
Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction



1
Department of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia
2
Department of Chemistry, DIT University, Uttarakhand 248009, India
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(7), 699; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070699
Submission received: 26 May 2022
/
Revised: 19 June 2022
/
Accepted: 22 June 2022
/
Published: 25 June 2022
(This article belongs to the Special Issue Kinetics and Mechanism of Catalytic Reactions—Integrity of Experiment and Theory)
Abstract
:The methyl (•CH3) + 3O2 radical is an important reaction in both atmospheric and combustion processes. We investigated potential energy surfaces for the effect of CO2 and H2O molecules on a •CH3+ O2 system. The mechanism for three reaction systems, i.e., for •CH3 + 3O2, •CH3 + 3O2 (+CO2) and •CH3 + 3O2 (+H2O), were explored using ab initio/DFT methods [CCSD(T)//M062X/6-311++G(3df,3pd)] in combination with a Rice−Ramsperger−Kassel−Marcus (RRKM)/master-equation (ME) simulation between a temperature range of 500 to 1500 K and a pressure range of 0.0001 to 10 atm. When a CO2 and H2O molecule is introduced in a •CH3 + 3O2 reaction, the reactive complexes, intermediates, transition states and post complexes become thermodynamically more favorable. The calculated rate constant for the •CH3 + 3O2 (3 × 10−15 cm3 molecule−1 s−1 at 1000 K) is in good agreement with the previously reported experimentally measured values (~1 × 10−15 cm3 molecule−1 s−1 at 1000 K). The rate constant for the effect of CO2 (3 × 10−16 cm3 molecule−1 s−1 at 1000 K) and H2O (2 × 10−17 cm3 molecule−1 s−1 at 1000 K) is at least one–two-order magnitude smaller than the free reaction (3 × 10−15 cm3 molecule−1 s−1 at 1000 K). The effect of CO2 and H2O on •CH3 + 3O2 shows non-RRKM behavior, however, the effect on •CH3 + 3O2 shows RRKM behavior. Our results also demonstrate that a single CO2 and H2O molecule has the potential to accelerate a gas-phase reaction at temperature higher than >1300 K and slow the reaction at a lower temperature. The result is unique and observed for the first time.
1. Introduction
Methyl (•CH3) radical is the most stable intermediate species in many combustion-reaction processes [1,2]. In the Earth’s atmosphere, •CH3 radical is formed by the oxidation of methane by several tropospheric oxidizing agents such as OH, NO3 radicals and the Cl atom. The kinetics of a •CH3 + O2 reaction have been a key topic in combustion chemistry for the last five decades [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. The reaction mechanism behind •CH3 oxidation is certainly an important part of the kinetic modeling of volatile organic compounds (VOCs). Several research groups have been proposed for the oxidation mechanism for the •CH3 + O2 reaction, as given below [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17];
•CH3 + O2 → CH2O + •OH (R1)
•CH3 + O2 → HCO• + H2O (R2)
•CH3 + O2 → •CH3O + O (R3)
In the first reaction (R1), •CH3 radical primarily reacts with the O2 radical lead in the formation of formaldehyde (CH2O) and the hydroxyl radical (OH), suggested as major products [4,8,9,11]. In the second reaction (R2), the formation of the formyl radical (HCO•) and H2O (R2) were suggested as minor products via a hydrogen-transfer reaction followed by O-O breaking [8,9]. •CH3 can also be oxidized to form an oxygen-centered methoxy radical (•CH3O) and an O atom via O-O breaking (R3) [11]. Many experimental and theoretical studies on the •CH3···3O2 reaction system have been performed under atmospheric and combustion conditions [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. Several research groups have used various experimental techniques, such as flash photolysis, gas chromatography, and molecular modulation spectrometry, to monitor the decay of reactants or products. By using a shock tube, the rate constants of reactions for R1 and R2 have been measured between 1000 and 3000 K [3,4,5,6,7,8,9,10,11,12]. The proposed rate constants show large discrepancies between the groups. Although vigorous experimental measurements have been carried out on the •CH3 + 3O2 reaction, surprisingly few theoretical calculations are available in the literature [8,9,10,11]. Zhu et al. [11] performed CCSD(T)/6-3111G(3df,2p)//B3LYP/6-311G(d,p) in combination with the RRKM/master equation (ME), to compute the potential energy-surface and rate constants. Their theoretically reported rate constants for R1–R3 reactions were in good agreement with the experimentally measured values. Zhu et al. [11] also suggested that reaction R3 is insignificant in their calculations due to the high energy barrier. Very recently, Zhang et al. [8] re-visited the chemical kinetics mechanism for the •CH3 + 3O2 reaction CCSD(T)-F12/cc-pVQZ-F12//QCISD/6-311++G(2df,2p), with RRKM/ME simulations. They reported rate constants in the temperature range from 300 to 2500 K and pressure from 0.01 to 100 atm. Their theoretically calculated rate constants were in good agreement with the recently measured values, within a factor of 2 at the temperature range of 1300–1500 K. They also used the kinetic parameters of the •CH3 + 3O2 reaction in the methane-ignition modeling and found good agreement with the experimental measurement. However, in their calculations, a significant difference was observed at a higher temperature for reactions R2 and R3. Therefore, the estimation of rate constants should not be based on extrapolation from the experimental measurements over a limited temperature range. In this study, we have re-investigated the chemical kinetic mechanism for the formation of the most important reaction (R1), using a high-level quantum chemical method with an advanced statistical-rate theory under combustion conditions.
It is well-known that CO2 is the most important greenhouse gas that is released into the atmosphere from fossil-fuel burning, deforestation and natural processes, such as respiration and volcanic eruptions [18]. Over the last few decades, CO2 emissions have been increasing on a global scale, leading to global warming and acid rain [18,19]. Many efforts have been made to reduce CO2 emissions by using carbon-capture technology and sequestration methods [18,19]. CO2 can reach a supercritical state and is used as a diluent in many combustion chambers [18]. It is well-known that CO2 molecules present in the atmosphere can affect many important reactions [9,17]. To understand the significance of CO2 in the two drastically different environments, it is necessary to understand the chemical kinetics mechanism for the effect of CO2 on the •CH3 + 3O2 reaction. Although several studies on the oxidation reaction of the •CH3 + 3O2 reaction are available, to the best of our knowledge only a few studies [9,17] theoretically investigated the catalytic effect of carbon dioxide (CO2) on the •CH3 + 3O2 and HO + CO reaction systems [9,17]. Masunov et al. [9] have investigated the mechanism for the •CH3 + 3O2 reaction with and the without the presence of CO2, using M11/6-311G** + GD3. They have found that the formation of a van der Waals (vdW) complex stabilized the transition states (TSs), which lowered the barrier heights. Based on thermodynamics data, they proposed that the effect of CO2 is significant under combustion conditions. They also suggested that the chemical kinetic analysis for the effect of CO2 on CH3 + O2 reaction is underway. Therefore, in this study, we have re-investigated the effect of CO2 on the •CH3 + 3O2 reaction using a similar level of theory in combination with an advanced statistical-rate theory, to understand the chemical kinetic behavior for the effect of CO2 on •CH3 + 3O2.
Over the last few years, numerous investigations have been made into the catalytic effect of a single H2O molecule on many atmospheric and combustion reaction systems, such as CH3OH+OH, HCHO + OH, C2H4 + OH, CH3CHO + OH, CH3C(=O), CH3C(=O)CH3 + OH and CH2NH + OH as well as CH3O + O2, OH + CH4, CH2OH + O2 and H2O2 + OH reactions [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. All of these studies indicate that a single H2O molecule makes complexes, transition states and post complexes thermodynamically more stable than a water free-reaction, due to the formation of more hydrogen-bonded species. The above studies suggested that the reaction-rate constant decreases due to the presence of a high water concentration. In our recent works, we proposed the catalytic effect of H2O molecule on the •CH2OH + O2 reaction system and suggested that the catalytic role of water is insignificant under atmospheric and combustion conditions [26]. The calculated rate constant for the •CH2OH + 3O2 reaction at room temperature (298 K) is in good agreement with the previously measured values at the same level of theories. We suggested that when H2O molecule is added to the reaction, the formation of CH2O and HO2 is dominated under higher temperature conditions.
There are no theoretical chemical kinetic details for the catalytic effect of the CO2 and H2O molecules on the •CH3 + 3O2 reaction in the gas phase. In this work, for the first time, we have investigated the rate constants for the effect of CO2 and H2O molecules on the most important atmospheric and combustion prototype reactions, i.e., •CH3 + 3O2. The temperature and pressure-dependent rate constants are computed in the temperature range between 500 and 1500 K and 0.0001 to 10 atm, using the RRKM/ME simulation for all three reaction systems. To assess the accuracy of our results, we have compared the energies and rate constants for the •CH3 + 3O2 and •CH3 + 3O2 + (CO2) reaction systems with the available literature values. We hope this study is potentially important to design and analyze the possible forms of nonpetroleum-based fuel or fuel components in combustion engines.
2. Computational Methodology
2.1. Electronic-Structure Calculations
All the quantum chemical calculations were carried out with the Gaussian 09 suite of programs [37]. All the species involved in the •CH3 + O2, •CH3 + 3O2 (+CO2) and •CH3 + O2 (+H2O) reactions were optimized using the hybrid-density functional theory, i.e., M062X [designated as [M0] [38]] with Pople basis set 6-311++G(3df,3pd) [designated as [p] (see Table S1)]. We have used a reasonably high polarized and diffuse function of the Pople basis set 6-311++G(3df,3pd), which is reasonably good for the system studies here. We have used 6-311++G(3df,3pd) for a similar system in our earlier calculations [26,27,28], which has been used by many other researchers for the rate constants’ calculation. To add the correction from the van der Waal interaction on M0-p, the Grimme empirical dispersion correction” GD3”, was used. The vibrational frequency of each species was calculated to estimate the zero-point corrections (ZPE) and the vibrational partition functions and density of the states. All the optimized structures have real vibrational frequency except the transition states, which have one imaginary mode (see the Table S2). The vibrational mode, which corresponds to the Hindered Rotor (HR), was treated as HR approximation in the densum-input file. To further improve the accuracy of energy, the single-point energy calculations were performed at CCSD(T)/6-311++G(3df,3pd) [designated CC-p] level [28,29,30,31,32,33]. The RCCSD(T) method was used for closed-shell species, such as CH2O and H2O, CO2, and for all open-shell species the UCCSD(T) method was used. The combination of CC-p and M0-p generally gives the result that is accurate of ~1 kcal mol−1, which is ~20% error in the computed rate constant at 1000 K. Several research groups have already used M0-p with CC-p to calculate the energies and rate constants. The results reported by them were in good agreement with the experimentally measured values [28,30]. The entrance channels for the addition reactions have no intrinsic energy barriers. For a better description of the wave function “GUESS=MIX” keyword was used for the addition reactions. The “GUESS=MIX” option mixes the HOMO and LUMO orbitals to break spatial symmetry. The “GUESS=MIX” option was only used for geometry optimization.
2.2. Chemical Kinetics Calculations
For pressure-dependent reactions, the energy-dependent specific unimolecular rate constant k(E) was calculated using RRKM statistical rate theory given by [39] Equation (1):
The details of each term in the Equation (1) are given in the Chemical Kinetics Section of Supporting Information and MultiWell user manual [39].
At each temperature and pressure, master equation simulations were initiated using the chemical-activation energy distribution, which is appropriate for recombination reactions [39]. The pressure-dependent total rate constants ) for •CH3 + O2, •CH3 + O2 (+CO2) and •CH3 + O2 (+H2O) were calculated using [39] Equation (2):
The MultiWell code was used to calculate the branching fraction (f) for each reaction and calculate the unimolecular rate constants (kuni). The pressure-dependent rate constants were simulated using estimated energy-transfer parameters for the complexes and intermediates. The tunnelling corrections were implemented based on asymmetric Eckart potential. The approximated temperature-dependent exponential down <ΔE>down = 200 ∗ (T/300)0.85 cm−1 [28,32] was used to calculate the rate constants. The details about the energy-grain parameter to calculate the rate constant are given in the chemical kinetic section of the Supporting Information.
The equilibrium constant (Keq) for the formation of the complexes was calculated using THERMO code as given in Equation (3):
The equilibrium constant values were tabulated in Supporting Information Table S3.
3. Results and Discussion
3.1. Reaction Pathways for •CH3 + 3O2
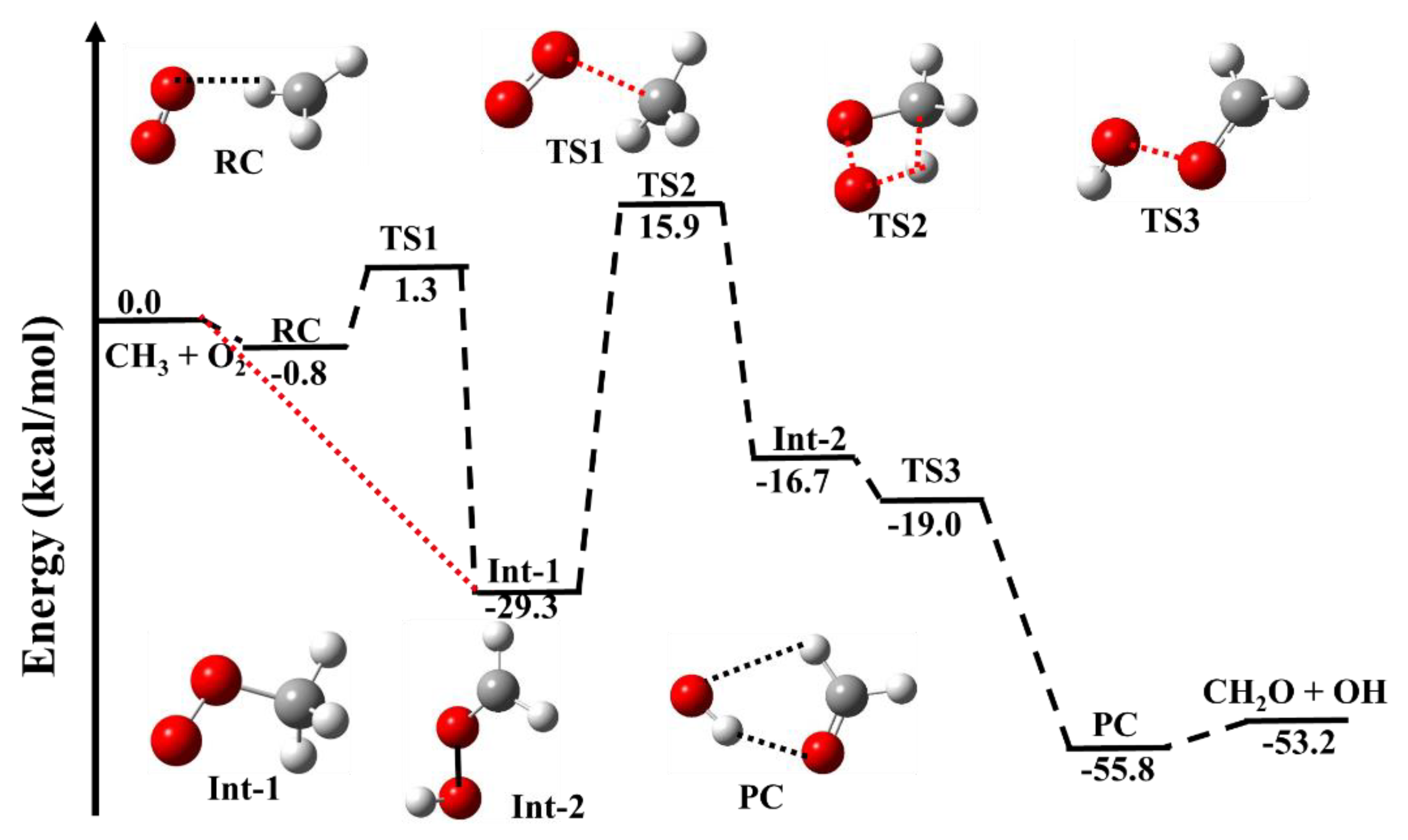
Figure 1 shows the zero−point corrected PES for the •CH3 + 3O2 reaction and the energies of reactants, pre−reactive complex (RC), intermediates (Ints), transition states (TSs,) post complexes (PCs) and products are tabulated in Table 1. The optimized geometries of reactants, RCs, Ints, PCs, TSs and products are given in Supporting Information Table S1. In the •CH3 radical, an unpaired electron resides on the carbon atom. When O2 molecule attacks the carbon atom of •CH3, the new C−O bond leads to the formation of an intermediate •OO-CH3 (Int-1). The pathway is shown using a red dashed line in Figure 1. Recently, Masunov et al. [9] proposed a mechanism for the formation of the reactive complex (RC) with ~1 kcal/mol energy, followed by a transition state TS1 leading to form Int-1. We have also optimized the RC and TS1 using the M0-p method, and our results are very similar to Masunov et al.’s values [9]. The computed stabilization energy of Int-1 (RO2) is −29.3 kcal mol−1, which is in very good agreement with the previously reported values [8,9]. In the RO2 (Int-1), the adduct can further rearrange to form QOOH (Int-2) via 1–3 hydrogen shift TS2 with +15.9 kcal mol−1, with respect to the reactants. The barrier height for this pathway is ~45 kcal mol−1 (relative to Int-1), which is in very good agreement with the previous studies. As discussed in the earlier studies, this step is the rate-determining step of the reaction and is most favored in the high-temperature range. The stabilization energy of Int-2 is calculated to be −16.7 kcal mol−1, which can further dissociate via a barrierless-transition state TS3 (−19.0 kcal mol−1). The stabilization of this TS is due to the formation of a five-membered hydrogen-bonded complex (PC). The calculated energy for Int-3 is −55.8 kcal mol−1, which subsequently leads to the formation of OH and CH2O. The calculated energies for TS1, TS2, Int-2, TS3, PC and HO + CH2O are also in good agreement with the previously reported values [9,11]. It is important to mention how the H-bonding interaction affects the energies of the TS and complexes. In the PC, the hydrogen bond is formed between the H-atom of the OH radical and the O-atom of the CH2O, whereas Int-1 and Int-2 are non-hydrogen bonded moieties. Therefore, the PC is the most stable among all these species.
Our calculated reaction energies for •CH3 + 3O2 → CH2O + OH are also in good agreement (−53.7 kcal/mol) with the most recent release thermochemical value (−52.1 kcal/mol) [40,41,42]. The literature’s thermochemical values (ATcT) are a new paradigm of how to develop the most accurate thermochemical data given by Argonne National Lab, Lemont, Illinois, USA [40,41,42]. As discussed in the previous section, the most important channel in the combustion condition for the •CH3 + 3O2 reaction is leading to form the CH2O, so we focused only on the R1 channel in the current study, and the energies of other pathways are neglected. To avoid any repetition, we have restricted our discussion to only reaction R1, as related discussion to the R2 and R3 channels can be found in the literature [9,11].
3.1.1. Reaction Pathways for •CH3 + 3O2 (+CO2)
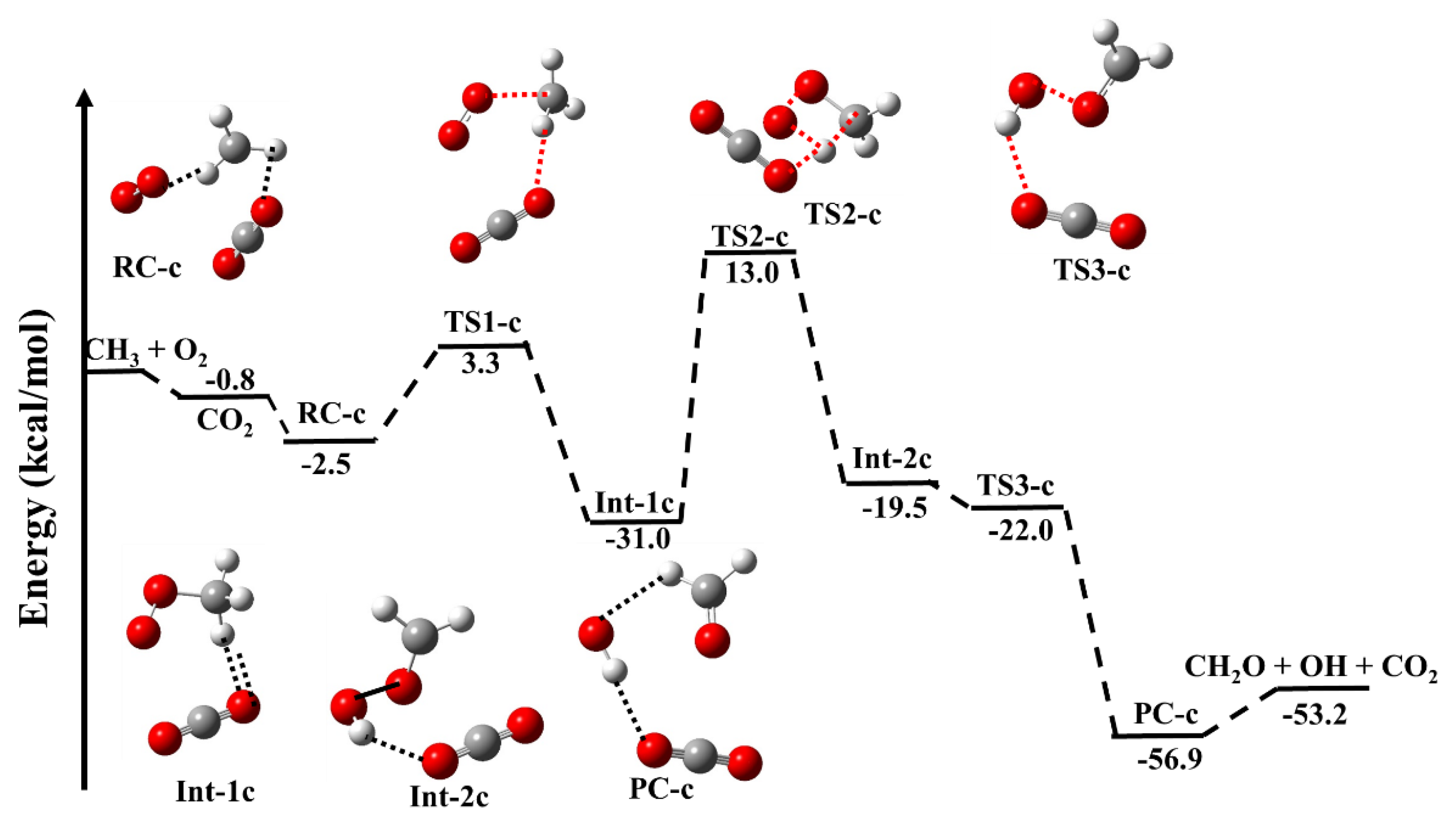
Figure 2 shows the PES for the CO2-catalyzed •CH3 + 3O2 reaction, and the energies are tabulated in Table 1. The optimized parameters of RC-c, Ints-c, TSs-c and PC-c are given in Table S1. As suggested in the previous work [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37], the possibilities of molecular collisions between O2, •CH3 and CO2 are very unlikely, therefore, we believe that •CH3 and O2 collide first to form an RC complex, and this complex collides with CO2 to form a three-molecular complex, i.e., RC-c. Based on our previous knowledge and the previous studies, we believe that the RC is more important (negative energy) than CH3···CO2 and •CO2···O2 (positive energy). Therefore, the complexes •CH3···CO2 and CO2···O2 are not considered in PES profile. As discussed in the earlier study [9], the CO2-catalyzed reaction proceeds via similar reaction channels as the uncatalyzed pathways, but the reaction mechanism becomes quite different, resulting in different energies than the uncatalyzed species. For more simplicity, we have used only the minimum energy structure in the current study. As shown in Figure 2, the binding energy of RC is −0.8 kcal mol−1 combined with CO2, leading to form a CO2 ···CH3···O2 (RC-c) with stabilization energy of −2.5 kcal mol−1. This value is in good agreement with previously reported values [9] (Table 1). The RC-c can further proceed by binding the diatomic oxygen to the methyl group, to form an intermediate CO2 ···CH3OO (Int-1c) via TS1-c. The calculated barrier height (~6 kcal/mol) is ~4 kcal/mol higher than the uncatalyzed reaction. Therefore, we can say that CO2 does not act as a catalyst for this pathway. It is also important to mention that CO2 acts as a catalyst for RC-c and Int-1c. This result is due to the formation of a hydrogen bond between the H-atom of CH3O2 and O-atom CO2 (2.6 Å). The Int-1c can further transfer to the Int-2c via an intramolecular hydrogen-atom transfer from the terminal oxygen of CH3OO via a four-membered cyclic-transition state with a high energy barrier. The barrier height (44 kcal/mol) for this pathway is 2 kcal/mol lower than the uncatalyzed pathway. In this pathway, the CO2 acting as a catalyst reduces the energy barrier by 2 kcal/mol. The intermediate Int-2c can further transfer to PC-c with a barrierless transition state TS3-c, which can dissociate into CH2O + OH + CO2.
The stabilization energy of Int-2c and PC-c are lower than the corresponding uncatalyzed structures (see Table 1). This can be also explained by the formation of hydrogen bonds. Int-2 (Figure 1) is a non-hydrogen-bonded structure, whereas the corresponding CO2-catalyzed analog Int-2c formed a strong hydrogen-bonded complex between the H-atom of OH and the O-atom of CO2 (2.1 Å). On the other hand, PC-c formed a seven-membered ring-like structure with two strong hydrogen bonds, between the terminal H-atom of CH2O and the O-atom of OH (2.5 Å) as well as the H-atom of OH and the O-atom of CO2 (2.0 Å), compared to the less stable five-membered ring-like structure with two hydrogen bonds in the uncatalyzed analog (PC). Similarly, TS3-c (−22.0 kcal mol−1) is more stable than the corresponding unanalyzed-transitions state (TS3), due to the hydrogen-bonding interaction. In general, the RC-c, TSs-c, Ints-c and PC-c are thermodynamically more favorable by 2 kcal/mol than the RC, TSs, Ints and PC species. The calculated energies of all the CO2-catalyzed structures are in good agreement with the recently reported values (see Table 1) [9].
3.1.2. Reaction Pathways for •CH3 + 3O2 (+H2O)
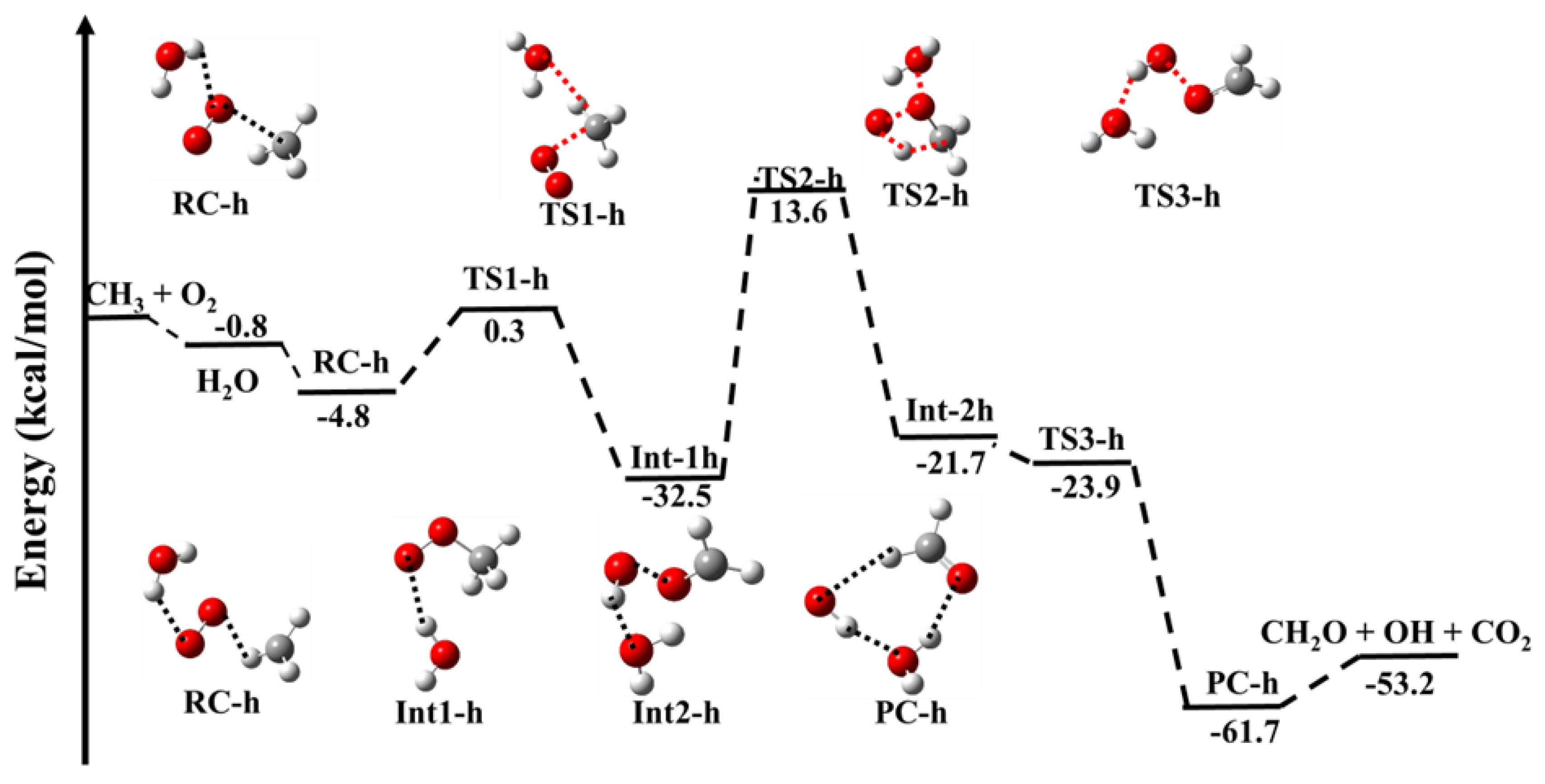
Figure 3 shows the zero-point corrected PES for the effect of H2O on the •CH3 + 3O2 reaction. The energies for effect of H2O on complex (RC-h), intermediates (Ints-h), TSs-h and PC-h are tabulated in Table 1. The geometrical parameters for all the species are tabulated in Supporting Information Tables S1 and S2. The effect of H2O on the •CH3 + 3O2 reaction proceeds via similar reaction channels, as discussed in the CO2-catalyzed reaction with different reaction energies. As mentioned earlier, the three molecular collisions are very uncommon. Therefore, we believe the RC formed first, which collides with H2O to form H2O···CH3···O2 (RC-h) with a stabilization energy of −4.8 kcal mol−1. This value is nearly 2 kcal/mol lower than the CO2-catalyzed reaction and 4 kcal/mol lower than the uncatalyzed reaction. This is due to the formation of a higher number of H-bonds in the RC-h. The RC-h can further proceed via TS1-h to form H2O···CH3OO (Int1-h). It is important to notice that the effect of H2O lowers the stabilization energy of Int-1h by 3 kcal mol−1, however, the effect of CO2 lowered the energy by ~1.5 kcal mol−1 compared to the uncatalyzed pathway. The result suggests the catalytic behavior of H2O is more significant than CO2. This is due to the formation of a strong hydrogen bond between the H atom of CH3O2 and the O atom of H2O (2.0 Å), in comparison with the formation of a weaker hydrogen bond (2.6 Å) in the case of CO2. Similarly, the barrier height of the H2O-catalyzed transition state (TS1-h) is 3 kcal mol−1 more stable than the CO2-catalyzed transition state (TS1-c); this difference can be seen by the formation of two hydrogen bonds in TS1-h between the O-atom of O2 and the H-atom of H2O (2.2 Å) as well as the O-atom of H2O and the H-atom of CH3 (2.6 Å), whereas, in the case of TS1-c, only one hydrogen bond is formed between the O-atom of CO2 and the H-atom of CH3 (3.0 Å). A hydrogen atom is transferred intramolecularly from Int-1c via a four-membered cyclic-transition state, TS2-h, with high energy. The barrier height of TS2-h is ~46 kcal/mol is higher than the barrier height of a similar transition state of a CO2-catalyzed species, i.e., TS2-c (~44 kcal/mol). It has been observed that the stabilization energies of H2O-catalyzed species, i.e., Int2-h and PC-h, are lower than the corresponding CO2-assisted reaction. These results are due to the formation of a stronger H-bond in the water environment than a weak interaction in the CO2 environment. In general, the reaction species in the presence of a H2O is thermodynamically more favorable by 3–5 kcal/mol than the uncatalyzed reaction and ~2 kcal/mol more favorable than the CO2-assisted reaction. The result of the water effect on the CH3 + O2 reaction is unique and has not been published in the literature.
3.2. Rate Constants
3.2.1. •CH3 + 3O2 Reaction
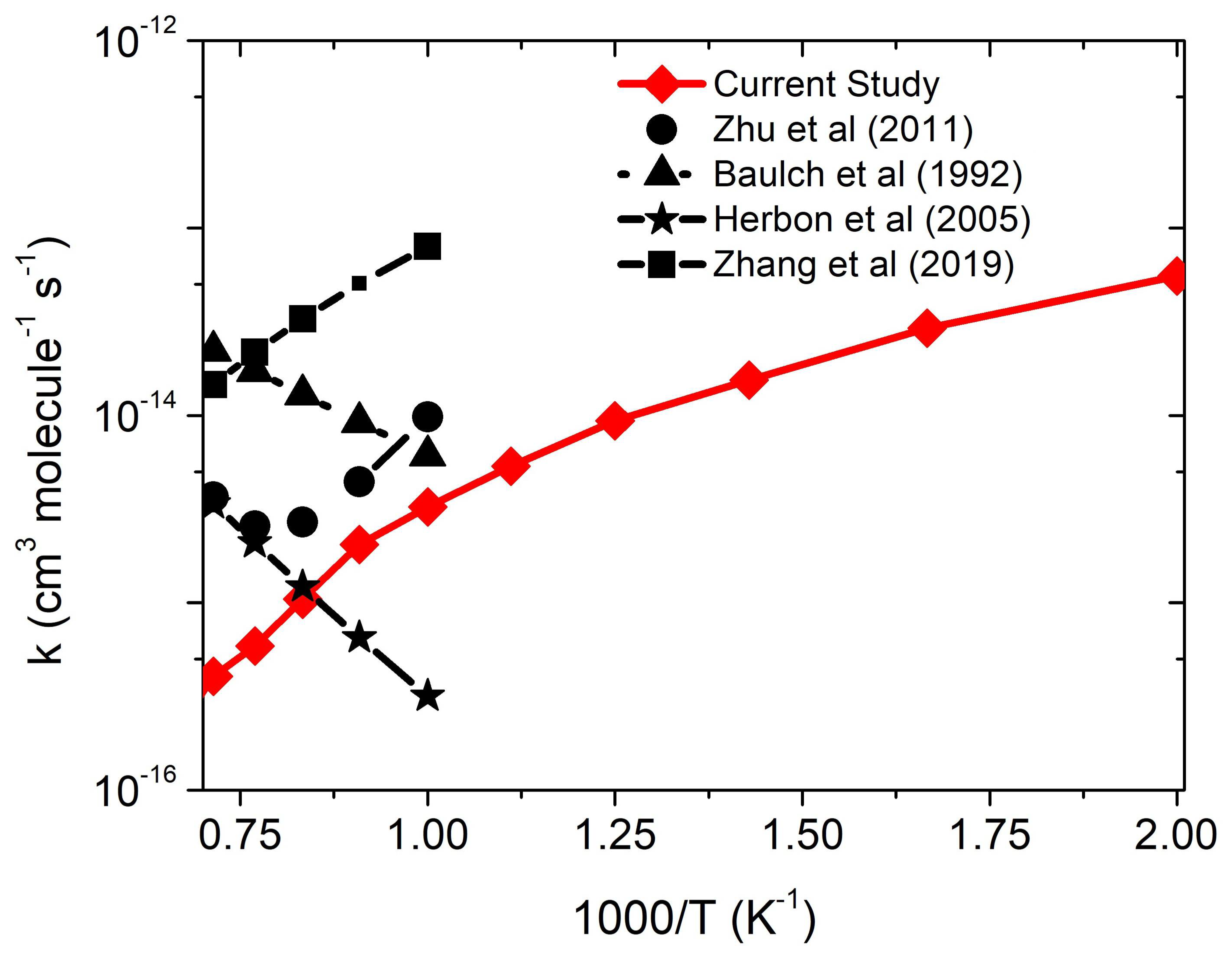
Figure 4 shows the rate constants for the •CH3 + 3O2 reaction obtained using the CC-p//M0-p method coupled with RRKM/ME simulations in the temperature range of 500–1500 K under a high pressure limit. The •O2-CH3 that is assumed to play important roles in the •CH3 + 3O2 reaction system is formed via the entrance channel, which has a well depth of ~30 kcal/mol. The rate constants for the formation of •O2-CH3 are pressure-dependent and negative-temperature dependent. This result is consistent with previously reported ones [8,11]. The rate constants are also calculated for the new reaction mechanism, i.e., R→RC→Int-1→Int-2→PC→CH2O + OH, as shown in Figure 1. In this case, we have observed that the reaction is independent of pressure, and the rate constant is at least two-order magnitude smaller than the R→Int-1→Int-2→PC→CH2O + OH channel. The proposed mechanism, based on Masunov et al. [9], is inconsistent with the previously reported rate constants [9]. Therefore, we believe that the mechanism via Int-1 is the most appropriate way to deal with the combustion-reaction system. Therefore, the formation of RC should be neglected from the •CH3 + 3O2 reaction channel. The formation of RC may not even be important in the atmospheric condition due to its short lifetime. The calculated rate constants are also compared with some of the previous studies, as shown in Figure 4. Our values are in good agreement with the value of Zhang et al. [8] at higher temperature. We have observed that the formation of CH2O+OH is almost negligible (<1300 K), which is also consistent with Zhang et al. [8] and Zhu et al. [11]. Due to the large number of data available for the •CH3 + 3O2 reaction, we restrict our discussion to only a few of the literature values to avoid repetition, plus the main focus of the current study is to compare the results of the catalytic effect of CO2 and H2O on the •CH3 + 3O2 reaction.
3.2.2. Effect of CO2 on •CH3 + 3O2 Reaction
The scheme for the formation of CH2O and OH from the •CH3 + O2 reactions in the presence of a CO2 catalyst can be expressed as:
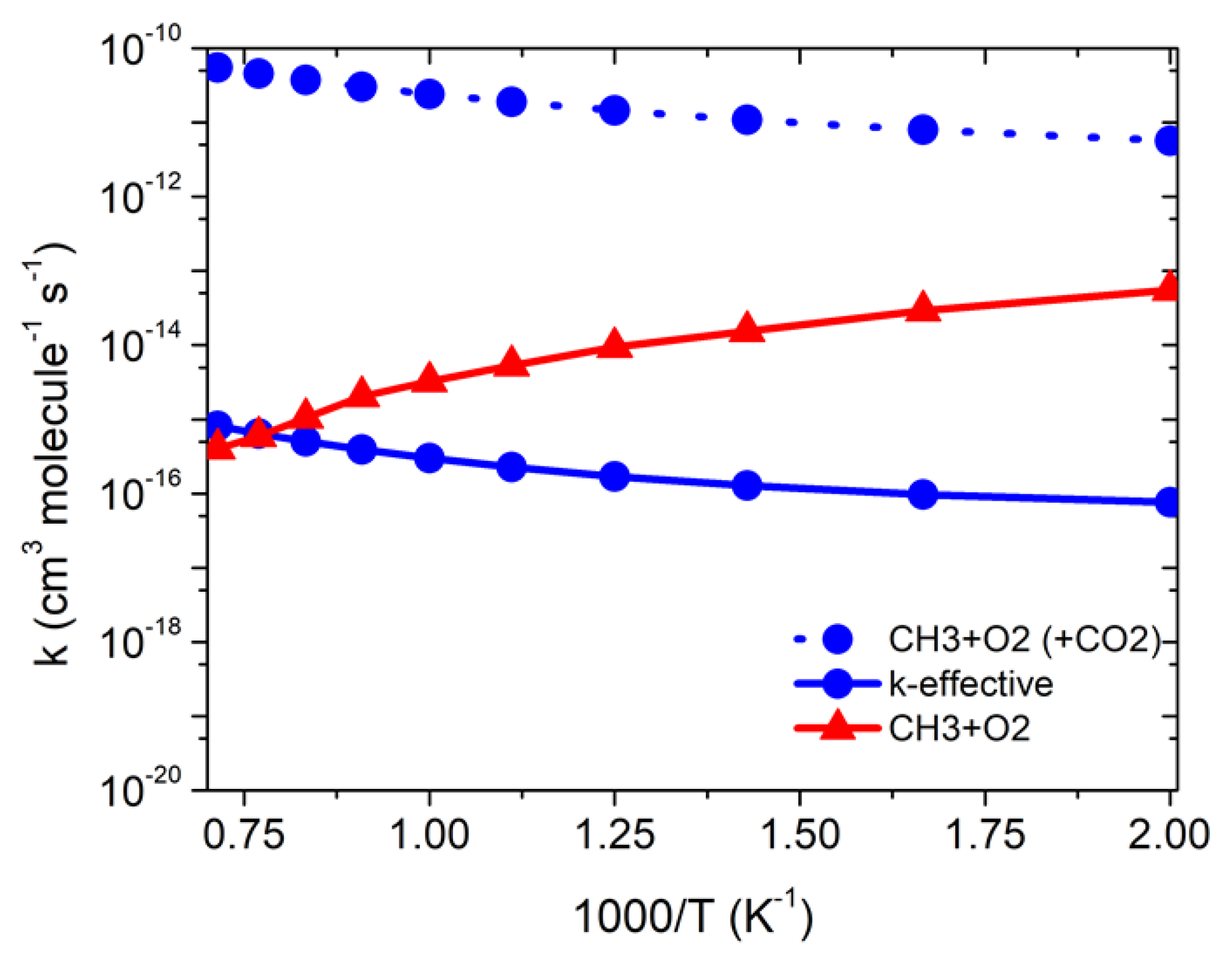
The high-pressure limit-rate constant was calculated using Equation (2), and the are the equilibrium constants of each reaction pathway involved in the •CH3 + O2 reaction. The rate constants for •CH3 + 3O2 (+CO2) in the temperature range of 500 to 1500 K are tabulated in Table 2 and shown in Figure 5. Our results show that the rate constants, without including the concentration of CO2, are higher than the •CH3 + 3O2 reaction in the temperature range of 500 to 1500 K. In general, the rate constants of the CO2-catalyzed reaction are higher than the •CH3 + 3O2 reaction in the entire temperature range (see Figure 5). As discussed in our previous studies [26,27,28,29,30,31,32] under pseudo-first-order conditions, the relative equilibrium concentrations strongly depend on the concentration of the excess CO2, and the correct equation to calculate the effective rate constant is The concentration of CO2 diluent was converted from the pressure and temperature using real-gas behavior, as tabulated in the NIST database [43], which is based on Masunov et al.’s study analysis [9]. Masunov et al. [9] suggested that the •CH3 + 3O2 reactions are catalyzed by CO2, based on the analysis of PES. They suggested that the details of the chemical kinetics mechanism and a comparison with the experimental verification and improvement of the chemical kinetic details are underway [9,44]. Due to the missing information of the rate constants for the effect of CO2 on the •CH3 + 3O2 reaction, we have investigated the effect of CO2 on •CH3 + 3O2, and the rate constants’ details are given in Table 2 and shown in Figure 5.
As expected, the effective rate constants are two-order magnitude smaller at temperature < 1000 K, with a one-order-magnitude difference between 1000–1200 K. The effective rate constants suggest that the catalytic effect may be pronounced at a high temperature > 1400 K, due to a high entropic factor. The new chemical kinetics results might help to understand and design the experimental measurement. Our chemical kinetics analysis also suggests that the catalytic effect of CO2 is dominated at higher temperature (>1400 K), which is consistent with the result of Masunov et al. [9], who suggested that the catalytic effect of CO2 may be important at >1000 K. It is interesting to mention that the formation CH2O is unfavorable in the RRKM/ME simulation, and the calculated rate constants are independent of the pressure, therefore, we can say that the effect of CO2 on the CH3 + O2 reaction shows a non-RRKM behavior, which can be treated using a simple canonical transition state method. However, the result reported in the current study in the high-pressure limit condition can be similar to a CTST method.
3.2.3. Effect of H2O on •CH3 + 3O2 Reaction
The formation of CH2O and OH from •CH3 + 3O2 reactions in the presence of H2O can be expressed as:
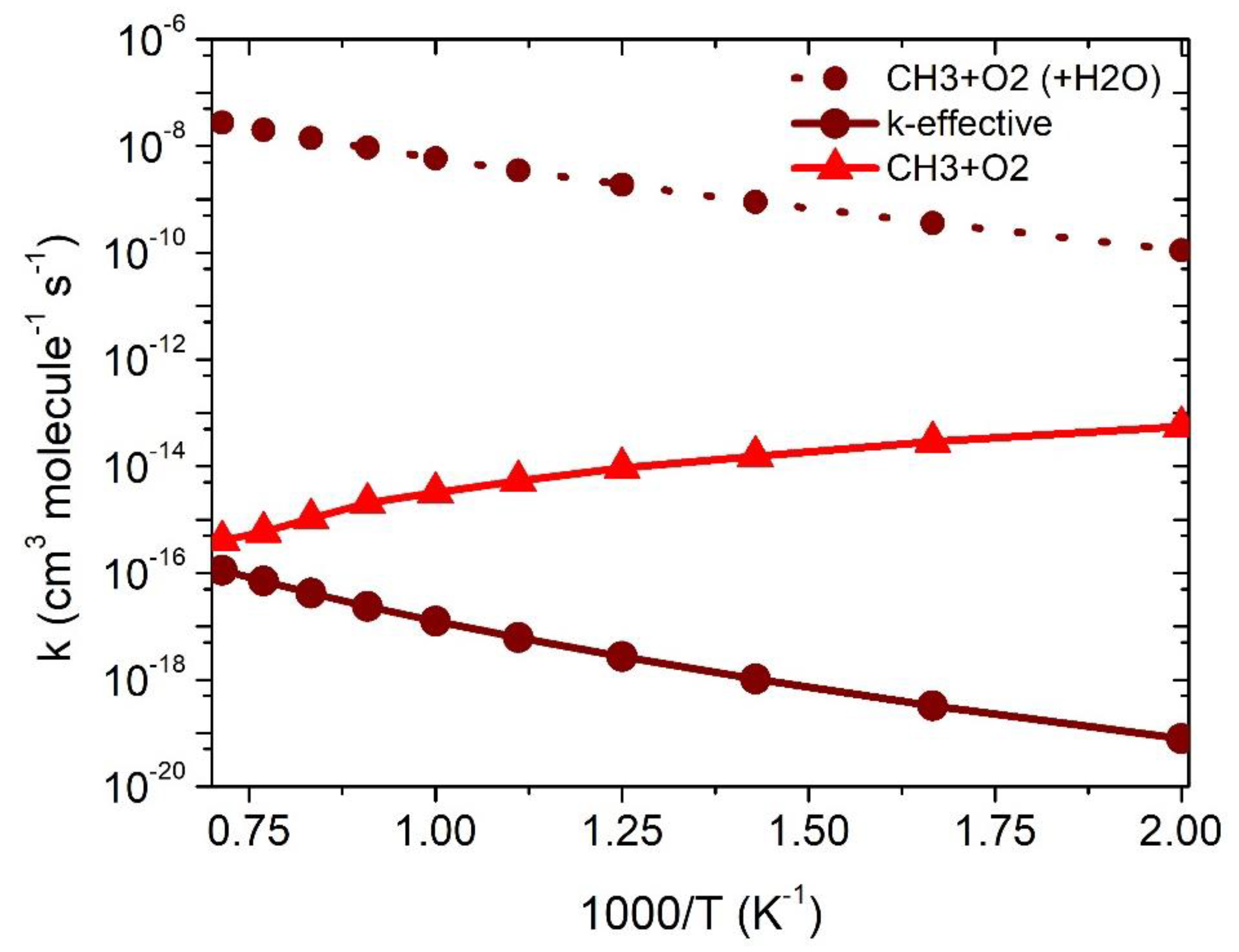
The bimolecular-rate constant can be calculated as given in Equation (2). The equilib rium constant and the constants of each reaction pathway involved in the •CH3 + O2 reaction were calculated and given in Supporting Information Table S3. Figure 6 shows the rate constants for •CH3 + 3O2 (+H2O), calculated in the temperature range of 500 to 1500 K. The rate constant for the RC + H2O reaction (6 × 10−9 cm3 molecule−1 s−1 at 1000 K) is ~six-order magnitude higher than the •CH3 + O2 reaction (3 × 10−15 cm3 molecule−1 s−1 at 1000 K. The rate constants for the effect of water are higher than the CH3 + O2 reaction in the entire temperature range of 500–1500 K (see Figure 6).
The total effective-rate constants were calculated using Where are the equilibrium constants of the •CH3 + O2 →RC reactions and [H2O] is the water concentration, as discussed in earlier studies [21,25]. The effective rate constant (7 × 10−17 cm3 molecule−1 s−1 at 1000 K) is ~two-order magnitude lower than the •CH3 + 3O2 reaction (~6 × 10−15 cm3 molecule−1 s−1 at 1000 K). Our result is also consistent with the previously reported value for a similar combustion-reaction system, i.e., •CH2OH + 3O2 [26]. The effective rate constant suggests that the catalytic effect may be pronounced at a high temperature >1500 K, and the catalytic effect of H2O may be important under a combustion condition >1500 K.
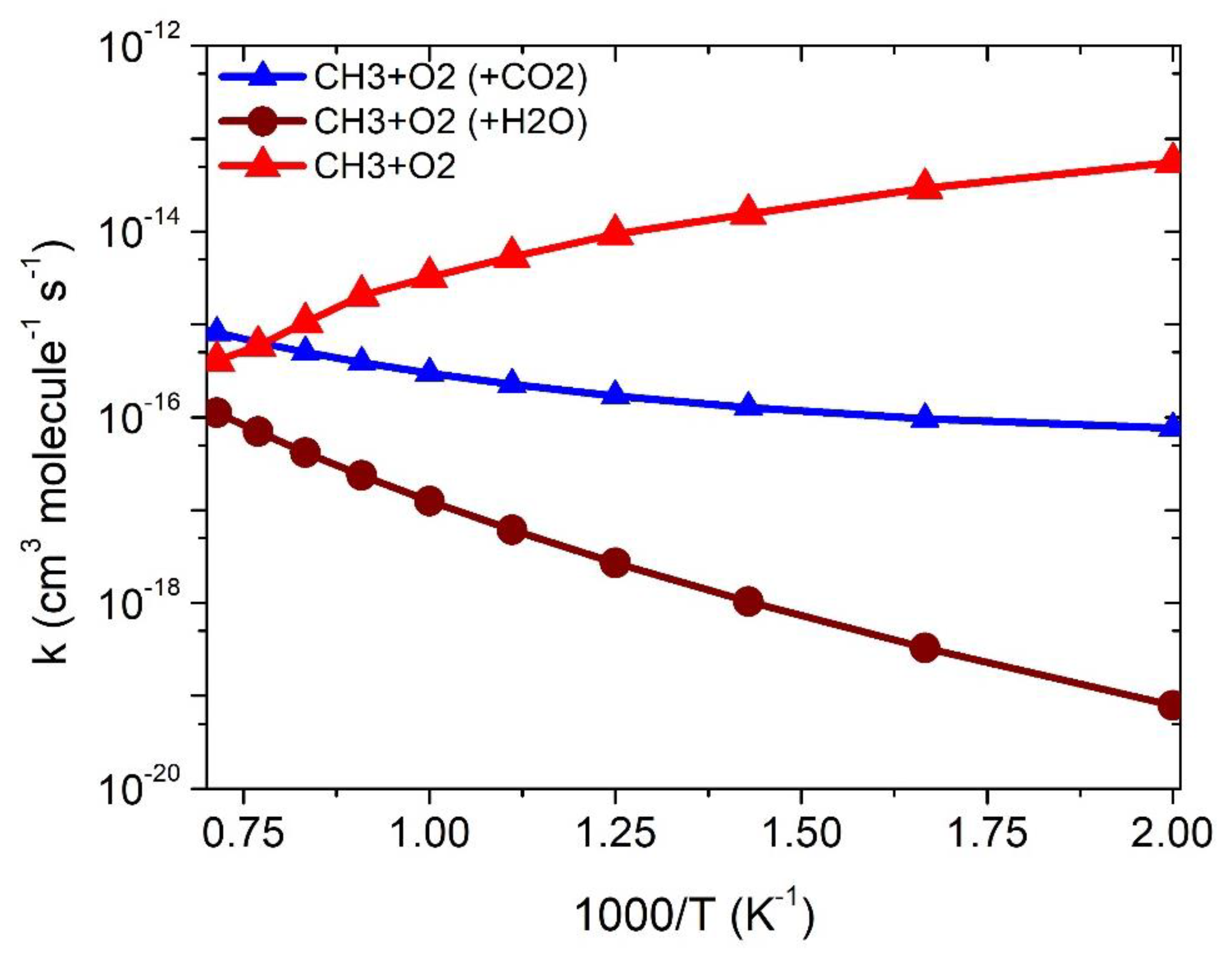
Figure 7 shows the comparison of the rate constants in all three reaction systems, and the rate constant values are given in Table 2. The rate constants for CH3 + O2 are negative-temperature-dependent, however, the rate constants for the effect of CO2 and H2O are positive-temperature-dependent. These results are due to fact that the TSs and RCs of CO2 and H2O have different values of energies, entropic factors and concentrations. In general, the effective rate constants for the CO2-assisted reactions are higher at >1400 K, while those for the H2O-assisted reactions are almost similar at >1500 K to the •CH3 + O2-reaction system (Figure 7). The rate constant at 1000 K is at least one–two-order magnitude smaller than •CH3 + O2. As a result, the effect of •CH3 + O2 catalyzed by CO2 and H2O is of only minor importance for the sink of •CH3 in a gas-phase-combustion reaction. Such results are interesting. We believe the present results provide insights into a better understanding of the gas-phase catalytic effect of CO2 and H2O molecules on the most important combustion-prototype molecule.
4. Conclusions
We have predicted the rate constants for the •CH3 + 3O2 reaction, and the effect of CO2 and H2O molecules on the •CH3 + 3O2 reaction using the CC-p/M06-p method, coupled with the RRKM/ME simulation, between a temperature range of 500 to 1500 K and a pressure range of 0.0001 to 10 atm. The results show that •CH3 + 3O2 leads to form CH2O and OH at a temperature > 1300 K, which is consistent with the previous studies. The calculated rate constants for •CH3 + 3O2 are in good agreement with the previously measured experimental values. When CO2 and H2O molecules are introduced to the •CH3 + 3O2 reaction, the reaction unfavored the formation of CH2O below 1300 K. The calculated rate constants for the effect of CO2 and H2O are independent of pressure and show positive-temperature dependence. The result suggests that the effect of CO2 and H2O shows a non-RRKM behavior, and the CH3 + O2 reaction shows an RRKM behavior. This result is unique, and it was observed for the first time. Our results also demonstrate that single CO2 and H2O molecules have the potential to accelerate a gas-phase reaction at a higher temperature > 1300 K and slow the reaction at a lower temperature.
Under the combustion condition, the kinetics of •CH3 + O2 (+CO2) are quite different from those of •CH3 + O2 (+H2O). This difference is possibly due to the reaction energies and concentrations of CO2 and H2O being different. These kinds of results are interesting, so experimental measurements are required to validate these findings. Based on current findings and previous findings, we can conclude that the main kinetic mechanism of •CH3 + 3O2 →CH2O + HO via •CH3OO (Int-1) and other mechanisms, such as the formation of RC via a TS1 to Int-1, is almost negligible. Such a weakly bound complex RC should not be included in the kinetic calculations under the combustion condition. In the case of the effect of H2O and CO2, the formations of RC-c and RC-h are important, and the catalytic effect has a negative effect under combustion conditions.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12070699/s1. The optimized geometries of all the species involved in •CH3 + 3O2, •CH3 + 3O2 (+CO2), and •CH3 + 3O2 (+H2O) are given in Table S1. The rot-vib parameter for all the species is given in Table S2, and the equilibrium constants are given in Table S3. The details of the chemical kinetic calculations are given in the Supporting Information.
Author Contributions
M.R.D. completed all the Gaussian calculations; M.A.A. and L.M.A.M. completed the chemical kinetic calculations; M.A.A. and M.R.D. prepared the draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia grant number [AN000532] and the APC was funded by Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia grant number [AN000532].
Data Availability Statement
All the quantum chemical data generated through this study are given in the Supporting Information.
Acknowledgments
Mohamad Akbar Ali (M.A.A.) gratefully acknowledges the Annual Funding track by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [Project No. AN000532].
Conflicts of Interest
The authors declare no competing interests.
References
- Srinivasan, N.K.; Su, M.-C.; Sutherland, J.W.; Michael, J.V. Reflected Shock Tube Studies of High-Temperature Rate Constants for OH + CH4 → CH3 + H2O and CH3 + NO2 → CH3O + NO. J. Phys. Chem. A 2005, 109, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Vaghjiani, G.L.; Ravishankara, A.R. New measurement of the rate coefficient for the reaction of OH with methane. Nature 1991, 350, 406–409. [Google Scholar] [CrossRef]
- Baulch, D.L.; Cobos, C.J.; Cox, R.A.; Esser, C.; Frank, P.; Just, T.; Kerr, J.A.; Pilling, M.J.; Troe, J.; Walker, R.W.; et al. Evaluated Kinetic Data for Combustion Modelling. J. Phys. Chem. Ref. Data 1992, 21, 411. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, N.K.; Su, M.-C.; Michael, J.V. CH3 + O2 → H2CO + OH Revisited. J. Phys. Chem. A 2007, 111, 11589–11591. [Google Scholar] [CrossRef] [PubMed]
- Herbon, J.T.; Hanson, R.K.; Bowman, C.T.; Golden, D.M. The reaction of CH3 + O2: Experimental determination of the rate coefficients for the product channels at high temperatures. Proc. Combust. Inst. 2005, 30, 955–963. [Google Scholar] [CrossRef]
- Hsu, D.S.Y.; Shaub, W.M.; Creamer, T.; Gutman, D.; Lin, M.C. Kinetic modeling of co-production from the reaction of CH3 with O2 in shock waves. Ber. Bunsenges. Phys. Chem. 1983, 87, 909–991. [Google Scholar] [CrossRef]
- Cobos, C.J.; Hippler, H.; Luther, K.; Ravishankara, A.R.; Troe, J. High-pressure falloffcurves and specific rate constants for the reaction CH3 + O2 ↔ CH3O2 ↔ CH3 + O. J. Phys. Chem. 1985, 89, 4332–4338. [Google Scholar] [CrossRef]
- Zhang, F.; Huang, C.; Xie, B.; Wu, X. Revisiting the chemical kinetics of CH3 + O2 and its impact on methane ignition. Combust. Flame 2019, 200, 125–134. [Google Scholar] [CrossRef]
- Masunov, A.E.; Wait, E.; Vasu, S.S. Quantum Chemical Study of CH3 + O2 Combustion Reaction System: Catalytic Effects of Additional CO2 Molecule. J. Phys. Chem. A 2017, 121, 5681–5689. [Google Scholar] [CrossRef]
- Pilling, M.J.; Smith, M.J.C. A laser flash photolysis study of the reaction CH3 + O2 → CH3O2 at 298 K. J. Phys. Chem. 1985, 89, 4713–4720. [Google Scholar] [CrossRef]
- Zhu, R.; Hsu, C.-C.; Lin, M.C. Ab initio study of the CH3 + O2 reaction: Kinetics, mechanism and product branching probabilities. J. Chem. Phys. 2011, 115, 195. [Google Scholar] [CrossRef]
- Tan, Y.; Douglas, M.A.; Thambimuthu, K.V. CO2 Capture Using Oxygen Enhanced Combustion Strategies for Natural Gas Power Plants. Fuel 2002, 81, 1007–1016. [Google Scholar] [CrossRef]
- Kumar, A.; Mallick, S.; Mishra, B.M.; Kumar, P. Effect of ammonia and formic acid on the CH3O + O2 reaction: A quantum chemical investigation. Phys. Chem. Chem. Phys. 2020, 22, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Lin, C.Y.; Wang, H.T.; Lin, M.C. A shock tube study of the oxidation of the methyl radical. AIP Conf. Proc. 1990, 208, 450–455. [Google Scholar]
- Zhang, T.; Zhang, Y.; Wen, M.; Tang, Z.; Long, B.; Yu, X.; Zhao, C.; Wang, W. Effects of water, ammonia and formic acid on HO2 + Cl reactions under atmospheric conditions: Competition between a stepwise route and one elementary step. RSC Adv. 2019, 9, 21544–21556. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, N.K.; Su, M.C.; Sutherland, J.W.; Michael, J.V. Reflected shock tube studies of high-temperature rate constants for CH3 + O2, H2CO + O2, and OH + O2. J. Phys. Chem. A 2005, 109, 7902–7914. [Google Scholar] [CrossRef]
- Masunov, A.E.; Elizabeth, E.; Wait, S.; Vasu, S. Catalytic Effect of Carbon Dioxide on Reaction OH + CO → H + CO2, in Supercritical Environment: Master Equation Study. J. Phys. Chem. A 2018, 122, 6355–6359. [Google Scholar] [CrossRef]
- Pryor, O.M.; Barak, S.; Koroglu, B.; Ninnemann, E.; Vasu, S.S. Measurements and Interpretation of Shock Tube Ignition Delay Times in Highly CO2 Diluted Mixtures Using Multiple Diagnostics. Combust. Flame 2017, 180, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Koroglu, B.; Pryor, O.; Lopez, J.; Nash, L.; Vasu, S.S. Shock Tube Ignition Delay Times and Methane Time-Histories Measurements During Excess CO2 Diluted Oxy-Methane Combustion. Combust. Flame 2016, 164, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Du, B.; Qin, Z. Catalytic effect of Water, Formic Acid, or Sulfuric Acid on the Reaction of Formaldehyde with OH Radicals. J. Phys. Chem. A 2014, 118, 4797–4807. [Google Scholar] [CrossRef]
- Jara-Toro, R.A.; Hernández, F.J.; Taccone, R.A.; Lane, S.I.; Pino, G.A. Water catalysis of the reaction between methanol and OH at 294 K and the atmospheric implications. Angew. Chem. Int. Ed. 2017, 56, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, L.G.; Varga, Z.; Xu, X.; Ren, W.; Truhlar, D.G. Water catalysis of the Reaction of Methanol with OH Radical in the Atmosphere is Negligible. Angew. Chem. Int. Ed. 2020, 59, 10826–10830. [Google Scholar] [CrossRef] [PubMed]
- Vöhringer-Martinez, E.; Hansmann, B.; Hernandez, H.; Francisco, J.S.; Troe, J.; Abel, B. Water Catalysis of a Radical-Molecule Gas-Phase Reaction. Science 2007, 315, 496–501. [Google Scholar] [CrossRef]
- Thomsen, D.; Kurtén, T.; Jørgensen, S.; Wallington, T.; Baggesen, S.; Aalling, C.; Kjaergaard, H. On the possible catalysis by single water molecules of gas-phase hydrogen abstraction reactions by OH radicals. Phys. Chem. Chem. Phys 2012, 14, 12992–12999. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.; Lin, J.J.; Takahashi, K.; Tomas, A.; Yu, L.; Kajii, Y.; Batut, S.; Schoemaecker, C.; Fittschen, C. Water vapor does not catalyze the reaction between methanol and OH Radicals. Angew. Chem. Int. Ed. 2019, 58, 5013–5017. [Google Scholar] [CrossRef]
- Dash, M.R.; Ali, M.A. Effect of a single water molecule on CH2OH + 3O2 reaction under atmospheric and combustion conditions. Phys. Chem. Chem. Phys. 2022, 24, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Balaganesh, M.; Lin, K.C. Catalytic effect of a single water molecule on the OH + CH2NH reaction. Phys. Chem. Chem. Phys 2018, 20, 4297–4307. [Google Scholar]
- Ali, M.A.; Ali, M.A. Effect of Water and Formic Acid on ·OH + CH4 Reaction: An Ab Initio/DFT Study. Catalysts 2022, 12, 133. [Google Scholar] [CrossRef]
- Inaba, S. Catalytic Role of H2O Molecules in Oxidation of CH3OH in Water. Catalysts 2018, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Balaganesh, M.; Jang, S. Can a single water molecule catalyze the OH+CH2CH2 and OH+CH2O reactions? Atmos. Environ. 2019, 207, 82–92. [Google Scholar] [CrossRef]
- Ali, M.A.; Balaganesh, M.; Al-Odail, F.A.; Lin, K.C. Effect of ammonia and water molecule on OH + CH3OH reaction under tropospheric condition. Sci. Rep. 2021, 11, 12185. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A. Computational studies on the gas phase reaction of methylenimine (CH2NH) with water molecules. Sci. Rep. 2020, 10, 10995. [Google Scholar] [CrossRef] [PubMed]
- Buszek, R.J.; Torrent-Sucarrat, M.; Anglada, J.M.; Francisco, J.S. Effects of a Single Water Molecule on the OH + H2O2 Reaction. J. Phys. Chem. A 2012, 116, 5821–5829. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R. On the Possible Catalytic Role of a Single Water Molecule in the Acetone + OH Gas Phase reaction: A Theoretical Pseudo-Second-order Kinetics Study. Theor. Chem. Acc. 2011, 129, 209–217. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Reyes, L.; Vivier-Bunge, A. Can a Single Water Molecule Really Catalyze the Acetaldehyde OH Reaction in Tropospheric Conditions? J. Phys. Chem. Lett. 2010, 1, 3112–3115. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, W.; Li, C.; Du, Y.; Lu, J. Catalytic effect of a single water molecule on the atmospheric reaction of HO2 + OH: Fact or fiction? A mechanistic and kinetic study. RSC Adv. 2013, 3, 7381–7391. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, T., Jr.; Vreven, K.N.; Kudin, J.C.; Burant, J.M.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Barker, J.R.; Nguyen, T.L.; Stanton, J.F.; Aieta, C.; Ceotto, M.; Gabas, F.; Kumar, T.J.D.; Li, C.G.L.; Lohr, L.L.; Maranzana, A.; et al. MultiWell-<Version>-Software Suite; Barker, J.R., Ed.; University of Michigan: Ann Arbor, MI, USA, 2016; Available online: https://multiwell.engin.umich (accessed on 24 May 2022).
- Ruscic, B.; Pinzon, R.E.; Morton, M.L.; von Laszevski, G.; Bittner, S.J.; Nijsure, S.G.; Amin, K.A.; Minkoff, M.; Wagner, A.F. Introduction to Active Thermochemical Tables: Several “Key” Enthalpies of Formation Revisited. J. Phys. Chem. 2004, 108, 9979–9997. [Google Scholar] [CrossRef]
- Ruscic, B.; Pinzon, R.E.; Von Laszewski, G.; Kodeboyina, D.; Burcat, A.; Leahy, D.; Montoy, D.; Wagner, A.F. Active Thermochemical Tables: Thermochemistry for the 21st Century. J. Phys. Conf. Ser. 2005, 16, 561–570. [Google Scholar] [CrossRef]
- Ruscic, B.; Bross, D.H. Active Thermochemical Tables (ATcT) Enthalpies of Formation Values Based on ver. 1.112r of the Thermochemical Network. 2021. Available online: ATcT.anl.gov (accessed on 24 May 2022).
- Lemmon, E.W.; McLinden, M.O.; Friend, D.G. Thermophysical Properties of Fluid Systems. In NIST Chemistry Webbook; NIST Standard Reference Database Number 69. Available online: https://atct.anl.gov/ (accessed on 24 May 2022).
- Metcalfe, W.K.; Burke, M.; Ahmed, S.S.; Curran, H.J. A Hierarchical and Comparative Kinetic Modeling Study of C1−C2 Hydrocarbon and Oxygenated Fuels. Int. J. Chem Kinet. 2013, 45, 638–675. [Google Scholar] [CrossRef]
Figure 1.
Zero−point corrected potential energy surface for •CH3 + 3O2 reaction calculated using CC−p//M0-p level. The red−dashed-line pathway is based on earlier studies.
Figure 1.
Zero−point corrected potential energy surface for •CH3 + 3O2 reaction calculated using CC−p//M0-p level. The red−dashed-line pathway is based on earlier studies.

Figure 2.
Zero−point corrected potential energy surface for •CH3+O2 (+CO2) reaction calculated using CC−p//M0−p level.
Figure 2.
Zero−point corrected potential energy surface for •CH3+O2 (+CO2) reaction calculated using CC−p//M0−p level.

Figure 3.
Zero−point corrected potential energy surface for .CH3 + O2 (+H2O) calculated using CC−p//M0−p level.
Figure 3.
Zero−point corrected potential energy surface for .CH3 + O2 (+H2O) calculated using CC−p//M0−p level.


Figure 5.
Rate constants for the effect of CO2 on •CH3 + 3O2 reaction in the temperature range of 500–1500 K.
Figure 5.
Rate constants for the effect of CO2 on •CH3 + 3O2 reaction in the temperature range of 500–1500 K.

Figure 6.
Rate constants for the effect of H2O on •CH3 +3O2 reaction in the temperature range of 500–1500 K.
Figure 6.
Rate constants for the effect of H2O on •CH3 +3O2 reaction in the temperature range of 500–1500 K.

Figure 7.
Comparison of the rate constants between •CH3 + 3O2, •CH3 + O2 (+CO2) and •CH3 + O2 (+H2O) reactions.
Figure 7.
Comparison of the rate constants between •CH3 + 3O2, •CH3 + O2 (+CO2) and •CH3 + O2 (+H2O) reactions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Enthalpies of reaction (∆Hrxn (0 K) in kcal mol−1 for •CH3 + O2 reaction.
| •CH3 + O2 → | This Work | Literature |
| OO···CH3 (RC) | −0.8 | −0.4 b |
| OO···CH3 (TS1) | 1.3 | 1.6 b |
| CH3OO (Int-1) | −29.3 | −30.4 a, −33.8 b |
| HOO···CH2 (TS2) | +15.9 | 15.0 a, 13.7 b |
| HO···CH2O (Int-2) | −16.7 | −18.5 a, −20.6 b |
| HO···CH2O (TS3) | −19.0 | −21.3 b |
| HO···CH2O (PC) | −55.8 | −58.2 b |
| CH2O + OH | −53.2 | −53.7, −52.10 c |
| •CH3 + O2 + CO2 → | This Work | Literature |
| CH3···O2 + CO2 | −0.8 | −0.4 b |
| CO2 ···CH3OO (RC-c) | −2.5 | −2.6 b |
| CO2 ···CH3OO (TS1-c) | 3.3 | −0.3 b |
| CO2 ···CH3OO (Int-1c) | −31.0 | −37.0 b |
| CO2 ···CH3OO (TS2-c) | 13.0 | 8.7 b |
| CO2 ···CH3OO (Int-2c) | −19.5 | −25.0 b |
| CO2 ···CH3OO (TS-3c) | −22.0 | −25.4 b |
| HO···CH2O···CO2 (PC-c) | −56.9 | −61.6 b |
| •CH3 + O2 + H2O → | This Work | |
| CH3···O2 + H2O | −0.8 | |
| H2O···CH3OO (RC-h) | −4.8 | |
| H2O···CH3OO (TS1-h) | 0.3 | |
| H2O ···CH3OO (Int-1h) | −32.5 | |
| H2O·····CH3OO (TS2-h) | 13.6 | |
| H2O···CH3OO (Int-2h) | −21.7 | |
| H2O·····CH3OO (TS3-h) | 13.6 | |
| HO···CH2O···H2O (PC-h) | −61.7 |
Table 2.
Calculated rate constants for •CH3 + O2, •CH3 + O2 +(CO2) and •CH3 + O2 (+H2O) in the temperature range of 500 to 1500 K.
Table 2.
Calculated rate constants for •CH3 + O2, •CH3 + O2 +(CO2) and •CH3 + O2 (+H2O) in the temperature range of 500 to 1500 K.
| Temp | kCH3+O2 | ||
|---|---|---|---|
| 500 | 5.5 × 10−14 | 7.6 × 10−17 | 1.7 × 10−19 |
| 600 | 2.9 × 10−14 | 9.6 × 10−17 | 6.4 × 10−19 |
| 700 | 1.5 × 10−14 | 1.3 × 10−16 | 1.8 × 10−18 |
| 800 | 9.4 × 10−15 | 1.7 × 10−16 | 4.5 × 10−18 |
| 900 | 5.3 × 10−15 | 2.3 × 10−16 | 9.5 × 10−18 |
| 1000 | 3.2 × 10−15 | 3.0 × 10−16 | 1.8 × 10−17 |
| 1100 | 2.1 × 10−15 | 3.9 × 10−16 | 3.4 × 10−17 |
| 1200 | 1.0 × 10−15 | 5.04 × 10−16 | 5.8 × 10−17 |
| 1300 | 5.9 × 10−16 | 6.41 × 10−16 | 9.5 × 10−17 |
| 1400 | 4.0 × 10−16 | 8.2 × 10−16 | 1.5 × 10−16 |
| 1500 | 2.1 × 10−16 | 1.02 × 10−15 | 2.3 × 10−16 |
| k = ATn exp(−B/T) | A = 2.8 × 1014 n = −9.1 B = 3516 | A = 2.5 × 10–33 n = 4.4 B = −1711 | A = 3.3 × 10–33 n = 5.4 B = 1338 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ali, M.A.; Dash, M.R.; Al Maieli, L.M. Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction. Catalysts 2022, 12, 699. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070699
AMA Style
Ali MA, Dash MR, Al Maieli LM. Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction. Catalysts. 2022; 12(7):699. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070699
Chicago/Turabian StyleAli, Mohamad Akbar, Manas Ranjan Dash, and Latifah Mohammed Al Maieli. 2022. "Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction" Catalysts 12, no. 7: 699. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070699
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.