
Unraveling the Reaction Mechanism of HCHO Catalytic Oxidation on Pristine Co3O4 (110) Surface: A Theoretical Study

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
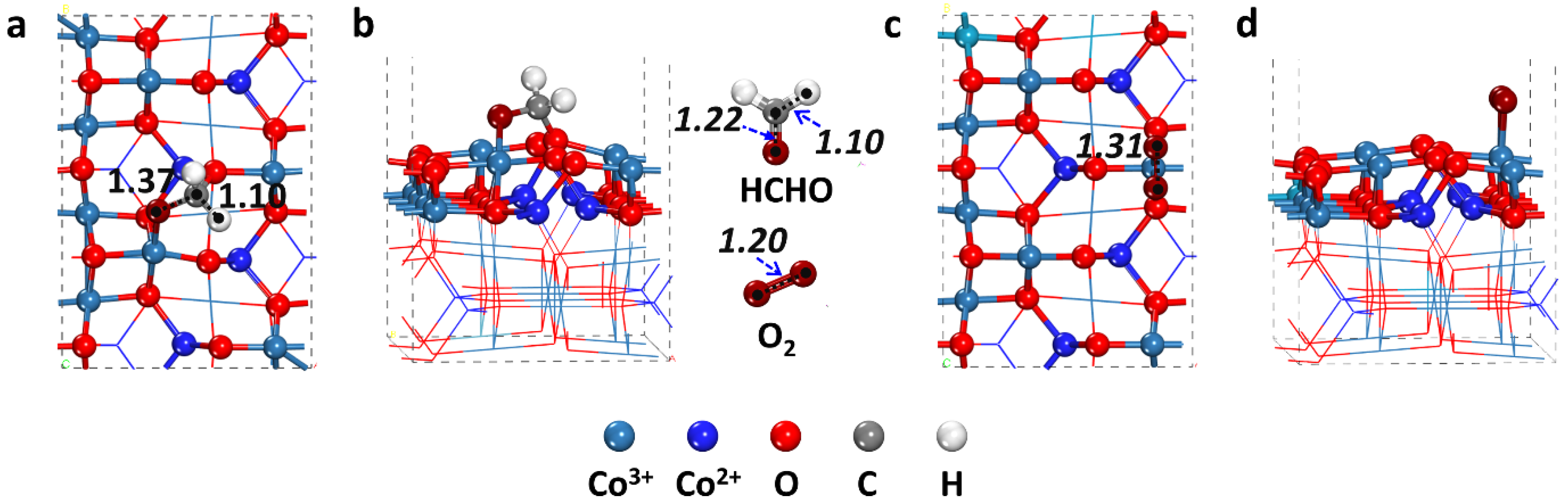
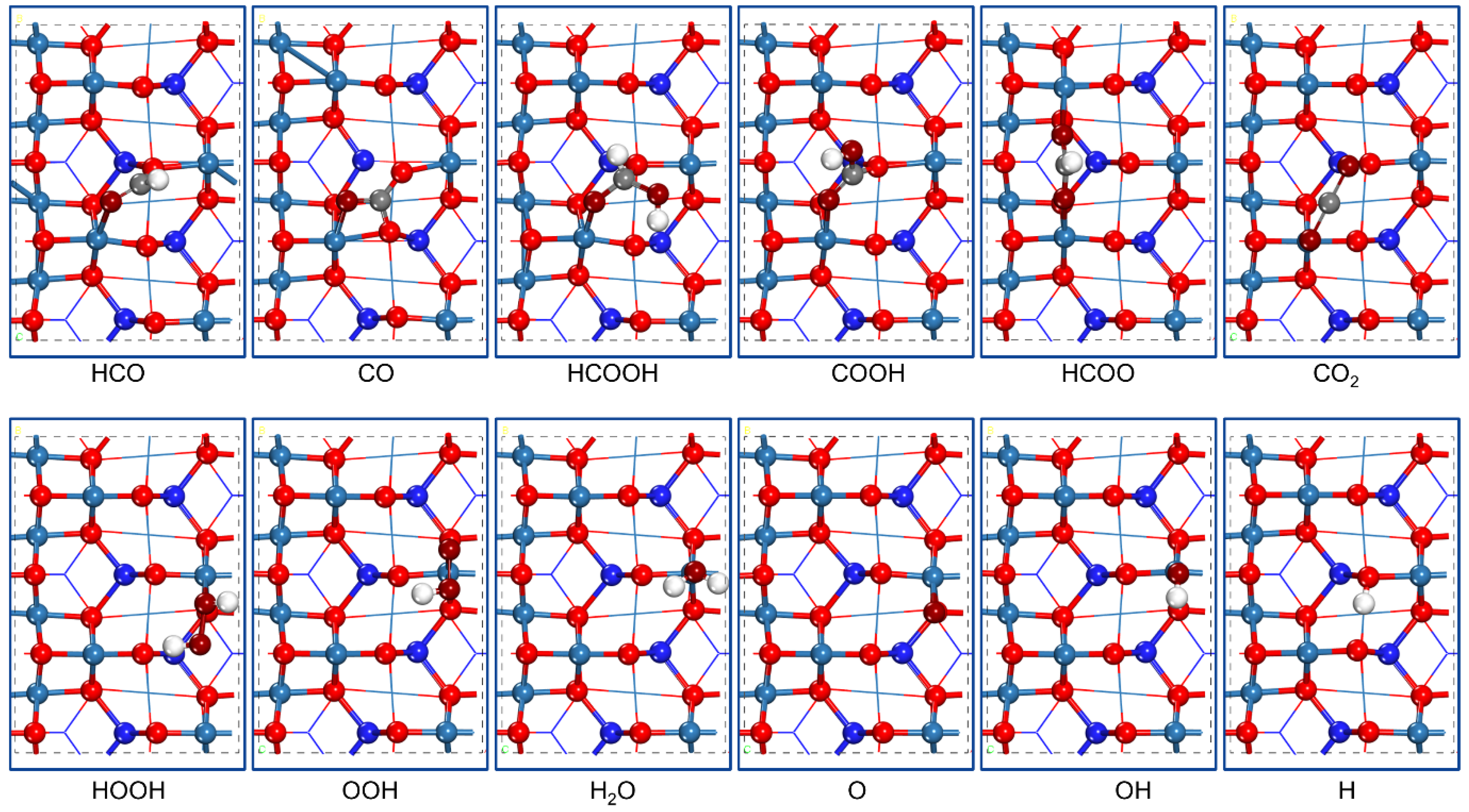
2.1. Possible Adsorption Species
2.2. Reactions Starting from HCHO and O2 Co-Adsorbing on Co3O4 (110) Surface
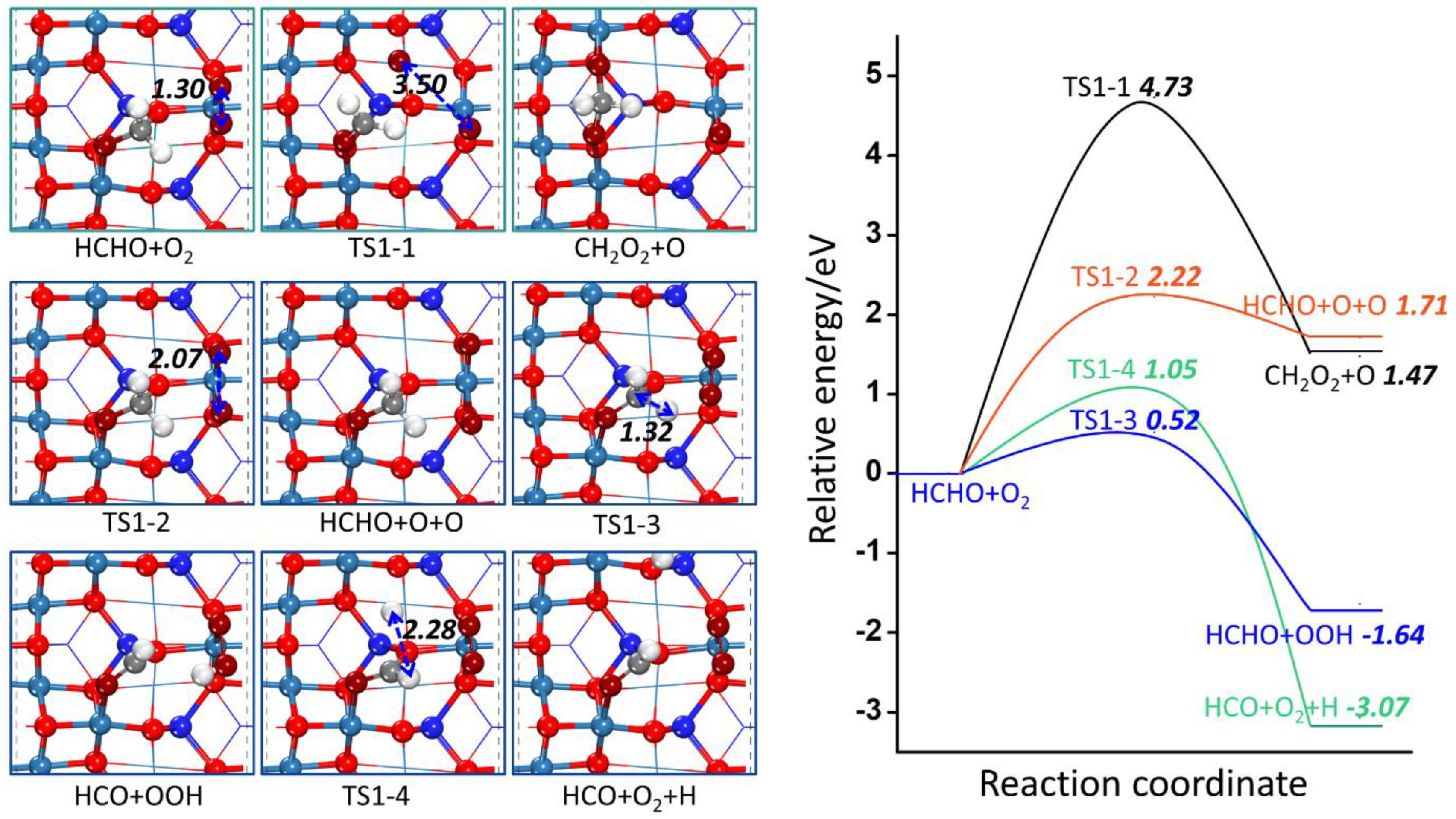
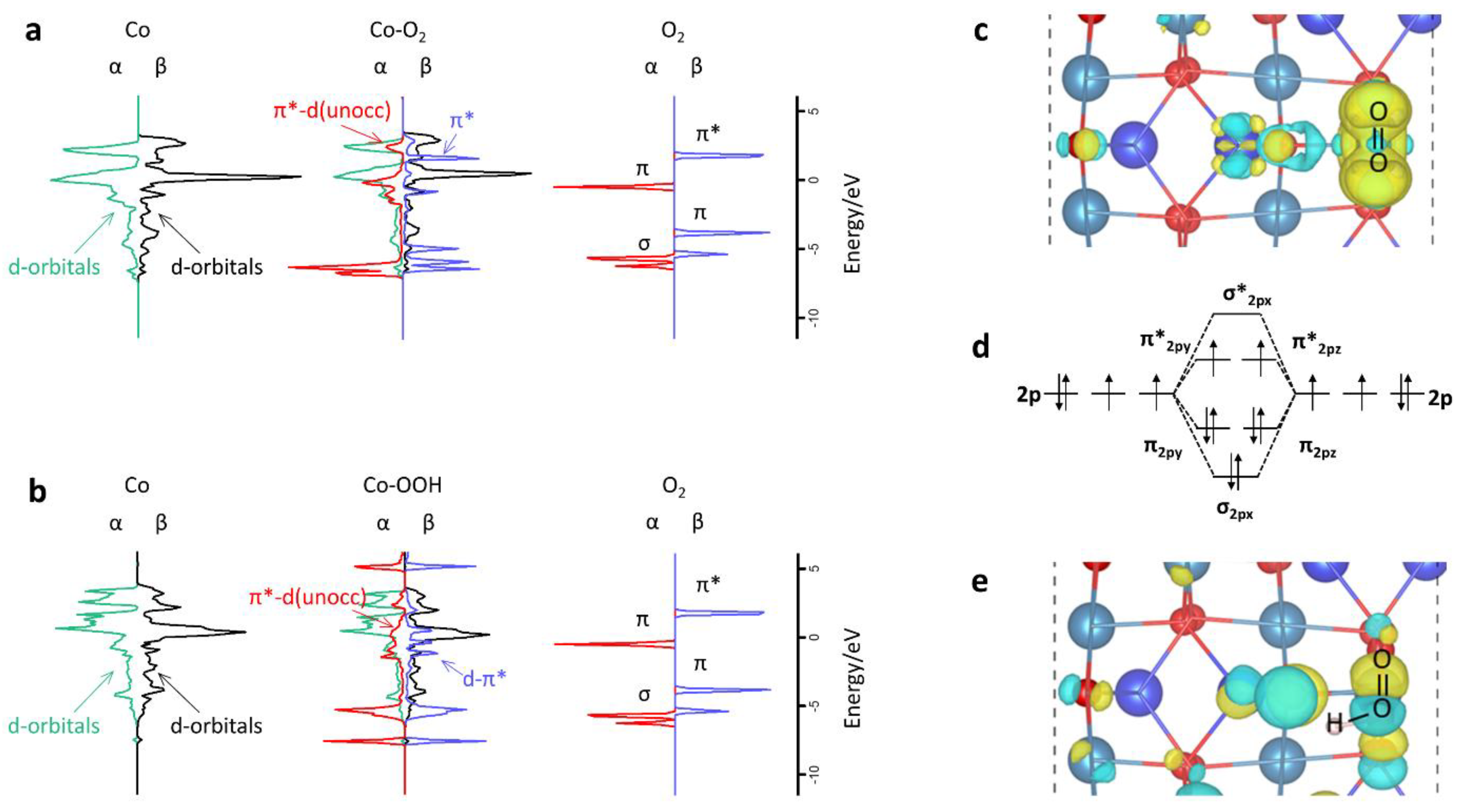
2.2.1. The Possible First Step of HCHO Oxidation
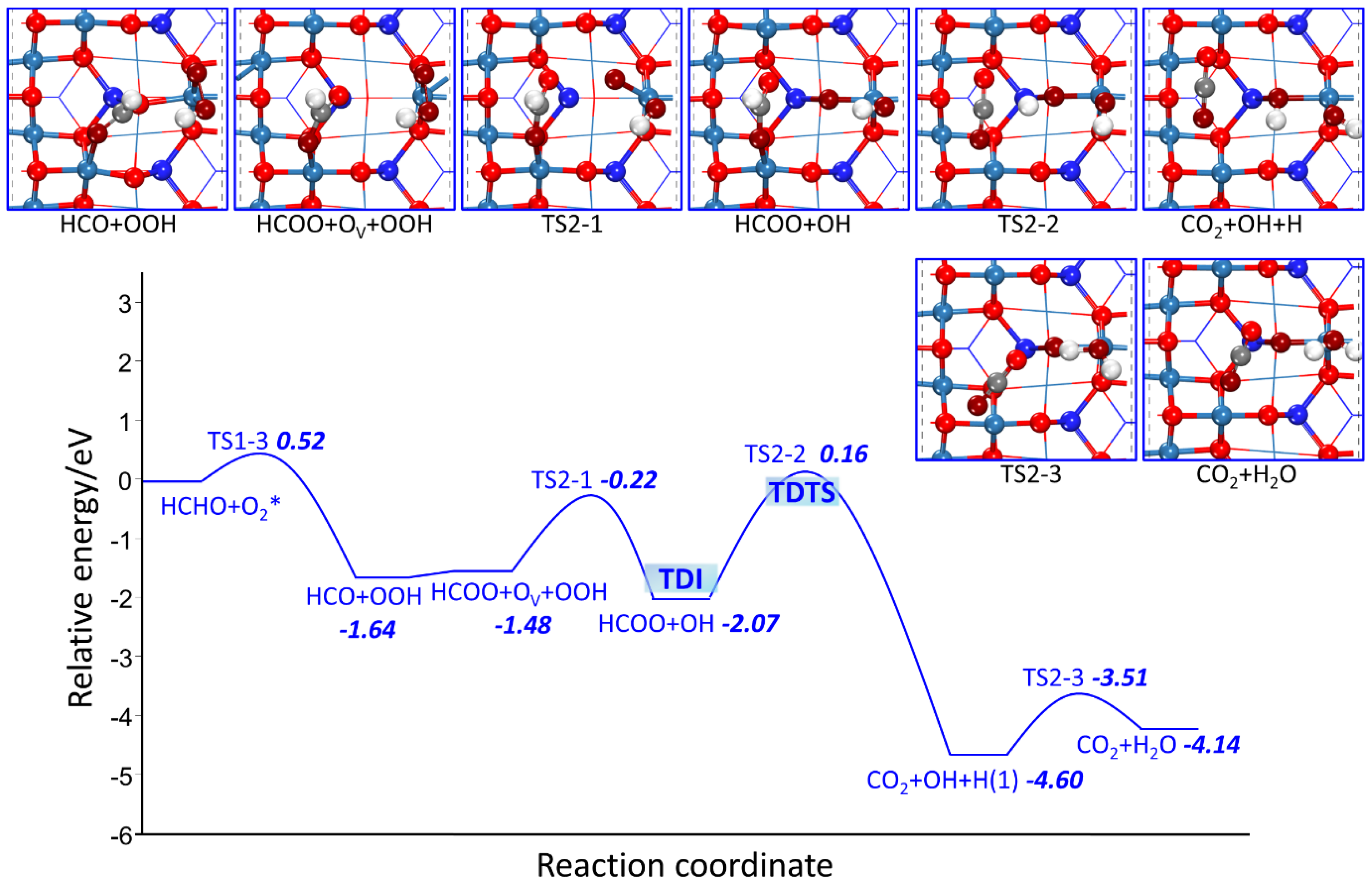
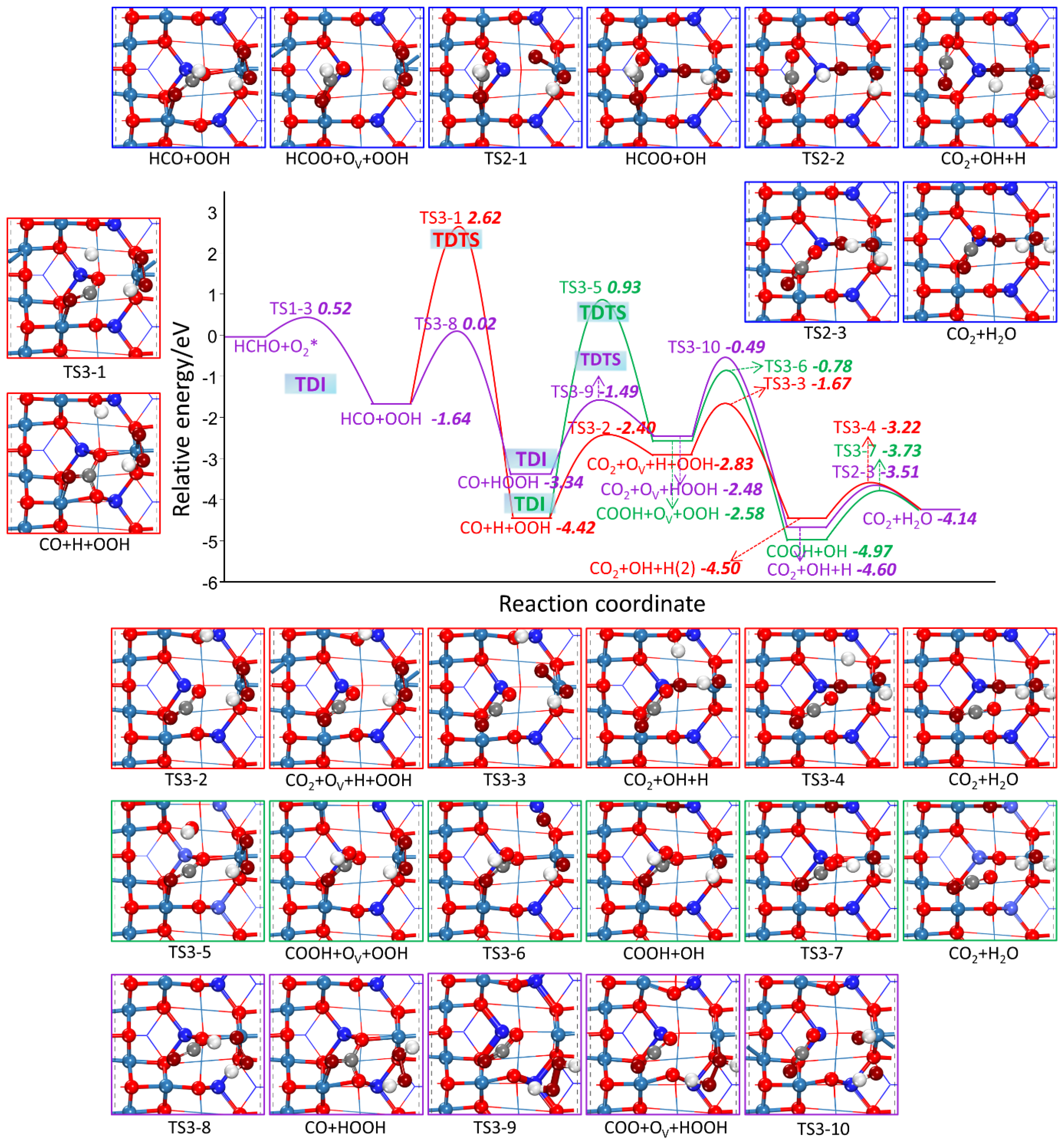
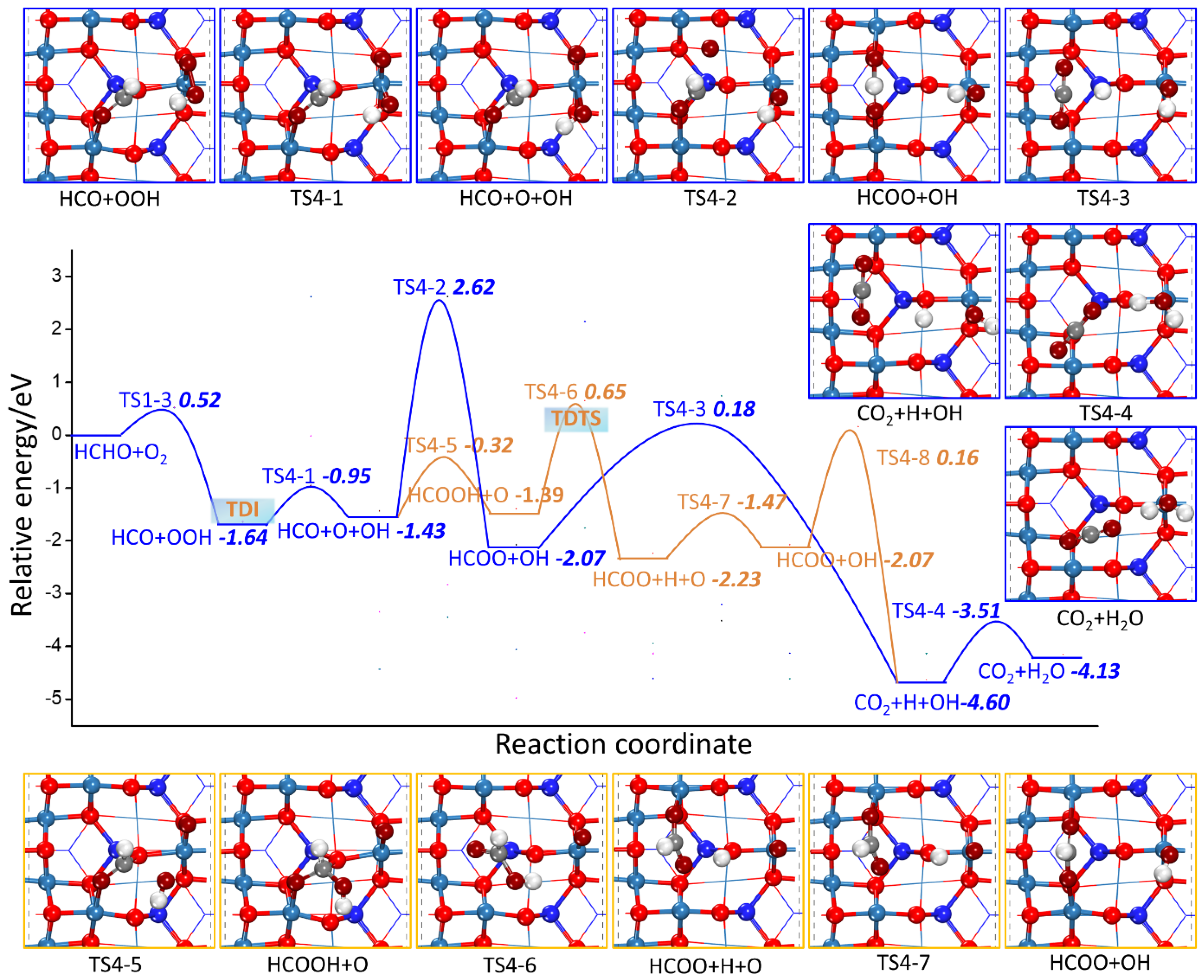
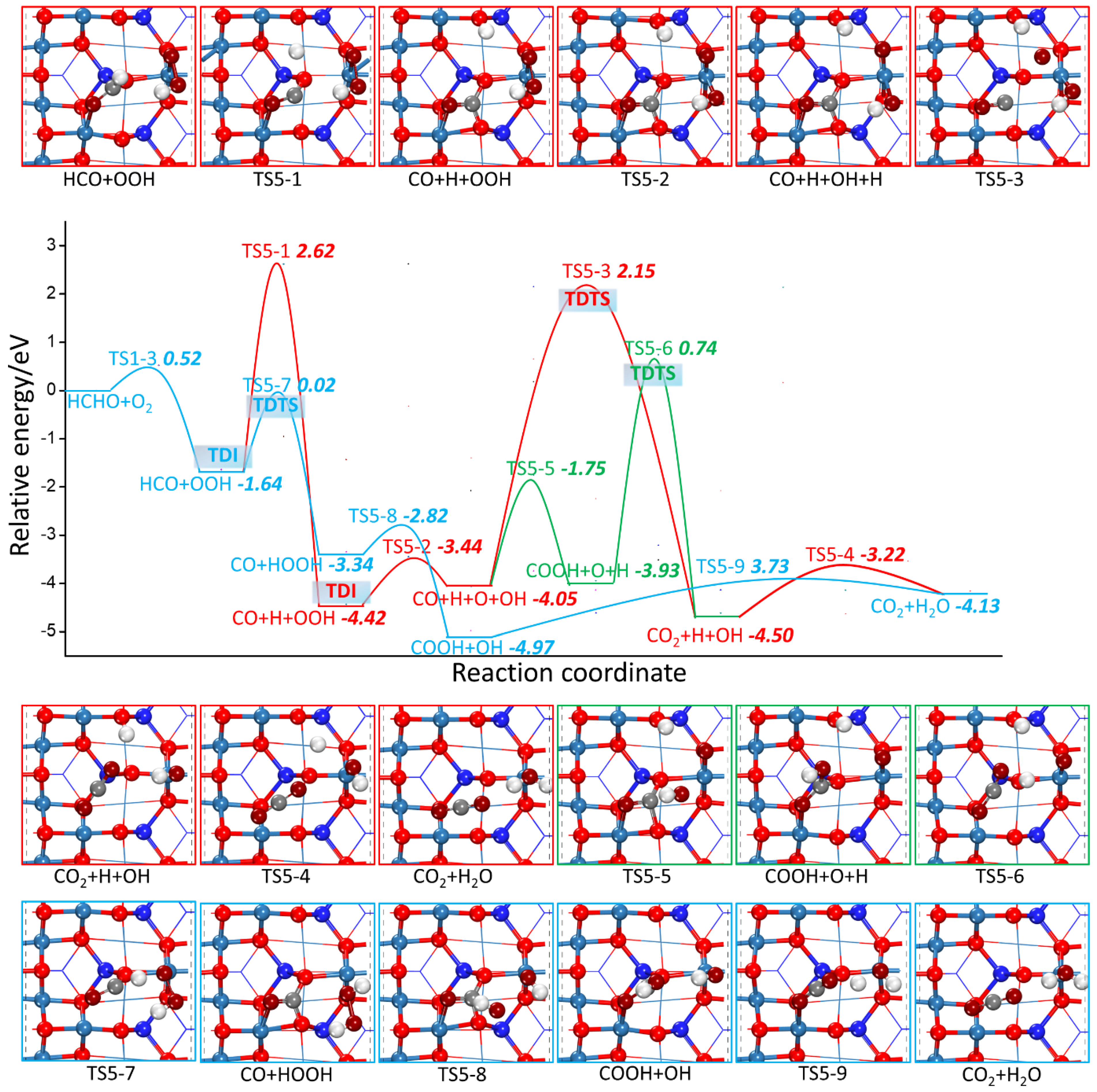
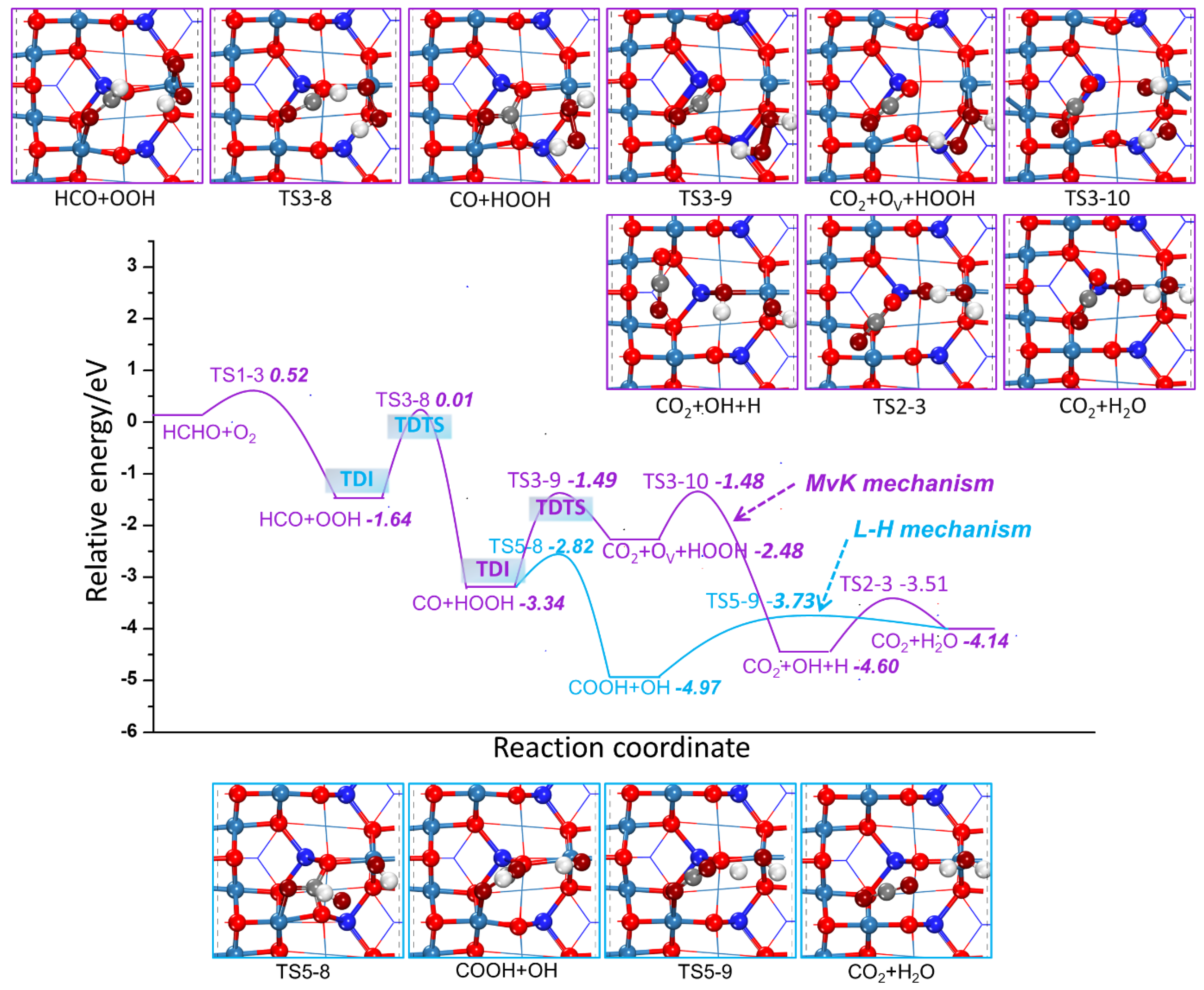
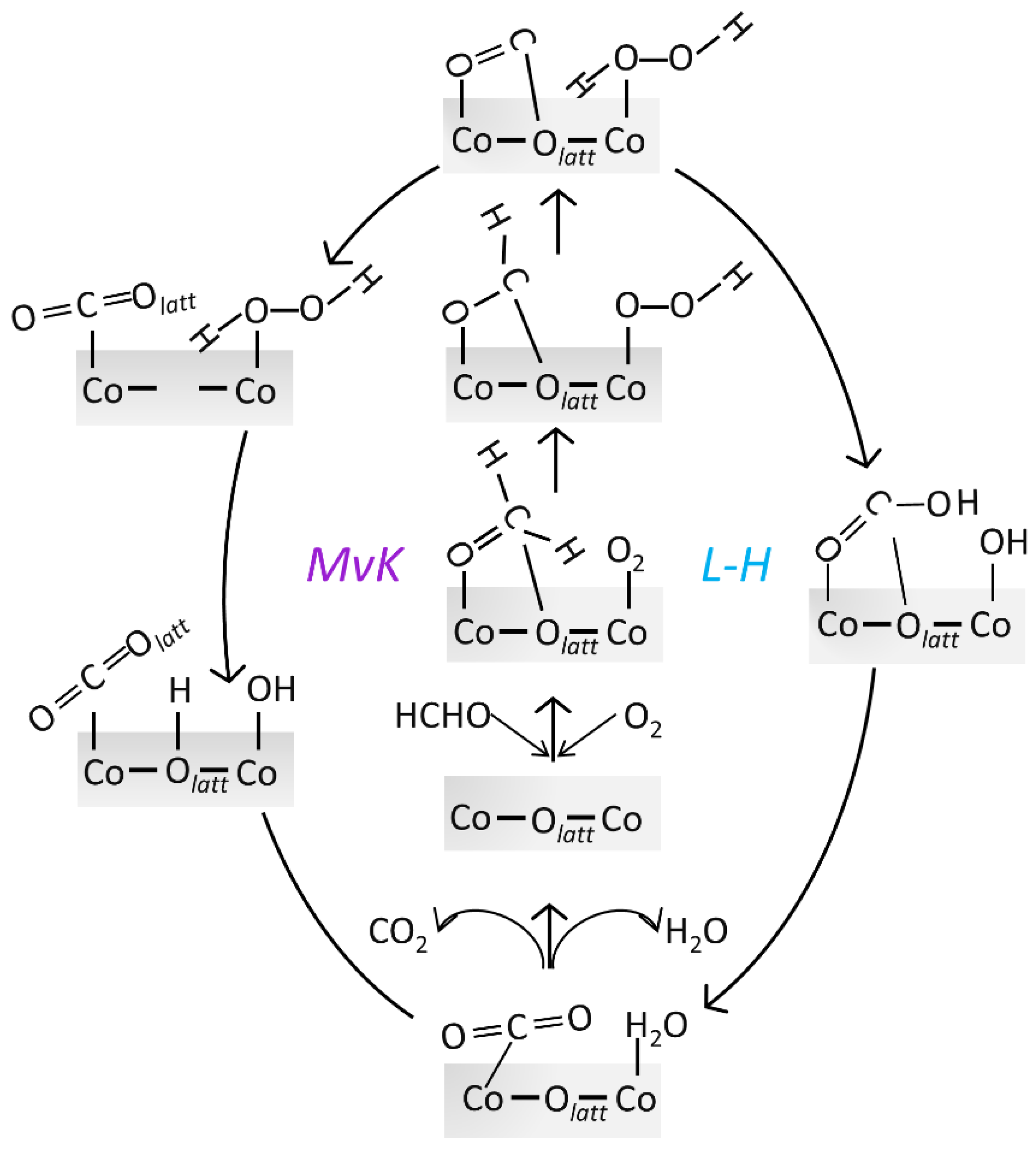
2.2.2. HCHO Catalytic Oxidation Mechanism on Co3O4 (110) Surface from HCO and OOH
- MvK Mechanism
- L-H Mechanism
2.2.3. Comparison of HCHO Catalytic Oxidation through MvK and L-H Mechanism
3. Computational Details
3.1. Surface Models
3.2. Computational Details
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Salthammer, T.; Mentese, S.; Marutzky, R. Formaldehyde in the Indoor Environment. Chem. Rev. 2010, 110, 2536–2572. [Google Scholar] [CrossRef]
- Huang, Y.; Ho, S.S.H.; Ho, K.F.; Lee, S.C.; Yu, J.Z.; Louie, P.K.K. Characteristics and health impacts of VOCs and carbonyls associated with residential cooking activities in Hong Kong. J. Hazard. Mater. 2011, 186, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Yu, J.; Jaroniec, M.; Tao, F.F. Room-temperature catalytic oxidation of formaldehyde on catalysts. Catal. Sci. Technol. 2016, 6, 3649–3669. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Jiang, C.; Zhou, P.; Zhang, P.; Yu, J. The effect of manganese vacancy in birnessite-type MnO2 on room-temperature oxidation of formaldehyde in air. Appl. Catal. B Environ. 2017, 204, 147–155. [Google Scholar] [CrossRef]
- Li, H.; Huang, T.; Lu, Y.; Cui, L.; Wang, Z.; Zhang, C.; Lee, S.; Huang, Y.; Cao, J.; Ho, W. Unraveling the mechanisms of room-temperature catalytic degradation of indoor formaldehyde and its biocompatibility on colloidal TiO2-supported MnOx–CeO2. Environ. Sci. Nano 2018, 5, 1130–1139. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Wang, W.; Chen, M.; Li, H.; Lee, S.-C.; Ho, W.; Huang, T.; Cao, J. Oxygen vacancy–engineered δ-MnOx/activated carbon for room-temperature catalytic oxidation of formaldehyde. Appl. Catal. B Environ. 2020, 278, 119294. [Google Scholar] [CrossRef]
- Zhu, D.; Chen, M.; Huang, Y.; Li, R.; Huang, T.; Cao, J.-j.; Shen, Z.; Lee, S.C. FeCo alloy encased in nitrogen-doped carbon for efficient formaldehyde removal: Preparation, electronic structure, and d-band center tailoring. J. Hazard. Mate. 2022, 424, 127593. [Google Scholar] [CrossRef]
- Zhang, C.; He, H.; Tanaka, K.-I. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature. Appl. Catal. B Environ. 2006, 65, 37–43. [Google Scholar] [CrossRef]
- Jabłońska, M.; Król, A.; Kukulska-Zając, E.; Tarach, K.; Girman, V.; Chmielarz, L.; Góra-Marek, K. Zeolites Y modified with palladium as effective catalysts for low-temperature methanol incineration. Appl. Catal. B Environ. 2015, 166–167, 353–365. [Google Scholar] [CrossRef]
- Chen, B.-B.; Zhu, X.-B.; Crocker, M.; Wang, Y.; Shi, C. FeOx-supported gold catalysts for catalytic removal of formaldehyde at room temperature. Appl. Catal. B Environ. 2014, 154–155, 73–81. [Google Scholar] [CrossRef]
- Bai, B.; Li, J. Positive Effects of K+ Ions on Three-Dimensional Mesoporous Ag/Co3O4 Catalyst for HCHO Oxidation. ACS Catal. 2014, 4, 2753–2762. [Google Scholar] [CrossRef]
- Bai, B.; Arandiyan, H.; Li, J. Comparison of the performance for oxidation of formaldehyde on nano-Co3O4, 2D-Co3O4, and 3D-Co3O4 catalysts. Appl. Catal. B Environ. 2013, 142–143, 677–683. [Google Scholar] [CrossRef]
- Huang, Y.; Long, B.; Tang, M.; Rui, Z.; Balogun, M.-S.; Tong, Y.; Ji, H. Bifunctional catalytic material: An ultrastable and high-performance surface defect CeO2 nanosheets for formaldehyde thermal oxidation and photocatalytic oxidation. Appl. Catal. B Environ. 2016, 181, 779–787. [Google Scholar] [CrossRef]
- Zeng, L.; Song, W.; Li, M.; Zeng, D.; Xie, C. Catalytic oxidation of formaldehyde on surface of HTiO2/HCTiO2 without light illumination at room temperature. Appl. Catal. B Environ. 2014, 147, 490–498. [Google Scholar] [CrossRef]
- Li, H.; Ho, W.; Cao, J.; Park, D.; Lee, S.-C.; Huang, Y. Active Complexes on Engineered Crystal Facets of MnOx–CeO2 and Scale-Up Demonstration on an Air Cleaner for Indoor Formaldehyde Removal. Environ. Sci. Technol. 2019, 53, 10906–10916. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cui, L.; Lu, Y.; Huang, Y.; Cao, J.; Park, D.; Lee, S.-C.; Ho, W. In Situ Intermediates Determination and Cytotoxicological Assessment in Catalytic Oxidation of Formaldehyde: Implications for Catalyst Design and Selectivity Enhancement under Ambient Conditions. Environ. Sci. Technol. 2019, 53, 5230–5240. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Huang, Y.; Zhu, D.; Ho, W.; Cao, J.; Lee, S. Improved Oxygen Activation over a Carbon/Co3O4 Nanocomposite for Efficient Catalytic Oxidation of Formaldehyde at Room Temperature. Environ. Sci. Technol. 2021, 55, 4054–4063. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Huang, Y.; Zhu, D.; Ho, W.; Lee, S.; Cao, J. A Review of Co3O4-based Catalysts for Formaldehyde Oxidation at Low Temperature: Effect Parameters and Reaction Mechanism. Aerosol Sci. Eng. 2020, 4, 147–168. [Google Scholar] [CrossRef]
- Ma, C.; Wang, D.; Xue, W.; Dou, B.; Wang, H.; Hao, Z. Investigation of Formaldehyde Oxidation over Co3O4−CeO2 and Au/Co3O4−CeO2 Catalysts at Room Temperature: Effective Removal and Determination of Reaction Mechanism. Environ. Sci. Technol. 2011, 45, 3628–3634. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Zhang, L.; Jiang, D. Surface oxygen vacancies on Co3O4 mediated catalytic formaldehyde oxidation at room temperature. Catal. Sci. Technol. 2016, 6, 3845–3853. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, Z.; Cheng, B.; Jiang, C. Co3O4 nanorod-supported Pt with enhanced performance for catalytic HCHO oxidation at room temperature. Appl. Surf. Sci. 2017, 404, 426–434. [Google Scholar] [CrossRef]
- Zha, K.; Sun, W.; Huang, Z.; Xu, H.; Shen, W. Insights into High-Performance Monolith Catalysts of Co3O4 Nanowires Grown on Nickel Foam with Abundant Oxygen Vacancies for Formaldehyde Oxidation. ACS Catal. 2020, 10, 12127–12138. [Google Scholar] [CrossRef]
- Cai, Y.; Xu, J.; Guo, Y.; Liu, J. Ultrathin, Polycrystalline, Two-Dimensional Co3O4 for Low-Temperature CO Oxidation. ACS Catal. 2019, 9, 2558–2567. [Google Scholar] [CrossRef]
- Bae, J.; Shin, D.; Jeong, H.; Choe, C.; Choi, Y.; Han, J.W.; Lee, H. Facet-Dependent Mn Doping on Shaped Co3O4 Crystals for Catalytic Oxidation. ACS Catal. 2021, 11, 11066–11074. [Google Scholar] [CrossRef]
- Xiao, M.; Yu, X.; Guo, Y.; Ge, M. Boosting Toluene Combustion by Tuning Electronic Metal–Support Interactions in In Situ Grown Pt@Co3O4 Catalysts. Environ. Sci. Technol. 2022, 56, 1376–1385. [Google Scholar] [CrossRef]
- Deng, J.; Song, W.; Chen, L.; Wang, L.; Jing, M.; Ren, Y.; Zhao, Z.; Liu, J. The effect of oxygen vacancies and water on HCHO catalytic oxidation over Co3O4 catalyst: A combination of density functional theory and microkinetic study. Chem. Eng. J. 2019, 355, 540–550. [Google Scholar] [CrossRef]
- Wang, X.; Ying, J.; Mai, Y.; Zhang, J.; Chen, J.; Wen, M.; Yu, L. MOF-derived metal oxide composite Mn2Co1Ox/CN for efficient formaldehyde oxidation at low temperature. Catal. Sci. Technol. 2019, 9, 5845–5854. [Google Scholar] [CrossRef]
- Chen, J.; Huang, M.; Chen, W.; Tang, H.; Jiao, Y.; Zhang, J.; Wang, G.; Wang, R. Defect Engineering and Synergistic Effect in Co3O4 Catalysts for Efficient Removal of Formaldehyde at Room Temperature. Ind. Eng. Chem. Res. 2020, 59, 18781–18789. [Google Scholar] [CrossRef]
- Wang, N.; Xu, Z.; Luo, T.; Yan, Z.; Jin, M.; Shi, L. Pt Anchored on Mn(Co)CO3/MnCo2O4 Heterostructure for Complete Oxidation of Formaldehyde at Room Temperature. ChemistrySelect 2020, 5, 10537–10548. [Google Scholar] [CrossRef]
- Jiang, Q.G.; Ao, Z.M.; Li, S.; Wen, Z. Density functional theory calculations on the CO catalytic oxidation on Al-embedded graphene. RSC Adv. 2014, 4, 20290–20296. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Selloni, A. Electronic states and magnetic structure at the Co3O4 (110) surface: A first-principles study. Phys. Rev. B 2012, 85, 085306. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Eads (eV) | Adsorption/Configuration | Distance (Å) | Bonding Details | |
|---|---|---|---|---|---|
| Bond | Length (Å) | ||||
| HCHO | 1.28 | C-O2c, O-Co3+ | 1.467, 1.898 | C-O | 1.372 |
| HCO | 3.45 | C-O2c, O-Co3+ | 1.315, 2.222 | C-O | 1.250 |
| CO | 3.57 | C-two O2c | 1.281, 1.349 | C-O | 1.283 |
| HCOOH | 0.76 | C-O2c, O-Co3+ | 1.455, 1.969 | C-O | 1.355 |
| COOH | 3.71 | C-O2c, O-Co3+ | 1.303, 2.025 | C-O/C-O | 1.260/1.355 |
| HCOO | 2.23 | O-Co2+, O-Co2+ | 2.006, 2.104 | C-O/C-O | 1.260/1.279 |
| CO2 | 0.01 | away from the surface | — | C-O | 1.181 |
| HOOH | 1.13 | O-Co3+ | 2.077 | O-O | 1.483 |
| OOH | 1.26 | O-Co3+ | 1.909 | O-O | 1.455 |
| O2 | 0.85 | O-Co3+ | 1.930, 1.937 | O-O | 1.310 |
| H2O | 0.74 | O-Co3+ | 2.140 | O-H | 0.979 |
| OH | 2.35 | O-Co3+ | 1.849 | O-H | 0.979 |
| O | 2.87 | O-Co3+ | 1.750 | — | — |
| H | 3.74 | H-O2c | 1.002 | — | — |
| Reactions | Co3O4 (110) Surface | ||
|---|---|---|---|
| Ea/eV | ΔE/eV | ||
| R1-1 | HCHO + O2 → CH2O2 + O | 4.73 | 1.47 |
| R1-2 | HCHO + O2 → HCHO + O + O | 2.22 | 1.71 |
| R1-3 | HCHO + O2 → HCO + OOH | 0.52 | −1.64 |
| R1-4 | HCHO + O2 → HCO + H + O2 | 1.05 | −3.07 |
| HCO + Olatt + OOH → HCOO + OV + OOH | - | 0.16 | |
| R2-1 | HCOO + OV + OOH → HCOO + OH + Olatt | 1.25 | −0.60 |
| R2-2 | HCOO + OH→ CO2 + H + OH | 2.24 | −2.55 |
| R2-3 | CO2 + H + OH → CO2 + H2O | 1.09 | 0.48 |
| R3-1 | HCO + OOH → CO + H + OOH | 4.26 | −2.78 |
| R3-2 | CO + Olatt + H + OOH → CO2 + OV + H + OOH | 2.02 | 1.59 |
| R3-3 | CO2 + OV + H + OOH → CO2 + OH + H + Olatt | 1.16 | −1.77 |
| R3-4 | CO2 + H + OH → CO2 + H2O | 1.40 | 0.48 |
| R3-5 | CO + Olatt + H + OOH → COOH + OV + OOH | 5.36 | 1.84 |
| R3-6 | COOH + OV + OOH → COOH + OH + Olatt | 1.80 | −2.39 |
| R3-7 | COOH + OOH → CO2 + H2O | 1.24 | 0.84 |
| R3-8 | HCO + OOH → CO + HOOH | 1.62 | −1.70 |
| R3-9 | CO + Olatt + HOOH → CO2 + OV + HOOH | 1.85 | 0.86 |
| R3-10 | CO2+ OV +HOOH →CO2 + Olatt H + OH | 1.00 | −2.12 |
| R4-1 | HCO + OOH → HCO + O + OH | 0.68 | 0.21 |
| R4-2 | HCO+ O + OH → HCOO + OH | 4.05 | −0.65 |
| R4-3 | HCOO + OH → CO2 + H + OH | 2.24 | −2.55 |
| R4-4 | CO2 + H + OH → CO2 + H2O | 1.09 | 0.48 |
| R4-5 | HCO + O + OH → HCOOH + O | 1.11 | 0.04 |
| R4-6 | HCOOH + O → HCOO + H + O | 2.05 | −0.84 |
| R4-7 | HCOO + H + O → HCOO + OH | 0.76 | 0.16 |
| R4-8 | HCOO + OH → CO2 + H + OH | 2.24 | −2.55 |
| R5-1 | HCO + OOH → CO + H + OOH | 0.98 | 0.37 |
| R5-2 | CO + H + OOH → CO + H + O + OH | 4.26 | −2.78 |
| R5-3 | CO + H + O + OH → CO2 + OH + H | 6.20 | −0.55 |
| R5-4 | CO2 + OH + H → CO2 + H2O | 1.09 | 0.48 |
| R5-5 | CO + H + O + OH → COOH + O + H | 2.30 | 0.12 |
| R5-6 | COOH + O + H → CO2 + H + OH | 4.68 | −0.67 |
| R5-7 | HCO + OOH → CO + HOOH | 1.62 | −1.70 |
| R5-8 | CO + HOOH → COOH + OH | 0.52 | −1.63 |
| R5-9 | COOH + OH → CO2 + H2O | 1.24 | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Huang, T.; Huang, Y.; Chen, M.; Lee, S.-c.; Ho, W.; Cao, J. Unraveling the Reaction Mechanism of HCHO Catalytic Oxidation on Pristine Co3O4 (110) Surface: A Theoretical Study. Catalysts 2022, 12, 560. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050560
Li R, Huang T, Huang Y, Chen M, Lee S-c, Ho W, Cao J. Unraveling the Reaction Mechanism of HCHO Catalytic Oxidation on Pristine Co3O4 (110) Surface: A Theoretical Study. Catalysts. 2022; 12(5):560. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050560
Chicago/Turabian StyleLi, Rong, Tingting Huang, Yu Huang, Meijuan Chen, Shun-cheng Lee, Wingkei Ho, and Junji Cao. 2022. "Unraveling the Reaction Mechanism of HCHO Catalytic Oxidation on Pristine Co3O4 (110) Surface: A Theoretical Study" Catalysts 12, no. 5: 560. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050560