
The Role of Nickel and Brønsted Sites on Ethylene Oligomerization with Ni-H-Beta Catalysts

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Method
2.1. Catalyst Synthesis
2.2. Catalyst Characterization
2.2.1. Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES)
2.2.2. Pyridine Adsorption
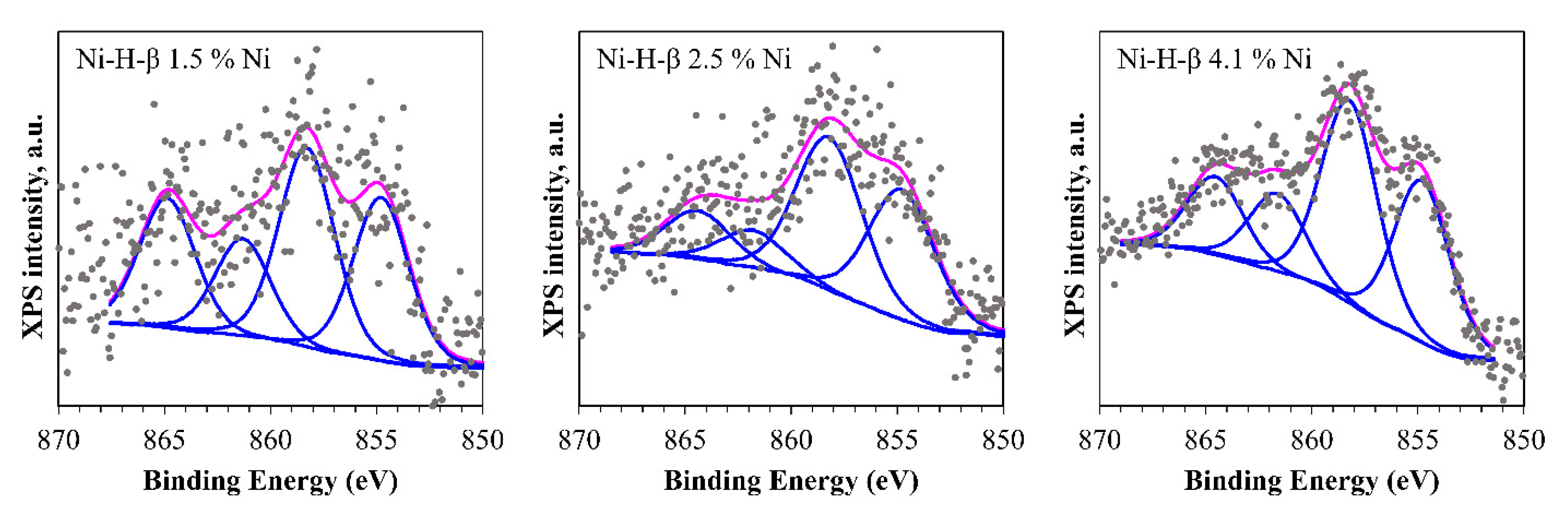
2.2.3. X-ray Photoelectron Spectroscopy (XPS)
2.2.4. X-ray Powder Diffraction (XRD)
2.3. Ethylene Adsorption Experiments
2.4. Ethylene Oligomerization Experiments
3. Results and Discussion
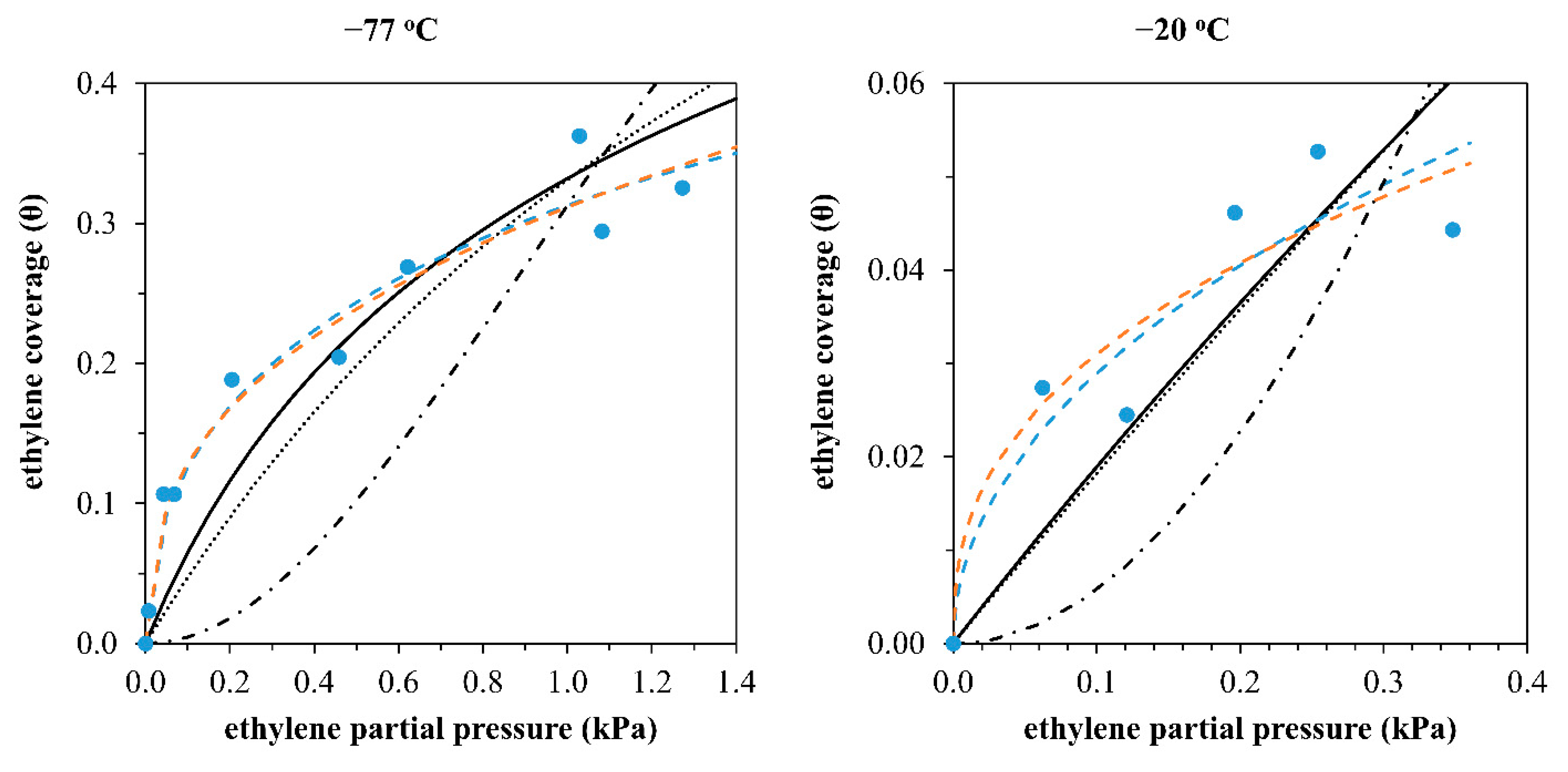
3.1. Ethylene Adsorption Study on Ni-H-Beta Catalyst
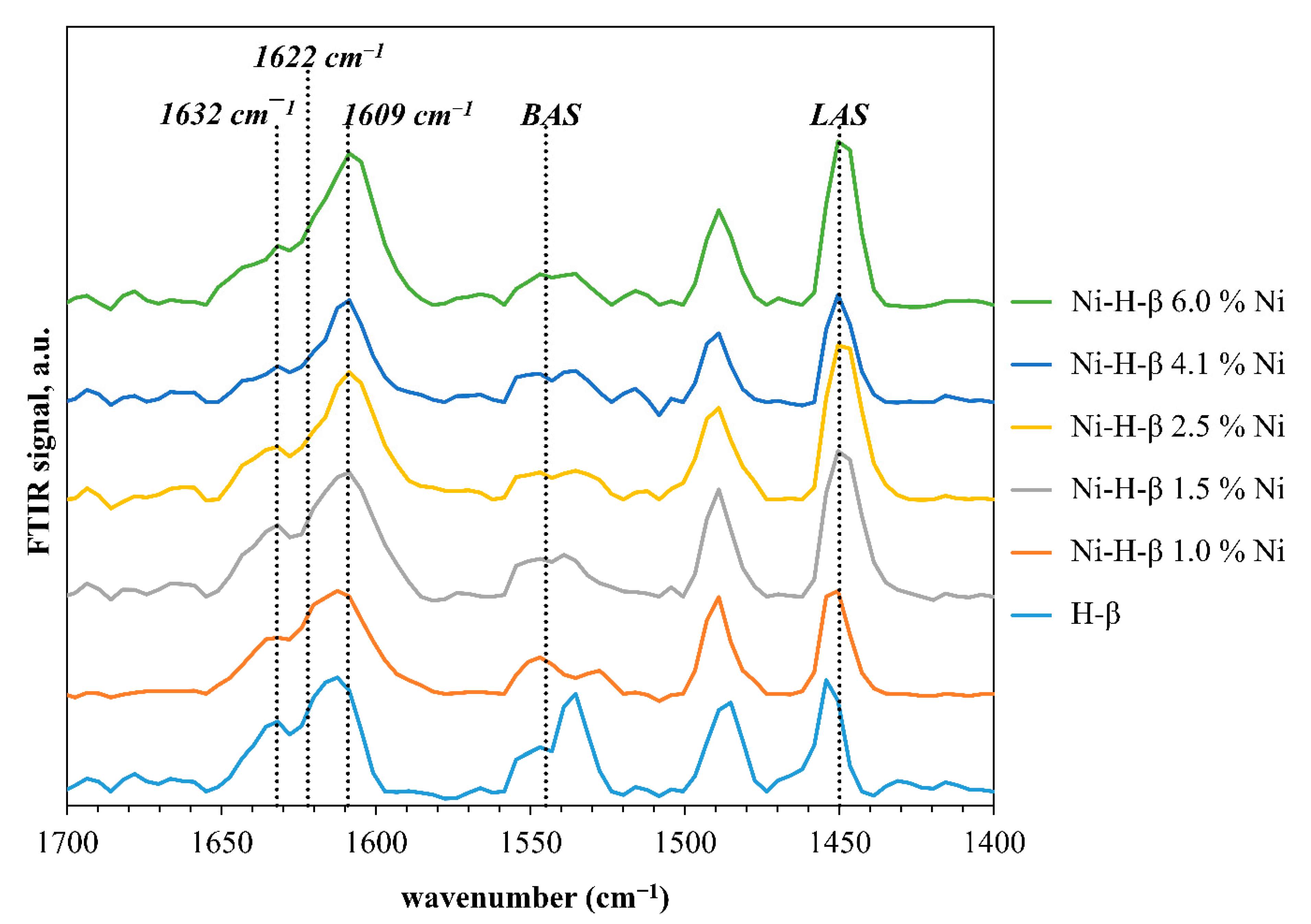
3.2. Physicochemical Properties of Ni-H-Beta Catalysts as a Function of Nickel Loading
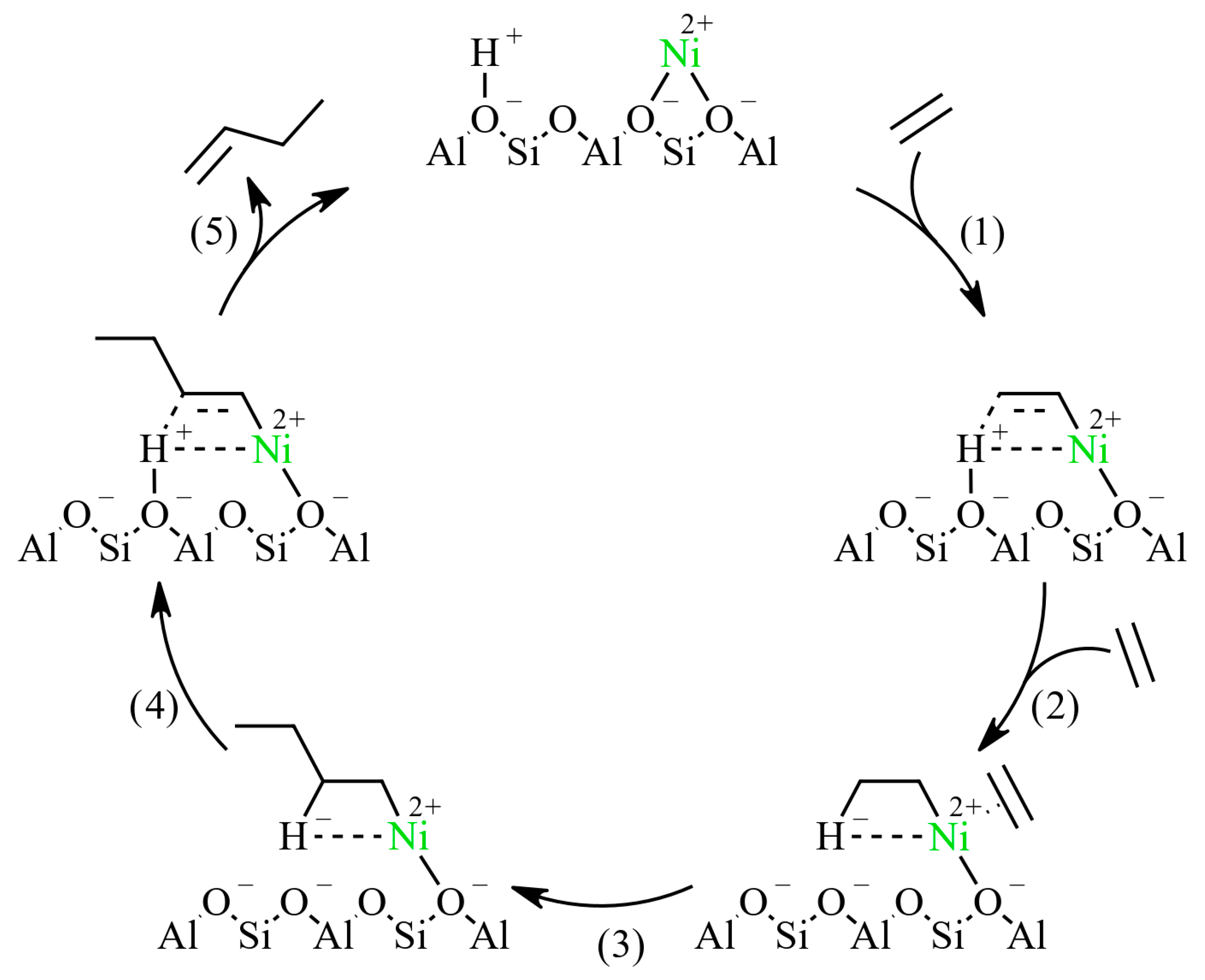
3.3. Effect of Nickel and Brønsted Sites on Ethylene Oligomerization with Ni-H-Beta Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’connor, C.; Kojima, M. Alkene oligomerization. Catal. Today 1990, 6, 329–349. [Google Scholar] [CrossRef]
- Finiels, A.; Fajula, F.; Hulea, V. Nickel-based solid catalysts for ethylene oligomerization-a review. Catal. Sci. Technol. 2014, 4, 2412–2426. [Google Scholar] [CrossRef]
- McGuinness, D.S. Olefin oligomerization via metallacycles: Dimerization, trimerization, tetramerization, and beyond. Chem. Rev. 2011, 111, 2321–2341. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.; Komurcu, M.; Ganjkhanlou, Y.; Brogaard, R.Y.; Lu, L.; Jens, K.J.; Berlier, G.; Olsbye, U. Ethene oligomerization on nickel microporous and mesoporous-supported catalysts: Investigation of the active sites. Catal. Today 2018, 299, 154–163. [Google Scholar] [CrossRef]
- Moussa, S.; Concepción, P.; Arribas, M.A.; Martinez, A. Nature of active nickel sites and initiation mechanism for ethylene oligomerization on heterogeneous Ni-beta catalysts. ACS Catal. 2018, 8, 7b03970. [Google Scholar] [CrossRef]
- Andrei, R.D.; Popa, M.I.; Fajula, F.; Hulea, V. Heterogeneous oligomerization of ethylene over highly active and stable Ni-AlSBA-15 mesoporous catalysts. J. Catal. 2015, 323, 76–84. [Google Scholar] [CrossRef]
- Skupińska, J. Oligomerization of α-Olefins to Higher Oligomers. Chem. Rev. 1991, 91, 613–648. [Google Scholar] [CrossRef]
- Derouane, E.G. Catalysts for Fine Chemical Synthesis, 4th ed.; Derouane, E.G., Roberts, S.M., Kozhevnikov, I.V., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; ISBN 9780471490548. [Google Scholar]
- Toch, K.; Thybaut, J.W.; Arribas, M.A.; Martínez, A.; Marin, G.B. Steering linear 1-alkene, propene or gasoline yields in ethene oligomerization via the interplay between nickel and acid sites. Chem. Eng. Sci. 2017, 173, 49–59. [Google Scholar] [CrossRef]
- Toch, K.; Thybaut, J.W.; Marin, G.B. Ethene oligomerization on Ni-SiO2-Al2O3: Experimental investigation and Single-Event MicroKinetic modeling. Appl. Catal. A Gen. 2015, 489, 292–304. [Google Scholar] [CrossRef]
- Martínez, A.; Arribas, M.A.; Concepción, P.; Moussa, S. New bifunctional Ni-H-Beta catalysts for the heterogeneous oligomerization of ethylene. Appl. Catal. A Gen. 2013, 467, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Lacarriere, A.; Robin, J.; Świerczyński, D.; Finiels, A.; Fajula, F.; Luck, F.; Hulea, V. Distillate-range products from non-oil-based sources by catalytic cascade reactions. ChemSusChem 2012, 5, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, M.; Finiels, A.; Fajula, F.; Hulea, V. Nature of the active sites in ethylene oligomerization catalyzed by ni-containing molecular sieves: Chemical and IR spectral investigation. J. Phys. Chem. C 2009, 113, 20360–20364. [Google Scholar] [CrossRef]
- Lallemand, M.; Finiels, A.; Fajula, F.; Hulea, V. Continuous stirred tank reactor for ethylene oligomerization catalyzed by NiMCM-41. Chem. Eng. J. 2011, 172, 1078–1082. [Google Scholar] [CrossRef]
- Lallemand, M.; Finiels, A.; Fajula, F.; Hulea, V. Catalytic oligomerization of ethylene over Ni-containing dealuminated Y zeolites. Appl. Catal. A Gen. 2006, 301, 196–201. [Google Scholar] [CrossRef]
- Lallemand, M.; Rusu, O.A.; Dumitriu, E.; Finiels, A.; Fajula, F.; Hulea, V. NiMCM-36 and NiMCM-22 catalysts for the ethylene oligomerization: Effect of zeolite texture and nickel cations/acid sites ratio. Appl. Catal. A Gen. 2008, 338, 37–43. [Google Scholar] [CrossRef]
- Hulea, V.; Fajula, F. Ni-exchanged AlMCM-41-An efficient bifunctional catalyst for ethylene oligomerization. J. Catal. 2004, 225, 213–222. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Xu, D.; Sun, C.; Ni, L.; Fu, W.; Zeng, S.; Jiang, S.; Zhang, Z.; Qiu, S. Synthesis and properties of MFI zeolites with microporous, mesoporous and macroporous hierarchical structures by a gel-casting technique. New J. Chem. 2016, 40, 4398–4405. [Google Scholar] [CrossRef]
- Moussa, S.; Arribas, M.A.; Concepción, P.; Martínez, A. Heterogeneous oligomerization of ethylene to liquids on bifunctional Ni-based catalysts: The influence of support properties on nickel speciation and catalytic performance. Catal. Today 2016, 277, 78–88. [Google Scholar] [CrossRef]
- Heveling, J.; Nicolaides, C.P.; Scurrell, M.S. Identification of novel catalysts and conditions for the highly efficient and stable heterogeneous oligomerization of ethylene. J. Chem. Soc. Chem. Commun. 1991, 126–127. [Google Scholar] [CrossRef]
- Heveling, J.; Nicolaides, C.P.; Scurrell, M.S. Catalysts and conditions for the highly efficient and stable heterogeneous oligomerization of ethylene. App. Cata. A Gen. 1998, 173, 1–9. [Google Scholar] [CrossRef]
- Heydenrych, M.D.; Nicolaides, C.P.; Scurrell, M.S. Oligomerization of ethene in a slurry reactor using a nickel(II)-exchanged silica-alumina catalyst. J. Catal. 2001, 197, 49–57. [Google Scholar] [CrossRef]
- Hartmann, M.; Pöppl, A.; Kevan, L. Ethylene dimerization and butene isomerization in nickel-containing MCM-41 and AlMCM-41 mesoporous molecular sieves: An electron spin resonance and gas chromatography study. J. Phys. Chem. 1996, 100, 9906–9910. [Google Scholar] [CrossRef]
- Jan, O.; Resende, F.L.P. Liquid hydrocarbon production via ethylene oligomerization over Ni-HΒ. Fuel Process. Technol. 2018, 179, 269–276. [Google Scholar] [CrossRef]
- Jan, O.; Song, K.; Dichiara, A.B.; Resende, F.L.P. Ethylene Oligomerization over Ni-Hβ Heterogeneous Catalysts. Ind. Eng. Chem. Res. 2018, 57, 10241–10250. [Google Scholar] [CrossRef]
- Jan, O.; Song, K.; Dichiara, A.; Resende, F.L.P. Oligomerization of supercritical ethylene over nickel-based silica-alumina catalysts. Chem. Eng. Sci. 2019, 197, 212–222. [Google Scholar] [CrossRef]
- Seufitelli, G.V.S.; Resende, F.L.P. Study of the catalytic reactions of ethylene oligomerization in subcritical and supercritical media over a NiBEA catalyst. Appl. Catal. A Gen. 2019, 576, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Brogaard, R.Y.; Olsbye, U. Ethene Oligomerization in Ni-Containing Zeolites: Theoretical Discrimination of Reaction Mechanisms. ACS Catal. 2016, 6, 1205–1214. [Google Scholar] [CrossRef]
- Forget, S.; Olivier-Bourbigou, H.; Delcroix, D. Homogeneous and Heterogeneous Nickel-Catalyzed Olefin Oligomerization: Experimental Investigation for a Common Mechanistic Proposition and Catalyst Optimization. ChemCatChem 2017, 9, 2408–2417. [Google Scholar] [CrossRef]
- Metzger, E.D.; Comito, R.J.; Hendon, C.H.; Dincǎ, M. Mechanism of single-site molecule-like catalytic ethylene dimerization in Ni-MFU-4l. J. Am. Chem. Soc. 2017, 139, 757–762. [Google Scholar] [CrossRef]
- Joshi, R.; Zhang, G.; Miller, J.T.; Gounder, R. Evidence for the Coordination-Insertion Mechanism of Ethene Dimerization at Nickel Cations Exchanged onto Beta Molecular Sieves. ACS Catal. 2018, 8, 11407–11422. [Google Scholar] [CrossRef]
- Brogaard, R.Y.; Kømurcu, M.; Dyballa, M.M.; Botan, A.; Van Speybroeck, V.; Olsbye, U.; De Wispelaere, K. Ethene Dimerization on Zeolite-Hosted Ni Ions: Reversible Mobilization of the Active Site. ACS Catal. 2019, 9, 5645–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.; Saxena, A.; Gounder, R. Mechanistic insights into alkene chain growth reactions catalyzed by nickel active sites on ordered microporous and mesoporous supports. Catal. Sci. Technol. 2020, 10, 7101–7123. [Google Scholar] [CrossRef]
- Cossee, P. Ziegler-Natta catalysis I. Mechanism of polymerization of α-olefins with Ziegler-Natta catalysts. J. Catal. 1964, 3, 80–88. [Google Scholar] [CrossRef]
- Arlman, E.J.; Cossee, P. Ziegler-Natta catalysis III. Stereospecific polymerization of propene with the catalyst system TiCl3AlEt3. J. Catal. 1964, 3, 99–104. [Google Scholar] [CrossRef]
- Ehrmaier, A.; Liu, Y.; Peitz, S.; Jentys, A.; Chin, Y.H.C.; Sanchez-Sanchez, M.; Bermejo-Deval, R.; Lercher, J. Dimerization of Linear Butenes on Zeolite-Supported Ni2+. ACS Catal. 2019, 9, 315–324. [Google Scholar] [CrossRef]
- Brückner, A.; Bentrup, U.; Zanthoff, H.; Maschmeyer, D. The role of different Ni sites in supported nickel catalysts for butene dimerization under industry-like conditions. J. Catal. 2009, 266, 120–128. [Google Scholar] [CrossRef]
- Ng, F.T.T.; Creaser, D.C. Ethylene dimerization over modified nickel exchanged Y-zeolite. Appl. Catal. A Gen. 1994, 119, 327–339. [Google Scholar] [CrossRef]
- Seufitelli, G.V.S.; Park, J.J.W.; Tran, P.N.; Dichiara, A.; Resende, F.L.P.; Gustafson, R. Kinetics of ethylene oligomerization over Ni-H-Beta catalysts. J. Catal. 2021, 401, 40–53. [Google Scholar] [CrossRef]
- Escobar, M.A.; Trofymchuk, O.S.; Rodriguez, B.E.; Lopez-Lira, C.; Tapia, R.; Daniliuc, C.; Berke, H.; Nachtigall, F.M.; Santos, L.S.; Rojas, R.S. Lewis Acid Enhanced Ethene Dimerization and Alkene Isomerization-ESI-MS Identification of the Catalytically Active Pyridyldimethoxybenzimidazole Nickel(II) Hydride Species. ACS Catal. 2015, 5, 7338–7342. [Google Scholar] [CrossRef]
- Kuhn, P.; Sémeril, D.; Matt, D.; Chetcuti, M.J.; Lutz, P. Structure–reactivity relationships in SHOP-type complexes: Tunable catalysts for the oligomerisation and polymerisation of ethylene. J. Chem. Soc. Dalt. Trans. 2007, 5, 515–528. [Google Scholar] [CrossRef]
- Speiser, F.; Braunstein, P.; Saussine, L. Catalytic ethylene dimerization and oligomerization: Recent developments with nickel complexes containing P,N-chelating ligands. Acc. Chem. Res. 2005, 38, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Elev, I.V.; Shelimov, B.N.; Kazansky, V.B. The Role of Ni+ Ions in the activity of NiCaY Zeolite catalysts for Ethylene Dimerization. J. Catal. 1984, 89, 470–477. [Google Scholar] [CrossRef]
- Davydov, A.A.; Kantcheva, M.; Chepotko, M.L. FTIR spectroscopic study on nickel(II)-exchanged sulfated alumina: Nature of the active sites in the catalytic oligomerization of ethene. Catal. Lett. 2002, 83, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Cai, T. Studies of a new alkene oligomerization catalyst derived from nickel sulfate. Catal. Today 1999, 51, 153–160. [Google Scholar] [CrossRef]
- Agirrezabal-Telleria, I.; Iglesia, E. Stabilization of active, selective, and regenerable Ni-based dimerization catalysts by condensation of ethene withinordered mesopores. J. Catal. 2017, 352, 505–514. [Google Scholar] [CrossRef]
- Beucher, R.; Hulea, V.; Cammarano, C. Kinetic and mechanistic insights into Ni-AlKIT-6 catalyzed ethylene oligomerization. React. Chem. Eng. 2022, 7, 133–141. [Google Scholar] [CrossRef]
- Jin, F.; Yan, Y.; Wu, G. Ethylene oligomerization over H- and Ni-form aluminosilicate composite with ZSM-5 and MCM-41 structure: Effect of acidity strenght, nickel site, and porosity. Catal. Today 2020, 355, 148–161. [Google Scholar] [CrossRef]
- Chen, L.; Li, G.; Wang, Z.; Li, S.; Zhang, M.; Li, X. Ethylene oligomerization over nickel supported silica-alumina catalysts with high selectivity for C10+ products. Catalysts 2020, 10, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Koninckx, E.; Mendes, P.S.F.; Thybaut, J.W.; Broadbelt, L.J. Ethylene oligomerization on nickel catalysts on a solid acid support: From New mechanistic insights to tunable bifunctionality. Appl. Catal. A Gen. 2021, 624, 118296. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef] [Green Version]
- Vannice, M.A. Modeling Reactions on Uniform (Ideal) Surfaces. In Kinetics of Catalytic Reactions; Springer: Boston, MA, USA, 2005; pp. 141–207. [Google Scholar]
- Foo, K.Y.; Hameed, B.H. Insights into the modeling of adsorption isotherm systems. Chem. Eng. J. J. 2010, 156, 2–10. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Mayadevi, S. Sorption Isotherms of Methane, Ethane, Ethylene, and Carbon Dioxide on ALPO-5 and SAPO-5. Langmuir 1996, 12, 980–986. [Google Scholar] [CrossRef]
- Hayward, D.O.; Trapnell, B. Chemisorption; Butterworths: London, UK, 1964. [Google Scholar]
- Rideal, E.K. Surface Chemistry; Cambridge England University Press: Cambridge, UK, 1930. [Google Scholar]
- Zeldowitsch, J. Adsorption site energy distribution. Acta Phys. Chim. URSS 1934, 1.1, 961–973. [Google Scholar]
- Halsey, G.; Taylor, H.S. The Adsorption of Hydrogen on Tungsten Powders. J. Chem. Phys. 1947, 15, 624–630. [Google Scholar] [CrossRef]
- Halsey, G.D. The Role of Surface Heterogeneity in Adsorption. Adv. Cata. 1952, 4, 259–269. [Google Scholar]
- Burnham, K.P.; Anderson, D.R. Model Selection and Inference: A Practical Information-Theoretic Approach, 2nd ed.; Springer: New York, NY, USA, 2002; ISBN 0387953647. [Google Scholar]
- Lee, M.; Yoon, J.W.; Kim, Y.; Yoon, J.S.; Chae, H.J.; Han, Y.H.; Hwang, D.W. Ni/SIRAL-30 as a heterogeneous catalyst for ethylene oligomerization. Appl. Catal. A Gen. 2018, 562, 87–93. [Google Scholar] [CrossRef]
- Vázquez, M.I.; Corma, A.; Fornés, V. Characterization of NiO supported on zeolite Y, by pyridine adsorption. Zeolites 1986, 6, 271–274. [Google Scholar] [CrossRef]
- Baran, R.; Kamińska, I.I.; Śrębowata, A.; Dzwigaj, S. Selective hydrodechlorination of 1,2-dichloroethane on NiSiBEA zeolite catalyst: Influence of the preparation procedure on a high dispersion of Ni centers. Microporous Mesoporous Mater. 2013, 169, 120–127. [Google Scholar] [CrossRef]
- Domnick, R.; Held, G.; Witte, P.; Steinrück, H.P. The transition from oxygen chemisorption to oxidation of ultra-thin Ni layers on Cu(111). J. Chem. Phys. 2001, 115, 1902–1908. [Google Scholar] [CrossRef]
- Scherrer, P.; Debye, P. Werk Übergeordnetes Werk. Nachr. Ges. Wiss. Göttingen Math. physik. Klasse 1918, 2, 101–120. [Google Scholar]
- Espinoza, R.L.; Nicolaides, C.P.; Korf, C.J.; Snel, R. Catalytic oligomerization of ethene over nickel-exchanged amorphous silica-alumina; Effect of the nickel concentration. Appl. Catal. 1987, 31, 259–266. [Google Scholar] [CrossRef]
- Sadek, R.; Chalupka, K.A.; Mierczynski, P.; Maniukiewicz, W.; Rynkowski, J.; Gurgul, J.; Lasoń-Rydel, M.; Casale, S.; Brouri, D.; Dzwigaj, S. The catalytic performance of Ni-Co/Beta zeolite catalysts in Fischer-Tropsch synthesis. Catalysts 2020, 10, 112. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
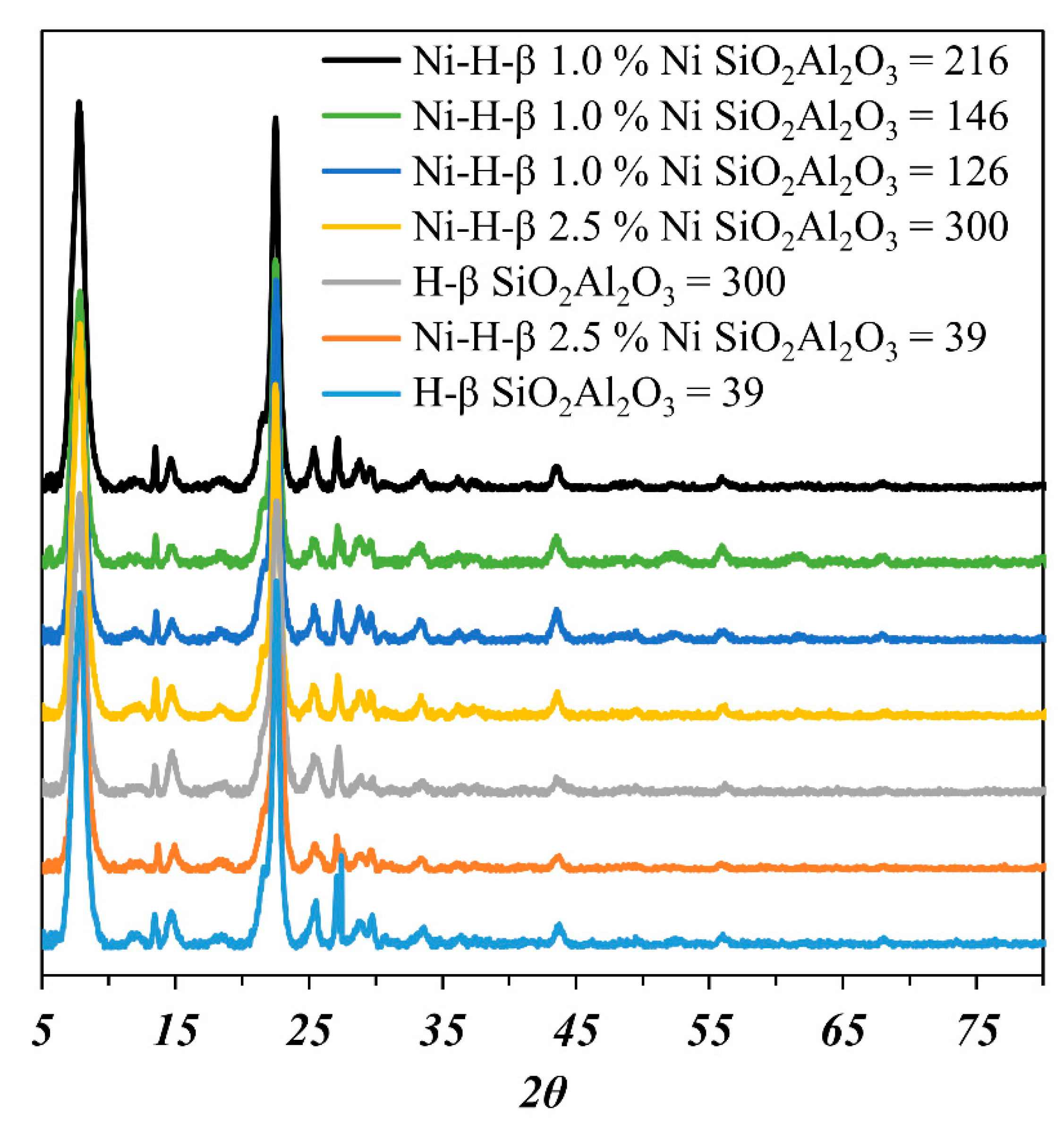{kind=link}
| Model Description | Mechanism 1 | Number of Molecules | Number of Sites | Adsorption Reaction | Langmuir Isotherm 2 |
|---|---|---|---|---|---|
| Dissociation over one site | A | 1 | 1 | ||
| Simple adsorption over one site | B | 1 | 1 | ||
| Simple adsorption over two sites | - | 1 | 2 | ||
| Dissociation over two sites | 1 | 2 | |||
| Association over one site | C | 2 | 1 |
| Dissociation or Simple Adsorption over One Site | Simple Adsorption over Two Sites | Dissociation over Two Sites | Association over One Site | |
|---|---|---|---|---|
| Mechanism | A | B | B | C |
| AIC | −95 | −102 | −124 | −78 |
| AICc | −94 | −101 | −123 | −77 |
| Δ | 29 | 22 | 0 | 46 |
| Probabilities | ||||
| Dissociation or Simple adsorption over one site | Simple adsorption over two sites | Dissociation over two sites | Association over one site | |
| Mechanism | A | B | B | C |
| weight (w) | 0.0% | 0.0% | 100.0% | 0.0% |
| Nickel Loading (wt. %) | LAS (µmol/g) | ΔLAS (µmol/g) | BAS (µmol/g) | |ΔLAS-BAS| (µmol/g) | BET (m2/g) 1 | Relative Grain Size of NiO Crystals (nm) | Conversion (%) 2 | |
|---|---|---|---|---|---|---|---|---|
| (Ni2+) | (H+) | |||||||
| 0.0 | - | 150 | 0 | 150 | - | - | - | - |
| 1.0 | 1.2 | 207 | 57 | 121 | 64 | 631 | 11 | 2.0 |
| 1.5 | 1.2 | 253 | 103 | 70 | 33 | 619 | - | 2.4 |
| 2.5 | 1.4 | 249 | 99 | 84 | 15 | 575 | - | 4.7 |
| 4.1 | 1.3 | 213 | 63 | 54 | 9 | 543 | 13 | 8.1 |
| 6.0 | 1.1 | 208 | 58 | 71 | 13 | 552 | 10 | 5.5 |
| s.d. 3 | - | - | - | - | - | - | - | 0.2 |
| Selectivity (wt. %) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| SiO2/Al2O3 | Nickel Loading (wt. %) | LAS (µmol/g) | ΔLAS (µmol/g) | BAS (µmol/g) | |ΔLAS-BAS| (µmol/g) | Conversion (%) 1 | C4 | C6 | C8 |
| 126 | 1.0 | 82 | 63 | 56 | 7 | 1.1 | 46.2 | 7.1 | 10.2 |
| 146 | 1.0 | 100 | 90 | 68 | 22 | 0.7 | 41.9 | 7.7 | 11.6 |
| 216 | 1.0 | 59 | 41 | 47 | 6 | 0.5 | 40.7 | 7.1 | 9.4 |
| 25 | 2.5 | 249 | 100 | 84 | 16 | 3.8 | 39.9 | 28.9 | 17.8 |
| 39 | 2.5 | 210 | 158 | 96 | 62 | 2.7 | 42.5 | 28.3 | 19.3 |
| 300 | 2.5 | 58 | 29 | 58 | 29 | 0.5 | 45.2 | 21.8 | 19.8 |
| s.d. 2 | - | - | - | - | - | 0.2 | 6.3 | 3.9 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seufitelli, G.V.S.; Park, J.J.W.; Tran, P.N.; Dichiara, A.; Resende, F.L.P.; Gustafson, R. The Role of Nickel and Brønsted Sites on Ethylene Oligomerization with Ni-H-Beta Catalysts. Catalysts 2022, 12, 565. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050565
Seufitelli GVS, Park JJW, Tran PN, Dichiara A, Resende FLP, Gustafson R. The Role of Nickel and Brønsted Sites on Ethylene Oligomerization with Ni-H-Beta Catalysts. Catalysts. 2022; 12(5):565. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050565
Chicago/Turabian StyleSeufitelli, Gabriel V. S., Jason J. W. Park, Phuong N. Tran, Anthony Dichiara, Fernando L. P. Resende, and Rick Gustafson. 2022. "The Role of Nickel and Brønsted Sites on Ethylene Oligomerization with Ni-H-Beta Catalysts" Catalysts 12, no. 5: 565. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050565