interaction and Enhanced Electrocatalytic Activity. J. Am. Chem. Soc. 2012, 134, 10237–10243. [Google Scholar] [CrossRef]
- Kauffman, D.R.; Alfonso, D.; Matranga, C.; Ohodnicki, P.; Deng, X.; Siva, R.C.; Zeng, C.; Jin, R. Probing active site chemistry with differently charged Au25q nanoclusters (q = −1, 0, +1). Chem. Sci. 2014, 5, 3151–3157. [Google Scholar] [CrossRef]
- Vickers, J.W.; Alfonso, D.; Kauffman, D.R. Electrochemical carbon dioxide reduction at nanostructured gold, copper, and alloy materials. Energy Technol. 2017, 5, 775–795. [Google Scholar] [CrossRef] [Green Version]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Linqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonnson, H.J. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Hansen, H.A.; Varley, J.B.; Peterson, A.A.; Nørskov, J.K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.-J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Effeciency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comp. Mat. Sci. 1996, 6, 15–50. [Google Scholar]
- Perdew, J.P.; Burke, K.; Enzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High Precision Sampling for Brillouin Zone Integration for Metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Matthew, K.; Sundararaman, R.; Lechtworth-Weaver, K.; Arias, T.A.; Hennig, R.J. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. Chem. Phys. 2014, 140, 84106. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Acevedo, O.; Kacprzak, K.A.; Akola, J.; Hakkinen, H. Quantum size effects in ambient CO oxidation catalysed by ligand-protected gold clusters. Nat. Chem. 2010, 2, 329–334. [Google Scholar] [CrossRef]
- Alfonso, D.; Kauffman, D.; Matranga, C.J. Active sites of ligand-protected Au25 nanoparticle catalysts for CO2 electroreduction to CO. Chem. Phys. 2016, 144, 184705. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Abroshan, H.; Chen, Y.; Jin, R.; Kim, H.J. Experimental and mechanistic understanding of aldehyde hydrogenation using Au25 nanoclusters with Lewis acids: Unique sites for catalytic reactions. J. Am. Chem. Soc. 2015, 137, 14295–14304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kaziz, S.; Li, H.; Hevia, M.G.; Wodka, D.; Mazet, C.; Burgi, T.; Barrabés, N. Modulation of Active Sites in Supported Au38(SC2H4Ph)24 Cluster Catalysts: Effect of Atmosphere and Support Material. J. Phys. Chem. C 2015, 119, 11193–11199. [Google Scholar] [CrossRef]
- Austin, N.; Zhao, S.; McKone, J.; Jin, R.; Mpourmpakis, G. Elucidating the active sites for CO2 electroreduction on ligand-protected Au25 nanoclusters. Catal. Sci. Technol. 2018, 8, 3795–3805. [Google Scholar] [CrossRef]
- Varley, J.; Hansen, H.; Ammitzbøll, N.L.; Grabow, L.; Peterson, A.; Rossmeisl, J.; Nørskov, J. Ni-Fe-S cubanes in CO2 reduction electrocatalysis: A DFT study. ACS Catal. 2013, 3, 2640–2643. [Google Scholar] [CrossRef]
- Chan, K.; Tsai, C.; Hansen, H.A.; Norskov, J. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem 2014, 6, 1899. [Google Scholar] [CrossRef]
- Li, S.; Alfonso, D.; Nagarajan, A.V.; House, S.D.; Yang, J.C.; Kauffman, D.R.; Mpourmpakis, G.; Jin, R. Monopalladium substitution in gold nanoclusters enhances CO2 electroreduction activity and selectivity. ACS Catal. 2020, 10, 12011–12016. [Google Scholar] [CrossRef]
- Li, S.; Nagarajan, A.V.; Alfonso, D.R.; Sun, M.; Kauffman, D.R.; Mpourmpakis, G.; Jin, R. Boosting CO2 electrochemical reduction with atomically precise surface modification on gold nanoclusters. Angew. Chem. Int. Ed. 2021, 60, 6351–6356. [Google Scholar] [CrossRef]
- Zhuang, S.; Chen, D.; Liao, L.; Zhao, Y.; Xia, N.; Zhang, W.; Wang, C.; Yang, J.; Wu, Z. Hard-sphere random close-packed Au47Cd2(TBBT)31 nanoclusters with a Faradaic efficiency of up to 96% for electrocatalytic CO2 reduction to CO. Angew. Chem. Int. Ed. 2020, 59, 3073–3077. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, L.; Li, L.; Dong, H.; Chen, S.; Zhu, W.; Hu, C.; Deng, W.; Zhao, Z.-J.; Gong, J. Ultrathin Pd-Au shells with controllable alloying degree on Pd nanocubes toward carbon dioxide reduction. J. Am. Chem. Soc. 2019, 141, 4791–4794. [Google Scholar] [CrossRef]
- Sun, K.; Ji, Y.; Liu, Y.; Wang, Z. Synergies between electronic and geometric effects of Mo-doped Au nanoparticles for effective CO2 electrochemical reduction. J. Mater. Chem. A 2020, 8, 12291–12295. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, Y.; Zhou, Z.; Cai, F.; Zhao, X.; Huang, W.; Li, Y.; Zhu, J.; Liu, P.; Yang, F. Enhancing CO2 electroreduction with the metal—Oxide interface. J. Am. Chem. Soc. 2017, 139, 5652–5655. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, D.R.; Alfonso, D.R.; Tafen, D.N.; Wang, C.; Zhou, Y.; Yu, Y.; Lekse, J.W.; Deng, X.; Espinoza, V.; Trindell, J. Selective electrocatalytic reduction of CO2 into CO at small, thiol-capped Au/Cu nanoparticles. J. Phys. Chem. C 2018, 122, 27991–28000. [Google Scholar] [CrossRef]
- Nie, X.; Esopi, M.R.; Janik, M.J.; Asthagiri, A. Selectivity of CO2 Reduction on Copper Electrode: The Role of Kinetics of Elementary Steps. Angew. Chem. Int. Ed. 2013, 52, 2459–2462. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, J.; Peng, H.; Hong, X.; Chan, K.; Nørskov, J.K. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 2017, 8, 15438. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
(A) Potential-dependent FE for CO on Au25(SR)18− nanoclusters, Au nanoparticles of 2 and 5 nm size, and bulk gold electrode. (B) Potential dependent FE for CO and H2 formation on Au25(SR)18− nanoclusters. Adapted from Reference [39]. Copyright 2012 by the American Chemical Society.
Figure 1.
(A) Potential-dependent FE for CO on Au25(SR)18− nanoclusters, Au nanoparticles of 2 and 5 nm size, and bulk gold electrode. (B) Potential dependent FE for CO and H2 formation on Au25(SR)18− nanoclusters. Adapted from Reference [39]. Copyright 2012 by the American Chemical Society.

Figure 2.
CO2 adsorbed on Au25(SCH3)18− nanocluster. Adapted from Reference [39]. Copyright 2012 by the American Chemical Society.
Figure 2.
CO2 adsorbed on Au25(SCH3)18− nanocluster. Adapted from Reference [39]. Copyright 2012 by the American Chemical Society.

Figure 3.
Free energy profile of the CO2 reduction to CO in the intact Au25(SCH3)18− nanocluster. Adapted from Reference [52]. Copyright by The American Institure of Physics (2016).
Figure 3.
Free energy profile of the CO2 reduction to CO in the intact Au25(SCH3)18− nanocluster. Adapted from Reference [52]. Copyright by The American Institure of Physics (2016).

Figure 4.
Free energy profile of the CO2 reduction to CO on the dethiolated Au25(SCH3)18− nanocluster Adapted from Reference [52]. Copyright by the American Institute of Physics (2016).
Figure 4.
Free energy profile of the CO2 reduction to CO on the dethiolated Au25(SCH3)18− nanocluster Adapted from Reference [52]. Copyright by the American Institute of Physics (2016).

Figure 5.
STEM images of (A) Au25 and (B) Pd1Au24. Adapted from Ref. [58]. Copyright by the American Chemical Society (2020).
Figure 5.
STEM images of (A) Au25 and (B) Pd1Au24. Adapted from Ref. [58]. Copyright by the American Chemical Society (2020).

Figure 6.
(A) FE for CO production. (B) CO partial current density. (C) CO mass activity. (D) Stability test of Pd1Au24 at −0.8 V for 6 h. Au25 and Pd1Au24 represent Au25(SR)18 and Au24Pd(SR)18, respectively. Adapted from Reference [58]. Copyright by the American Chemical Society (2020).
Figure 6.
(A) FE for CO production. (B) CO partial current density. (C) CO mass activity. (D) Stability test of Pd1Au24 at −0.8 V for 6 h. Au25 and Pd1Au24 represent Au25(SR)18 and Au24Pd(SR)18, respectively. Adapted from Reference [58]. Copyright by the American Chemical Society (2020).

Figure 7.
Free energy diagram of for electrochemical reduction of (A) CO2RR and (B) HER on the bare metal site of the dethiolated Au25(SCH3)18 and Au24Pd(SCH3)18 nanoclusters. The free energies of (C) CO2RR and (D) HER on the sulfur site of the demethylated nanoclusters are included for comparison.
Figure 7.
Free energy diagram of for electrochemical reduction of (A) CO2RR and (B) HER on the bare metal site of the dethiolated Au25(SCH3)18 and Au24Pd(SCH3)18 nanoclusters. The free energies of (C) CO2RR and (D) HER on the sulfur site of the demethylated nanoclusters are included for comparison.

Figure 8.
(A) FE for CO production. (B) CO partial current density. (C) TOF of CO2RR. (D) FE for H2 production. The error bar represents the mean and variance for 3 runs to check the reproducibility. Au23 and Au19Cd2 represent Au23(SR)16− and Au19Cd2(SR)16−, respectively. Adapted from Reference [59]. Copyright by Wiley-VCH (2021).
Figure 8.
(A) FE for CO production. (B) CO partial current density. (C) TOF of CO2RR. (D) FE for H2 production. The error bar represents the mean and variance for 3 runs to check the reproducibility. Au23 and Au19Cd2 represent Au23(SR)16− and Au19Cd2(SR)16−, respectively. Adapted from Reference [59]. Copyright by Wiley-VCH (2021).

Figure 9.
Optimized structures of (A) Au23(SR)16− and (B) Au19Cd2(SR)16−, upon –R(CH3) removal, CO adsorption and CO removal. Atom colors: Au: yellow, Cd: pink, S: blue, C: black, H: white. Black circle demonstrates S active site. Reaction pathway for (C) CO2RR and (D) HER on the Au23(SR)16− (red lines) and Au19Cd2(SR)16− (black lines) upon –CH3 removal. Solid lines represent free energy without an applied potential, while dotted lines represent those with an applied potential of −1.0 V vs. RHE. Adapted from Reference [59]. Copyright by Wiley-VCH (2021).
Figure 9.
Optimized structures of (A) Au23(SR)16− and (B) Au19Cd2(SR)16−, upon –R(CH3) removal, CO adsorption and CO removal. Atom colors: Au: yellow, Cd: pink, S: blue, C: black, H: white. Black circle demonstrates S active site. Reaction pathway for (C) CO2RR and (D) HER on the Au23(SR)16− (red lines) and Au19Cd2(SR)16− (black lines) upon –CH3 removal. Solid lines represent free energy without an applied potential, while dotted lines represent those with an applied potential of −1.0 V vs. RHE. Adapted from Reference [59]. Copyright by Wiley-VCH (2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alfonso, D. High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites. Catalysts 2022, 12, 505. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050505
AMA Style
Alfonso D. High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites. Catalysts. 2022; 12(5):505. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050505
Chicago/Turabian StyleAlfonso, Dominic. 2022. "High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites" Catalysts 12, no. 5: 505. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050505
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.