
Recent Advances in the Synthesis of Five-Membered Cyclic Carbonates and Carbamates from Allylic or Propargylic Substrates and CO2

 , , , and
, , , and
Abstract
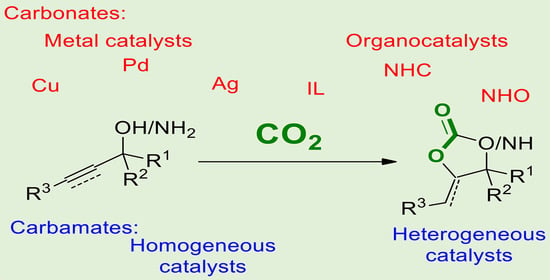
:
1. Introduction
2. Cyclic Carbonates
2.1. Five-Membered Cyclic Carbonates from Allylic Alcohols
2.2. α-Alkylidene Cyclic Carbonates from CO2 and Propargylic Alcohols
2.2.1. Metal Catalysts
Palladium
Silver
Copper
2.2.2. Ionic Liquids
2.2.3. Organocatalysts
N-Heterocyclic Carbenes
N-Heterocyclic Olefins
Other Organocatalysts
3. Cyclic Carbamates
3.1. Five-Membered Cyclic Carbamates from CO2 and Allylic or Homoallylic Amines
3.2. Five-Membered Cyclic Carbamates from Propargyl Amines
3.2.1. Homogeneous Catalysts
3.2.2. Heterogeneous Catalysts
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Trends in Atmospheric Carbon Dioxide. Available online: www.esrl.noaa.gov/gmd/ccgg/trends (accessed on 15 January 2022).
- U.S. Environmental Protection Agency. Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990–2018. 2020. Available online: https://www.epa.gov/sites/default/files/2020-04/documents/us-ghg-inventory-2020-main-text.pdf (accessed on 15 January 2021).
- Parry, M.L.; Canziani, O.F.; Palutikof, J.P.; van der Linden, P.J.; Hanson, C.E. Climate Change 2007: Impacts, Adaptation and Vulnerability. Contribution of Working Group II to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2007; p. 976. [Google Scholar]
- Climate Action. 2020 Climate & Energy Package. 2020. Available online: https://ec.europa.eu/clima/eu-action/climate-strategies-targets/2020-climate-energy-package_en (accessed on 15 January 2022).
- Kramer, G.J.; Haigh, M. No Quick Switch to Low-Carbon Energy. Nature 2009, 462, 568–569. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Dowell, N.M.; Minx, J.C.; Smith, P.; Williams, C.K. The Technological and Economic Prospects for CO2 Utilization and Removal. Nature 2019, 575, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Razzaq, A.; In, S.-I. Development of Graphene Based Photocatalysts for CO2 Reduction to C1 Chemicals: A Brief Overview. Catal. Today 2019, 335, 39–54. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The Renaissance of the Sabatier Reaction and Its Applications on Earth and in Space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Xu, L.; Xiu, Y.; Liu, F.; Liang, Y.; Wang, S. Research Progress in Conversion of CO2 to Valuable Fuels. Molecules 2020, 25, 3653. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Shimonishi, J.; Maeda, C.; Hasegawa, J. Bifunctional Porphyrin Catalysts for the Synthesis of Cyclic Carbonates from Epoxides and CO2: Structural Optimization and Mechanistic Study. J. Am. Chem. Soc. 2014, 136, 15270–15279. [Google Scholar] [CrossRef]
- Qu, Y.; Chen, Y.; Sun, J. Conversion of CO2 with Epoxides to Cyclic Carbonates Catalyzed by Amino Acid Ionic Liquids at Room Temperature. J. CO2 Util. 2022, 56, 101840. [Google Scholar] [CrossRef]
- Marciniak, A.A.; Lamb, K.J.; Ozorio, L.P.; Mota, C.J.A.; North, M. Heterogeneous Catalysts for Cyclic Carbonate Synthesis from Carbon Dioxide and Epoxides. Curr. Opin. Green Sustain. Chem. 2020, 26, 100365. [Google Scholar] [CrossRef]
- Lamb, K.J.; Ingram, I.D.V.; North, M.; Sengoden, M. Valorization of Carbon Dioxide into Oxazolidinones by Reaction with Aziridines. CGC 2019, 6, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Abdolmohammadi, S.; Hoseininezhad-Namin, M.S.; Behmagham, F.; Vessally, E. Carboxylative Cyclization of Propargylic Alcohols with Carbon Dioxide: A Facile and Green Route to α-Methylene Cyclic Carbonates. J. CO2 Util. 2020, 38, 220–231. [Google Scholar] [CrossRef]
- Schilling, W.; Das, S. Transition Metal-Free Synthesis of Carbamates Using CO2 as the Carbon Source. ChemSusChem 2020, 13, 6246–6258. [Google Scholar] [CrossRef] [PubMed]
- Arshadi, S.; Vessally, E.; Hosseinian, A.; Soleimani-amiri, S.; Edjlali, L. Three-Component Coupling of CO2, Propargyl Alcohols, and Amines: An Environmentally Benign Access to Cyclic and Acyclic Carbamates (A Review). J. CO2 Util. 2017, 21, 108–118. [Google Scholar] [CrossRef]
- Parker, H.L.; Sherwood, J.; Hunt, A.J.; Clark, J.H. Cyclic Carbonates as Green Alternative Solvents for the Heck Reaction. ACS Sustain. Chem. Eng. 2014, 2, 1739–1742. [Google Scholar] [CrossRef]
- Scrosati, B.; Hassoun, J.; Sun, Y.-K. Lithium-Ion Batteries. A Look into the Future. Energy Environ. Sci. 2011, 4, 3287. [Google Scholar] [CrossRef]
- Guerin, W.; Diallo, A.K.; Kirilov, E.; Helou, M.; Slawinski, M.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Enantiopure Isotactic PCHC Synthesized by Ring-Opening Polymerization of Cyclohexene Carbonate. Macromolecules 2014, 47, 4230–4235. [Google Scholar] [CrossRef]
- Rollin, P.; Soares, L.K.; Barcellos, A.M.; Araujo, D.R.; Lenardão, E.J.; Jacob, R.G.; Perin, G. Five-Membered Cyclic Carbonates: Versatility for Applications in Organic Synthesis, Pharmaceutical, and Materials Sciences. Appl. Sci. 2021, 11, 5024. [Google Scholar] [CrossRef]
- Cardillo, G.; Orena, M.; Porzi, G.; Sandri, S. A New Regio- and Stereo-Selective Functionalization of Allylic and Homoallylic Alcohols. J. Chem. Soc. Chem. Commun. 1981, 10, 465. [Google Scholar] [CrossRef]
- Minakata, S.; Sasaki, I.; Ide, T. Atmospheric CO2 Fixation by Unsaturated Alcohols Using TBuOI under Neutral Conditions. Angew. Chem. Int. Ed. 2010, 49, 1309–1311. [Google Scholar] [CrossRef]
- Parrish, J.P.; Salvatore, R.N.; Jung, K.W. Perspectives on Alkyl Carbonates in Organic Synthesis. Tetrahedron 2000, 56, 8207–8237. [Google Scholar] [CrossRef]
- Gennen, S.; Grignard, B.; Tassaing, T.; Jérôme, C.; Detrembleur, C. CO2-Sourced α-Alkylidene Cyclic Carbonates: A Step Forward in the Quest for Functional Regioregular Poly(Urethane)s and Poly(Carbonate)s. Angew. Chem. Int. Ed. 2017, 56, 10394–10398. [Google Scholar] [CrossRef]
- Sugawara, Y.; Yamada, W.; Yoshida, S.; Ikeno, T.; Yamada, T. Carbon Dioxide-Mediated Catalytic Rearrangement of Propargyl Alcohols into α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2007, 129, 12902–12903. [Google Scholar] [CrossRef] [PubMed]
- Iritani, K.; Yanagihara, N.; Utimoto, K. Carboxylative Coupling of Propargylic Alcohols with Allyl Chloride. J. Org. Chem. 1986, 51, 5499–5501. [Google Scholar] [CrossRef]
- Uemura, K.; Shiraishi, D.; Noziri, M.; Inoue, Y. Preparation of Cyclic Carbonates from Alkadienols, CO2, and Aryl or Vinylic Halides Catalyzed by a Palladium Complex. BCSJ 1999, 72, 1063–1069. [Google Scholar] [CrossRef]
- Sun, S.; Wang, B.; Gu, N.; Yu, J.-T.; Cheng, J. Palladium-Catalyzed Arylcarboxylation of Propargylic Alcohols with CO2 and Aryl Halides: Access to Functionalized α-Alkylidene Cyclic Carbonates. Org. Lett. 2017, 19, 1088–1091. [Google Scholar] [CrossRef]
- Yamada, W.; Sugawara, Y.; Cheng, H.M.; Ikeno, T.; Yamada, T. Silver-Catalyzed Incorporation of Carbon Dioxide into Propargylic Alcohols. Eur. J. Org. Chem. 2007, 2007, 2604–2607. [Google Scholar] [CrossRef]
- Kikuchi, S.; Yoshida, S.; Sugawara, Y.; Yamada, W.; Cheng, H.-M.; Fukui, K.; Sekine, K.; Iwakura, I.; Ikeno, T.; Yamada, T. Silver-Catalyzed Carbon Dioxide Incorporation and Rearrangement on Propargylic Derivatives. BCSJ 2011, 84, 698–717. [Google Scholar] [CrossRef]
- Xie, S.; Gao, X.; Zhou, F.; Wu, H.; Zhou, J. Enantioselective Carboxylative Cyclization of Propargylic Alcohol with Carbon Dioxide under Mild Conditions. Chin. Chem. Lett. 2020, 31, 324–328. [Google Scholar] [CrossRef]
- Song, Q.-W.; Chen, W.-Q.; Ma, R.; Yu, A.; Li, Q.-Y.; Chang, Y.; He, L.-N. Bifunctional Silver(I) Complex-Catalyzed CO2 Conversion at Ambient Conditions: Synthesis of α-Methylene Cyclic Carbonates and Derivatives. ChemSusChem 2015, 8, 821–827. [Google Scholar] [CrossRef]
- Yuan, Y.; Xie, Y.; Zeng, C.; Song, D.; Chaemchuen, S.; Chen, C.; Verpoort, F. A Recyclable AgI/Oac− Catalytic System for the Efficient Synthesis of α-Alkylidene Cyclic Carbonates: Carbon Dioxide Conversion at Atmospheric Pressure. Green Chem. 2017, 19, 2936–2940. [Google Scholar] [CrossRef]
- Dabral, S.; Bayarmagnai, B.; Hermsen, M.; Schießl, J.; Mormul, V.; Hashmi, A.S.K.; Schaub, T. Silver-Catalyzed Carboxylative Cyclization of Primary Propargyl Alcohols with CO2. Org. Lett. 2019, 21, 1422–1425. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Z.; Zhang, F.; Liu, Z.; Yang, P.; Zhang, H.; Yu, B.; Zhao, Y.; Liu, Z. A Rose Bengal-Functionalized Porous Organic Polymer for Carboxylative Cyclization of Propargyl Alcohols with CO2. Chem. Commun. 2019, 55, 12475–12478. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Hupp, J.T.; Nguyen, S.T. Porous Organic Polymers in Catalysis: Opportunities and Challenges. ACS Catal. 2011, 1, 819–835. [Google Scholar] [CrossRef]
- Gu, Y.; Shi, F.; Deng, Y. Ionic Liquid as an Efficient Promoting Medium for Fixation of CO2: Clean Synthesis of α-Methylene Cyclic Carbonates from CO2 and Propargyl Alcohols Catalyzed by Metal Salts under Mild Conditions. J. Org. Chem. 2004, 69, 391–394. [Google Scholar] [CrossRef] [PubMed]
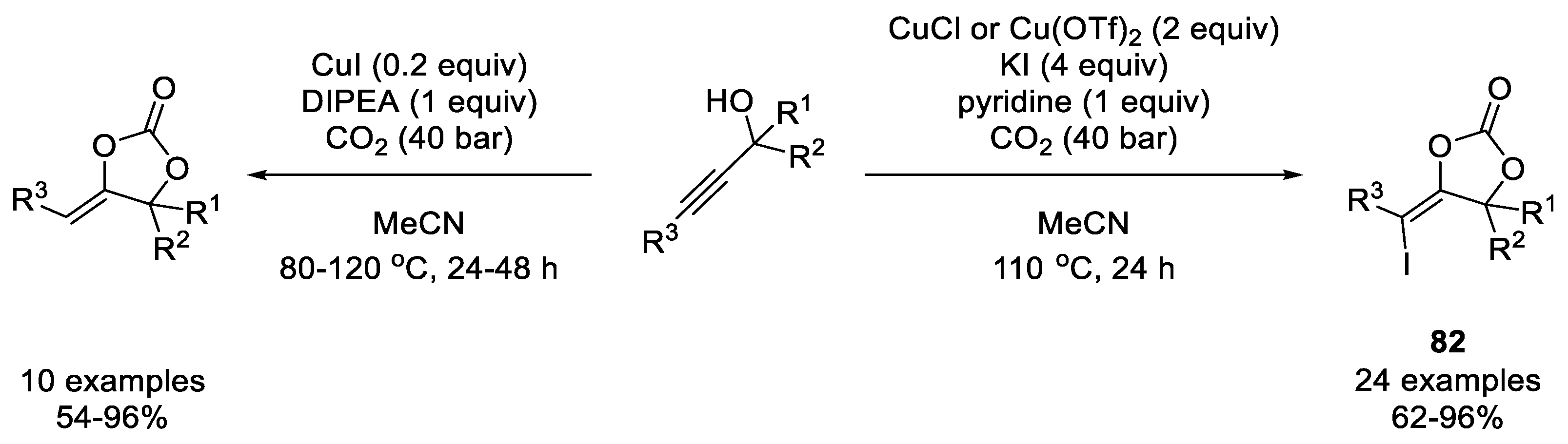
- Ouyang, L.; Tang, X.; He, H.; Qi, C.; Xiong, W.; Ren, Y.; Jiang, H. Copper-Promoted Coupling of Carbon Dioxide and Propargylic Alcohols: Expansion of Substrate Scope and Trapping of Vinyl Copper Intermediate. Adv. Synth. Catal. 2015, 357, 2556–2565. [Google Scholar] [CrossRef]
- Jiang, H.-F.; Wang, A.-Z.; Liu, H.-L.; Qi, C.-R. Reusable Polymer-Supported Amine-Copper Catalyst for the Formation of α-Alkylidene Cyclic Carbonates in Supercritical Carbon Dioxide. Eur. J. Org. Chem. 2008, 2008, 2309–2312. [Google Scholar] [CrossRef]
- Tian, H.-R.; Zhang, Z.; Liu, S.-M.; Dang, T.-Y.; Li, Z.; Lu, Y.; Liu, S.-X. A Highly Stable Polyoxovanadate-Based Cu(i)–MOF for the Carboxylative Cyclization of CO2 with Propargylic Alcohols at Room Temperature. Green Chem. 2020, 22, 7513–7520. [Google Scholar] [CrossRef]
- Qiu, J.; Zhao, Y.; Li, Z.; Wang, H.; Fan, M.; Wang, J. Efficient Ionic-Liquid-Promoted Chemical Fixation of CO2 into α-Alkylidene Cyclic Carbonates. ChemSusChem 2017, 10, 1120–1127. [Google Scholar] [CrossRef]
- Cervantes-Reyes, A.; Farshadfar, K.; Rudolph, M.; Rominger, F.; Schaub, T.; Ariafard, A.; Hashmi, A.S.K. Copper-Catalysed Synthesis of α-Alkylidene Cyclic Carbonates from Propargylic Alcohols and CO2. Green Chem. 2021, 23, 889–897. [Google Scholar] [CrossRef]
- Roy, M.M.D.; Rivard, E. Pushing Chemical Boundaries with N-Heterocyclic Olefins (NHOs): From Catalysis to Main Group Element Chemistry. Acc. Chem. Res. 2017, 50, 2017–2025. [Google Scholar] [CrossRef]
- Wang, Y.-B.; Wang, Y.-M.; Zhang, W.-Z.; Lu, X.-B. Fast CO2 Sequestration, Activation, and Catalytic Transformation Using N-Heterocyclic Olefins. J. Am. Chem. Soc. 2013, 135, 11996–12003. [Google Scholar] [CrossRef]
- Li, W.; Yang, N.; Lyu, Y. Theoretical Insights into the Catalytic Mechanism of N -Heterocyclic Olefins in Carboxylative Cyclization of Propargyl Alcohol with CO2. J. Org. Chem. 2016, 81, 5303–5313. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhou, R.; Han, F.; Feng, M.; Miao, C.; Zhang, S.; Ai, S. A CO2-Induced ROCO2 Na/ROCO2 H Buffer Solution Promoted the Carboxylative Cyclization of Propargyl Alcohol to Synthesize Cyclic Carbonates. Catal. Sci. Technol. 2020, 10, 736–741. [Google Scholar] [CrossRef]
- Ca’, N.D.; Gabriele, B.; Ruffolo, G.; Veltri, L.; Zanetta, T.; Costa, M. Effective Guanidine-Catalyzed Synthesis of Carbonate and Carbamate Derivatives from Propargyl Alcohols in Supercritical Carbon Dioxide. Adv. Synth. Catal. 2011, 353, 133–146. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, R.; Lu, X.-B. Isolable CO2 Adducts of Polarized Alkenes: High Thermal Stability and Catalytic Activity for CO2 Chemical Transformation. Adv. Synth. Catal. 2019, 361, 326–334. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [Green Version]
- Vacondio, F.; Silva, C.; Mor, M.; Testa, B. Qualitative Structure-Metabolism Relationships in the Hydrolysis of Carbamates. Drug Metab. Rev. 2010, 42, 551–589. [Google Scholar] [CrossRef]
- Brickner, S.J. Oxazolidinone Antibacterial Agents. Curr. Pharmeceutical Des. 1996, 2, 175–194. [Google Scholar]
- Introduction to Green Pharmaceutical Science: Fact, Fiction, and Future. In Scalable Green Chemistry; Koenig, S. (Ed.) Jenny Stanford Publishing: Singapore, 2013; pp. 21–44. ISBN 9780429099953. [Google Scholar]
- Cardillo, G.; Orena, M.; Sandri, S. Oxazolidin-2-Ones from Allylic Amines by Means of Iodine and Carbonate Anion on Polymeric Support. A Convenient Synthesis of (+-)-Propranolol. J. Org. Chem. 1986, 51, 713–717. [Google Scholar] [CrossRef]
- Toda, T.; Kitagawa, Y. Formation of Oxazolidinones and Oxazinanones by Reaction of Allylamines and of Homoallylamines with Carbon Dioxide and Iodine via Intramolecular Cyclization. Angew. Chem. Int. Ed. Engl. 1987, 26, 334–335. [Google Scholar] [CrossRef]
- García-Egido, E.; Fernández, I.; Muñoz, L. Convenient Synthesis of Oxazolidinones and Oxazinones from Allyl and Homoallyl Amines under Mild Conditions. Synth. Commun. 2006, 36, 3029–3042. [Google Scholar] [CrossRef]
- Kanzian, T.; Nigst, T.A.; Maier, A.; Pichl, S.; Mayr, H. Nucleophilic Reactivities of Primary and Secondary Amines in Acetonitrile. Eur. J. Org. Chem. 2009, 2009, 6379–6385. [Google Scholar] [CrossRef]
- Soldi, L.; Massera, C.; Costa, M.; Ca’, N.D. A Novel One-Pot Synthesis of Oxazolidinones through Direct Introduction of CO2 into Allylamine Derivatives. Tetrahedron Lett. 2014, 55, 1379–1383. [Google Scholar] [CrossRef]
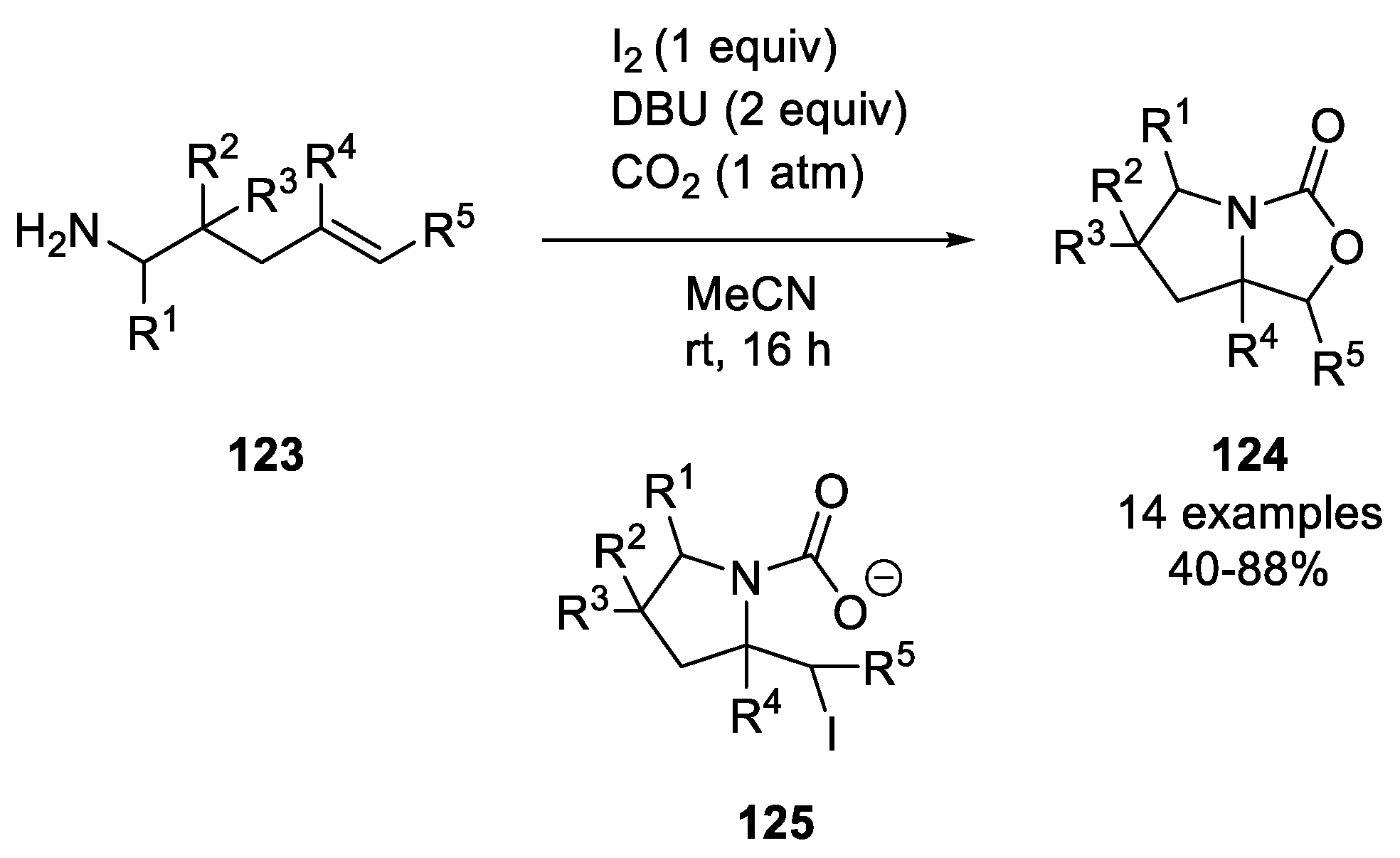
- Wang, S.; Zhang, X.; Cao, C.; Chen, C.; Xi, C. I2-Mediated Oxidative Bicyclization of 4-Pentenamines to Prolinol Carbamates with CO2 Incorporating Oxyamination of the C=C Bond. Green Chem. 2017, 19, 4515–4519. [Google Scholar] [CrossRef]
- Ye, J.-H.; Song, L.; Zhou, W.-J.; Ju, T.; Yin, Z.-B.; Yan, S.-S.; Zhang, Z.; Li, J.; Yu, D.-G. Selective Oxytrifluoromethylation of Allylamines with CO2. Angew. Chem. Int. Ed. 2016, 55, 10022–10026. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ye, J.-H.; Zhou, W.-J.; Zeng, X.; Yu, D.-G. Oxy-Alkylation of Allylamines with Unactivated Alkyl Bromides and CO2 via Visible-Light-Driven Palladium Catalysis. Org. Lett. 2018, 20, 3049–3052. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Fukui, K.; Kikuchi, S.; Yamada, T. Silver-Catalyzed Preparation of Oxazolidinones from Carbon Dioxide and Propargylic Amines. Chem. Lett. 2009, 38, 786–787. [Google Scholar] [CrossRef]
- Sekine, K.; Kobayashi, R.; Yamada, T. Silver-Catalyzed Three-Component Reaction of Propargylic Amines, Carbon Dioxide, and N -Iodosuccinimide for Stereoselective Preparation of (E)-Iodovinyloxazolidinones. Chem. Lett. 2015, 44, 1407–1409. [Google Scholar] [CrossRef]
- Takeda, Y.; Okumura, S.; Tone, S.; Sasaki, I.; Minakata, S. Cyclizative Atmospheric CO2 Fixation by Unsaturated Amines with t-BuOI Leading to Cyclic Carbamates. Org. Lett. 2012, 14, 4874–4877. [Google Scholar] [CrossRef]
- Yoshida, M.; Mizuguchi, T.; Shishido, K. Synthesis of Oxazolidinones by Efficient Fixation of Atmospheric CO2 with Propargylic Amines by Using a Silver/1,8-Diazabicyclo[5.4.0]Undec-7-Ene (DBU) Dual-Catalyst System. Chem. Eur. J. 2012, 18, 15578–15581. [Google Scholar] [CrossRef]
- García-Domínguez, P.; Fehr, L.; Rusconi, G.; Nevado, C. Palladium-Catalyzed Incorporation of Atmospheric CO2: Efficient Synthesis of Functionalized Oxazolidinones. Chem. Sci. 2016, 7, 3914–3918. [Google Scholar] [CrossRef] [Green Version]
- Brunel, P.; Monot, J.; Kefalidis, C.E.; Maron, L.; Martin-Vaca, B.; Bourissou, D. Valorization of CO2: Preparation of 2-Oxazolidinones by Metal–Ligand Cooperative Catalysis with SCS Indenediide Pd Complexes. ACS Catal. 2017, 7, 2652–2660. [Google Scholar] [CrossRef]
- Liu, X.; Wang, M.-Y.; Wang, S.-Y.; Wang, Q.; He, L.-N. In Situ Generated Zinc(II) Catalyst for Incorporation of CO2 into 2-Oxazolidinones with Propargylic Amines at Atmospheric Pressure. ChemSusChem 2017, 10, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, K.; He, L. Efficient and Recyclable Cobalt(II)/Ionic Liquid Catalytic System for CO2 Conversion to Prepare 2-Oxazolinones at Atmospheric Pressure. Chin. J. Chem. 2019, 37, 1223–1228. [Google Scholar] [CrossRef]
- Huang, W.-B.; Ren, F.-Y.; Wang, M.-W.; Qiu, L.-Q.; Chen, K.-H.; He, L.-N. Cu(II)-Catalyzed Phosphonocarboxylative Cyclization Reaction of Propargylic Amines and Phosphine Oxide with CO2. J. Org. Chem. 2020, 85, 14109–14120. [Google Scholar] [CrossRef] [PubMed]
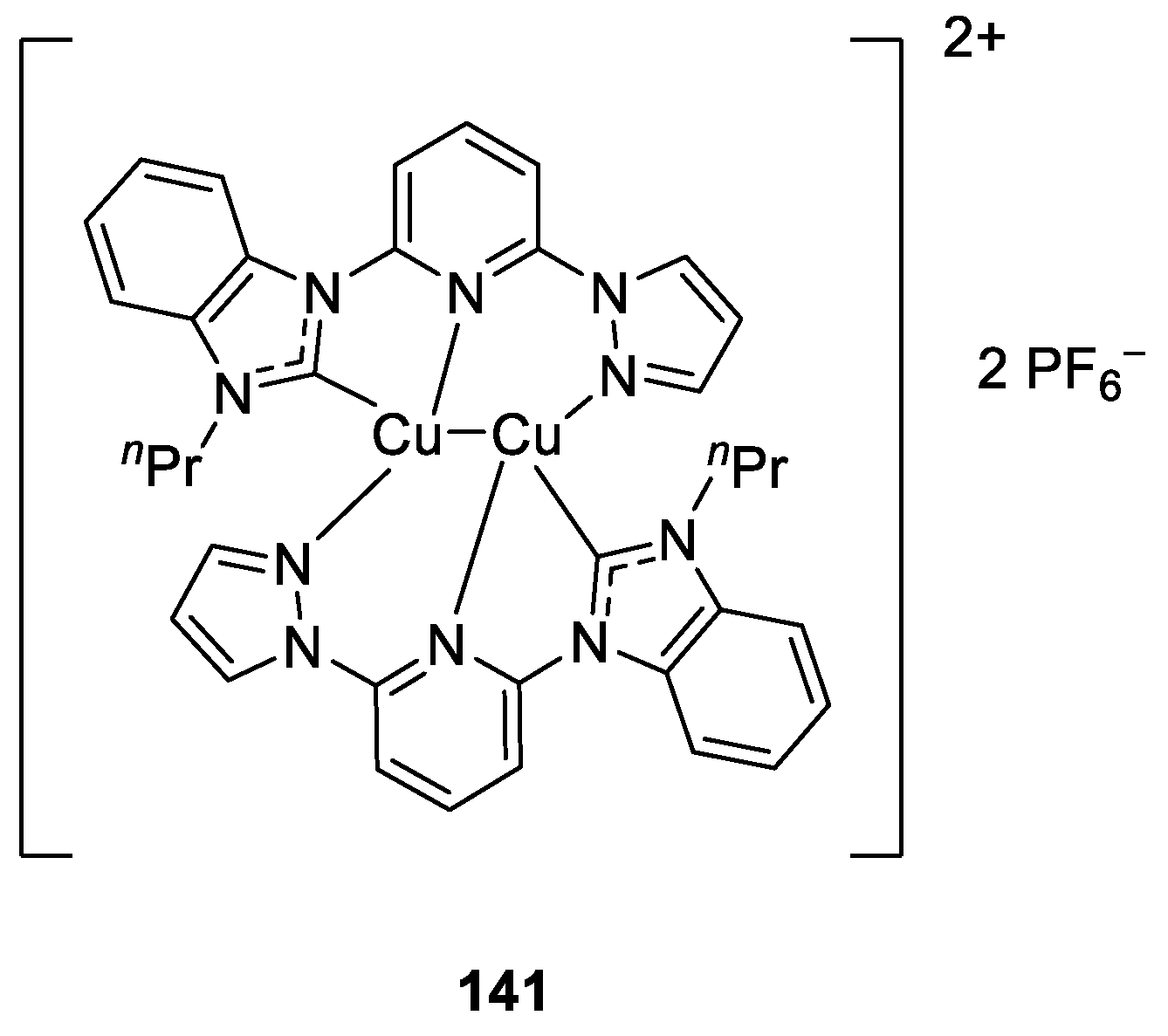
- Chen, F.; Tao, S.; Deng, Q.-Q.; Wei, D.; Liu, N.; Dai, B. Binuclear Tridentate Hemilabile Copper(I) Catalysts for Utilization of CO2 into Oxazolidinones from Propargylic Amines. J. Org. Chem. 2020, 85, 15197–15212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiu, J.; Li, Z.; Wang, H.; Fan, M.; Wang, J. An Experimental and Theoretical Study on the Unexpected Catalytic Activity of Triethanolamine for the Carboxylative Cyclization of Propargylic Amines with CO2. ChemSusChem 2017, 10, 2001–2007. [Google Scholar] [CrossRef]
- Fujii, A.; Choi, J.-C.; Fujita, K. Quaternary Ammonium Salt-Catalyzed Carboxylative Cyclization of Propargylic Amines with CO2. Tetrahedron Lett. 2017, 58, 4483–4486. [Google Scholar] [CrossRef]
- Cao, C.; Xia, S.; Song, Z.; Xu, H.; Shi, Y.; He, L.; Cheng, P.; Zhao, B. Highly Efficient Conversion of Propargylic Amines and CO2 Catalyzed by Noble-Metal-Free [Zn116] Nanocages. Angew. Chem. Int. Ed. 2020, 59, 8586–8593. [Google Scholar] [CrossRef]
- Matsuo, H.; Choi, J.-C.; Fujitani, T.; Fujita, K. Silica-Catalyzed Carboxylative Cyclization of Propargylic Amines with CO2. Tetrahedron Lett. 2020, 61, 152557. [Google Scholar] [CrossRef]







































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| R1, R2, R3 | R4 | Yield | Product |
| R1 = Bn, R2 = Ph, R3 = H | 1-Adamantyl (1-Ad) | 86% | 131a |
| R1 = H, R2 = Ph, R3 = H | 1-Ad | 46% (48 h) | 131b |
| R1 = Bn, R2 = o-Tol, R3 = H | 1-Ad | 56% (48 h) | 131c |
| R1 = Bn, R2 = p-FC6H4, R3 = H | 1-Ad | 44% (48 h) | 131d |
| R1 = Bn, R2 = Ph, R3 = H | Cyclopropyl | 55% (48 h) | 131e |
| R1 = Bn, R2 = Ph, R3 = H | Isopropyl | 75% (48 h) | 131f |
| R1 = Bn, R2 = Ph, R3 = H | 4-Tetrahydropyranyl (4-THP) | 78% (60 h) | 131g |
| R1 = Bn, R2 = Ph, R3 = H | 3-Oxetanyl | 64% | 131h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vranješević, F.; Kolympadi Markovic, M.; Matulja, D.; Ambrožić, G.; Sordo, J.Á.; Laclef, S.; Vrček, V.; Marković, D. Recent Advances in the Synthesis of Five-Membered Cyclic Carbonates and Carbamates from Allylic or Propargylic Substrates and CO2. Catalysts 2022, 12, 547. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050547
Vranješević F, Kolympadi Markovic M, Matulja D, Ambrožić G, Sordo JÁ, Laclef S, Vrček V, Marković D. Recent Advances in the Synthesis of Five-Membered Cyclic Carbonates and Carbamates from Allylic or Propargylic Substrates and CO2. Catalysts. 2022; 12(5):547. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050547
Chicago/Turabian StyleVranješević, Filip, Maria Kolympadi Markovic, Dario Matulja, Gabriela Ambrožić, José Ángel Sordo, Sylvain Laclef, Valerije Vrček, and Dean Marković. 2022. "Recent Advances in the Synthesis of Five-Membered Cyclic Carbonates and Carbamates from Allylic or Propargylic Substrates and CO2" Catalysts 12, no. 5: 547. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050547