
Cyano/Hydroxyl Groups Co-Functionalized g-C3N4 for Photocatalytic NO Removal: A Synergistic Strategy towards Inhibition of Toxic Intermediate NO2
 , ,
, ,
Abstract
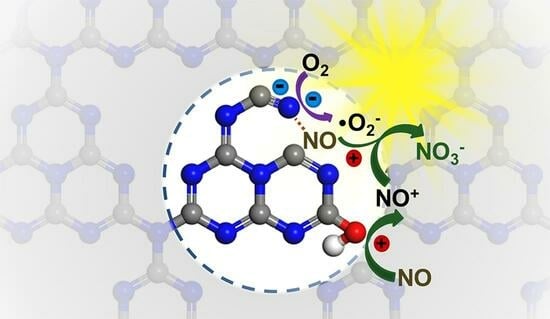
:
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. Chemical and Reagents
3.2. Preparation of the Photocatalysts
3.3. Characterization
3.4. Photocatalytic Performance Tests
3.5. Active Species Scavenging Experiments
3.6. Fluorophotometric Measurements of H2O2
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalysts | N/C | O/C |
|---|---|---|
| CN | 1.190 | 0.053 |
| DCN | 1.176 | 0.053 |
| DCNO-P | 1.173 | 0.063 |
| DCNO-R | 1.049 | 0.069 |

References
- Huang, Y.; Liang, Y.L.; Rao, Y.F.; Zhu, D.D.; Cao, J.-J.; Shen, Z.; Ho, W.K.; Lee, C.S. Environment-Friendly Carbon Quantum Dots/ZnFe2O4 Photocatalysts: Characterization, Biocompatibility, and Mechanisms for NO Removal. Environ. Sci. Technol. 2017, 51, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, L.B.; Patel, C.K. Nitric Oxide Air Pollution: Detection by Optoacoustic Spectroscopy. Science 1971, 173, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, S.H.; Schroder, M. Porous Metal-organic Frameworks as Emerging Sorbents for Clean Air. Nat. Rev. Chem. 2019, 3, 108–118. [Google Scholar] [CrossRef]
- Yu, J.J.; Jiang, Z.; Zhu, L.; Hao, Z.P.; Xu, Z.P. Adsorption/desorption Studies of NOx on Well-mixed Oxides Derived from Co-Mg/Al Hydrotalcite-like Compounds. J. Phys. Chem. B 2006, 110, 4291–4300. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Jirsak, T.; Liu, G.; Hrbek, J.; Dvorak, J.; Maiti, A. Chemistry of NO2 on Oxide Surfaces: Formation of NO3 on TiO2 (110) and NO2↔O Vacancy Interactions. J. Am. Chem. Soc. 2001, 123, 9597–9605. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Zhao, Z.W.; Sun, Y.J.; Zhang, Y.X.; Yan, S.; Wu, Z.B. An Advanced Semimetal-Organic Bi Spheres-g-C3N4 Nanohybrid with SPR-Enhanced Visible-Light Photocatalytic Performance for NO Purification. Environ. Sci. Technol. 2015, 49, 12432–12440. [Google Scholar] [CrossRef]
- Zhu, H.Z.; Nie, Z.G.; Hu, Y.F.; Wang, J.Y.; Bai, H.; Li, Y.; Guo, Q.J.; Wang, C.P. Experimental Study on Denitration Performance of Iron Complex-Based Absorption Solutions and Their Regeneration by Zn. Energy Fuels 2019, 33, 8998–9003. [Google Scholar] [CrossRef]
- Klose, W.; Rincon, S. Adsorption and Reaction of NO on Activated Carbon in the Presence of Oxygen and Water Vapour. Fuel 2007, 86, 203–209. [Google Scholar] [CrossRef]
- Lee, J.; Theis, J.R.; Kyriakidou, E.A. Vehicle Emissions Trapping Materials: Successes, Challenges, and the Path Forward. Appl. Catal. B-Environ. 2019, 243, 397–414. [Google Scholar] [CrossRef]
- Wang, X.Q.; Liu, Y.; Wu, Z.B. The Poisoning Mechanisms of Different Zinc Species on a Ceria-based NH3-SCR Catalyst and the Co-effects of Zinc and Gas-phase Sulfur/chlorine Species. J. Colloid. Interf. Sci. 2020, 566, 153–162. [Google Scholar] [CrossRef]
- Seneque, M.; Can, F.; Duprez, D.; Courtois, X. NOx Selective Catalytic Reduction (NOx-SCR) by Urea: Evidence of the Reactivity of HNCO, Including a Specific Reaction Pathway for NOx Reduction Involving NO + NO2. ACS Catal. 2016, 6, 4064–4067. [Google Scholar] [CrossRef]
- Ma, L.; Li, J.H.; Ke, R.; Fu, L.X. Catalytic Performance, Characterization, and Mechanism Study of Fe2(SO4)3/TiO2 Catalyst for Selective Catalytic Reduction of NOx by Ammonia. J. Phys. Chem. C 2011, 115, 7603–7612. [Google Scholar] [CrossRef]
- Fu, S.L.; Song, Q.; Yao, Q. Mechanism Study on the Adsorption and Reactions of NH3, NO, and O2 on the CaO Surface in the SNCR deNOx Process. Chem. Eng. J. 2016, 285, 137–143. [Google Scholar] [CrossRef]
- Zhou, M.; Dong, G.H.; Yu, F.K.; Huang, Y. The Deep Oxidation of NO Was Realized by Sr multi-site Doped g-C3N4 Via Photocatalytic Method. Appl. Catal. B-Environ. 2019, 256, 117825. [Google Scholar] [CrossRef]
- Ai, Z.H.; Ho, W.K.; Lee, S.C.; Zhang, L.Z. Efficient Photocatalytic Removal of NO in Indoor Air with Hierarchical Bismuth Oxybromide Nanoplate Microspheres under Visible Light. Environ. Sci. Technol. 2009, 43, 4143–4150. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.H.; Jacobs, D.L.; Zang, L.; Wang, C.Y. Carbon Vacancy Regulated Photoreduction of NO to N2 Over Ultrathin g-C3N4 Nanosheets. Appl. Catal. B-Environ. 2017, 218, 515–524. [Google Scholar] [CrossRef]
- Wang, X.C.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A Metal-free Polymeric Photocatalyst for Hydrogen Production from Water under Visible Light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef]
- Choi, C.H.; Lin, L.; Gim, S.; Lee, S.; Kim, H.; Wang, X.; Choi, W. Polymeric Carbon Nitride with Localized Aluminum Coordination Sites as a Durable and Efficient Photocatalyst for Visible Light Utilization. ACS Catal. 2018, 8, 4241–4256. [Google Scholar] [CrossRef]
- Dong, F.; Zhao, Z.W.; Xiong, T.; Ni, Z.L.; Zhang, W.; Sun, Y.J.; Ho, W.K. In Situ Construction of g-C3N4/g-C3N4 Metal-Free Heterojunction for Enhanced Visible-Light Photocatalysis. ACS Appl. Mater. Interfaces 2013, 5, 11392–11401. [Google Scholar] [CrossRef]
- Moon, G.H.; Fujitsuka, M.; Kim, S.; Majima, T.; Wang, X.; Choi, W. Eco-Friendly Photochemical Production of H2O2 through O2 Reduction over Carbon Nitride Frameworks Incorporated with Multiple Heteroelements. ACS Catal. 2017, 7, 2886–2895. [Google Scholar] [CrossRef]
- Dong, G.H.; Zhao, L.L.; Wu, X.X.; Zhu, M.S.; Wang, F. Photocatalysis Removing of NO Based on Modified Carbon Nitride: The Effect of Celestite Mineral Particles. Appl. Catal. B-Environ. 2019, 245, 459–468. [Google Scholar] [CrossRef]
- Luo, J.M.; Dong, G.H.; Zhu, Y.Q.; Yang, Z.; Wang, C.Y. Switching of Semiconducting Behavior from n-type to p-type Induced High Photocatalytic NO Removal Activity in g-C3N4. Appl. Catal. B-Environ. 2017, 214, 46–56. [Google Scholar] [CrossRef]
- Dong, X.A.; Li, J.Y.; Xing, Q.; Zhou, Y.; Huang, H.W.; Dong, F. The Activation of Reactants and Intermediates Promotes the Selective Photocatalytic NO Conversion on Electron-localized Sr-intercalated g-C3N4. Appl. Catal. B-Environ. 2018, 232, 69–76. [Google Scholar] [CrossRef]
- Huang, H.W.; Xiao, K.; Tian, N.; Dong, F.; Zhang, T.; Du, X.; Zhang, Y.H. Template-free Precursor-surface-etching Route to Porous, Thin g-C3N4 Nanosheets for Enhancing Photocatalytic Reduction and Oxidation Activity. J. Mater. Chem. A 2017, 5, 17452–17463. [Google Scholar] [CrossRef]
- Xiong, T.; Cen, W.L.; Zhang, Y.X.; Dong, F. Bridging the g-C3N4 Interlayers for Enhanced Photocatalysis. ACS Catal. 2016, 6, 2462–2472. [Google Scholar] [CrossRef]
- Ran, M.X.; Li, J.R.; Cui, W.; Li, Y.H.; Li, P.D.; Dong, F. Efficient and Stable Photocatalytic NO Removal on C Self-doped g-C3N4: Electronic Structure and Reaction Mechanism. Catal. Sci. Technol. 2018, 8, 3387–3394. [Google Scholar] [CrossRef]
- Liao, J.Z.; Cui, W.; Li, J.Y.; Sheng, J.P.; Wang, Z.M.; Dong, X.; Chen, P.; Jiang, G.; Wang, Z.; Dong, F. Nitrogen Defect Structure and NO Plus Intermediate Promoted Photocatalytic NO Removal on H2 Treated g-C3N4. Chem. Eng. J. 2020, 379, 122282. [Google Scholar] [CrossRef]
- Wang, J.D.; Cui, W.; Chen, R.M.; He, Y.; Yuan, C.; Sheng, J.; Li, J.; Zhang, Y.; Dong, F.; Sun, Y. OH/Na Co-functionalized Carbon Nitride: Directional Charge Transfer and Enhanced Photocatalytic Oxidation Ability. Catal. Sci. Technol. 2020, 10, 529–535. [Google Scholar] [CrossRef]
- Liu, G.M.; Huang, Y.; Lv, H.Q.; Wang, H.; Zeng, Y.; Yuan, M.; Meng, Q.G.; Wang, C.Y. Confining Single-atom Pd on g-C3N4 with Carbon Vacancies Towards Enhanced Photocatalytic NO Conversion. Appl. Catal. B-Environ. 2021, 284, 119683. [Google Scholar] [CrossRef]
- Fan, J.H.; Qin, H.H.; Jiang, S.M. Mn-doped g-C3N4 Composite to Activate Peroxymonosulfate for Acetaminophen Degradation: The Role of Superoxide Anion and Singlet Oxygen. Chem. Eng. J. 2019, 359, 723–732. [Google Scholar] [CrossRef]
- Geng, Y.X.; Chen, D.Y.; Li, N.J.; Xu, Q.F.; Li, H.; He, J.; Lu, J.M. Z-Scheme 2D/2D Alpha-Fe2O3/g-C3N4 Heterojunction for Photocatalytic Oxidation of Nitric Oxide. Appl. Catal. B-Environ. 2021, 280, 119409. [Google Scholar] [CrossRef]
- Yang, J.; Liang, Y.J.; Li, K.; Yang, G.; Wang, K.; Xu, R.; Xie, X.J. One-step Synthesis of Novel K+ and Cyano Groups Decorated Triazine-/heptazine-based g-C3N4 Tubular Homojunctions for Boosting Photocatalytic H2 Evolution. Appl. Catal. B-Environ. 2020, 262, 118252. [Google Scholar] [CrossRef]
- Shi, Y.H.; Li, J.S.; Wan, D.J.; Huang, J.H.; Liu, Y.D. Peroxymonosulfate-enhanced Photocatalysis by Carbonyl-modified g-C3N4 for Effective Degradation of the Tetracycline Hydrochloride. Sci. Total Environ. 2020, 749, 142313. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.Y.; Wang, L.J.; Zhang, Y.J.; Wen, C.F.; Wang, X.L.; Yang, H.G. Carboxyl Functionalized Graphite Carbon Nitride for Remarkably Enhanced Photocatalytic Hydrogen Evolution. Appl. Catal. B-Environ. 2020, 266, 118590. [Google Scholar] [CrossRef]
- Zhang, S.; Song, S.; Gu, P.C.; Ma, R.; Wei, D.; Zhao, G.; Wen, T.; Jehan, R.; Hu, B.W.; Wang, X.K. Visible-light-driven Activation of Persulfate Over Cyano and Hydroxyl Group Co-modified Mesoporous g-C3N4 for Boosting Bisphenol A Degradation. J. Mater. Chem. A 2019, 7, 5552–5560. [Google Scholar] [CrossRef]
- Zeng, Q.M.; Ni, J.P.; Mariotti, D.; Lu, L.Y.; Chen, H.; Ni, C.S. Plasma-treatment of Polymeric Carbon Nitride for Efficient NO Abatement under Visible Light. J. Phys. D Appl. Phys. 2022, 55, 354003. [Google Scholar] [CrossRef]
- Xiao, Y.T.; Tian, G.H.; Li, W.; Xie, Y.; Jiang, B.; Tian, C.; Zhao, D.Y.; Fu, H.G. Molecule Self-Assembly Synthesis of Porous Few-Layer Carbon Nitride for Highly Efficient Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 2508–2515. [Google Scholar] [CrossRef]
- Huang, Z.H.; Chen, H.; Zhao, L.; Fang, W.; He, X.; Li, W.X.; Tian, P. In Suit Inducing Electron-donating and Electron-withdrawing Groups in Carbon Nitride by One-step NH4Cl-assisted Route: A Strategy for High Solar Hydrogen Production Efficiency. Environ. Int. 2019, 126, 289–297. [Google Scholar] [CrossRef]
- She, X.J.; Liu, L.; Ji, H.Y.; Mo, Z.; Li, Y.; Huang, L.; Du, D.; Xu, H.; Li, H.M. Template-free Synthesis of 2D Porous Ultrathin Nonmetal-doped g-C3N4 Nanosheets with Highly Efficient Photocatalytic H2 Evolution from Water under Visible Light. Appl. Catal. B-Environ. 2016, 187, 144–153. [Google Scholar] [CrossRef]
- Dong, G.H.; Ho, W.K.; Wang, C.Y. Selective Photocatalytic N2 Fixation Dependent on g-C3N4 Induced by Nitrogen Vacancies. J. Mater. Chem. A 2015, 3, 23435–23441. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, Y.L.; Zhao, Z.K. Porous Defect-modified Graphitic Carbon Nitride via a Facile One-step Approach with Significantly Enhanced Photocatalytic Hydrogen Evolution under Visible Light Irradiation. Appl. Catal. B-Environ. 2018, 226, 1–9. [Google Scholar] [CrossRef]
- Song, X.P.; Yang, Q.; Jiang, X.H.; Yin, M.Y.; Zhou, L.M. Porous Graphitic Carbon Nitride Nanosheets Prepared under Self-producing Atmosphere for Highly Improved Photocatalytic Activity. Appl. Catal. B-Environ. 2017, 217, 322–330. [Google Scholar] [CrossRef]
- He, F.; Chen, G.; Yu, Y.G.; Zhou, Y.S.; Zheng, Y.; Hao, S. The Sulfur-bubble Template-mediated Synthesis of Uniform Porous g-C3N4 with Superior Photocatalytic Performance. Chem. Commun. 2015, 51, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Shi, W.L.; Zhu, C.; Li, H.; Kang, Z.H. CoO and g-C3N4 Complement Each Other for Highly Efficient Overall Water Splitting under Visible Light. Appl. Catal. B-Environ. 2018, 226, 412–420. [Google Scholar] [CrossRef]
- Guo, F.; Shi, W.L.; Wang, H.B.; Huang, H.; Liu, Y.; Kang, Z.H. Fabrication of a CuBi2O4/g-C3N4 p-n Heterojunction with Enhanced Visible Light Photocatalytic Efficiency Toward Tetracycline Degradation. Inorg. Chem. Front. 2017, 4, 1714–1720. [Google Scholar] [CrossRef]
- Yang, S.B.; Gong, Y.J.; Zhang, J.S.; Zhan, L.; Ma, L.; Fang, Z.; Vajtai, R.; Wang, X.C.; Ajayan, P.M. Exfoliated Graphitic Carbon Nitride Nanosheets as Efficient Catalysts for Hydrogen Evolution Under Visible Light. Adv. Mater. 2013, 25, 2452–2456. [Google Scholar] [CrossRef]
- Yang, J.; Liang, Y.J.; Li, K.; Yang, G.; Wang, K.; Xu, R.; Xie, X.J. Cyano and Potassium-rich g-C3N4 Hollow Tubes for Efficient Visible-light-driven Hydrogen Evolution. Catal. Sci. Technol. 2019, 9, 3342–3346. [Google Scholar] [CrossRef]
- Che, H.N.; Liu, L.H.; Che, G.B.; Dong, H.J.; Liu, C.B.; Li, C.M. Control of Energy Band, Layer Structure and Vacancy Defect of Graphitic Carbon Nitride by Intercalated Hydrogen Bond Effect of NO3- Toward Improving Photocatalytic Performance. Chem. Eng. J. 2019, 357, 209–219. [Google Scholar] [CrossRef]
- Gu, Z.Y.; Cui, Z.T.; Wang, Z.J.; Qin, K.S.; Asakura, Y.; Hasegawa, T.; Tsukuda, S.; Hongo, K.; Maezono, R.; Yin, S. Carbon Vacancies and Hydroxyls in Graphitic Carbon Nitride: Promoted Photocatalytic NO Removal Activity and Mechanism. Appl. Catal. B-Environ. 2020, 279, 119376. [Google Scholar] [CrossRef]
- Ho, W.K.; Zhang, Z.Z.; Xu, M.K.; Zhang, X.W.; Wang, X.X.; Huang, Y. Enhanced Visible-light-driven Photocatalytic Removal of NO: Effect on Layer Distortion on g-C3N4 by H2 Heating. Appl. Catal. B-Environ. 2015, 179, 106–112. [Google Scholar] [CrossRef]
- Liang, Q.H.; Li, Z.; Yu, X.L.; Huang, Z.H.; Kang, F.Y.; Yang, Q.H. Macroscopic 3D Porous Graphitic Carbon Nitride Monolith for Enhanced Photocatalytic Hydrogen Evolution. Adv. Mater. 2015, 27, 4634–4639. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.J.; Wang, L.; Zhuzhang, H.Y.; Wang, R.R.; Titirici, M.M.; Wang, X.C. Photocarving Nitrogen Vacancies in a Polymeric Carbon Nitride for Metal-free Oxygen Synthesis. Appl. Catal. B-Environ. 2019, 256, 117794. [Google Scholar] [CrossRef]
- Tang, J.L.; Liu, Y.S.; Hu, Y.J.; Huang, J.W.; Wang, B.; Yang, C.T.; Yang, G.C. Ultrafast NaN3-deflagration Induced Nitrogen Vacancy-enriched g-C3N4 for Tailoring Band Structures and Enhanced Photocatalytic Performance. J. Power Sources 2019, 434, 226731. [Google Scholar] [CrossRef]
- Ma, W.; Wang, N.; Guo, Y.; Yang, L.Q.; Lv, M.; Tang, X.; Li, S.T. Enhanced Photoreduction CO2 Activity on g-C3N4: By Synergistic Effect of Nitrogen Defective-enriched and Porous Structure, and Mechanism Insights. Chem. Eng. J. 2020, 388, 124288. [Google Scholar] [CrossRef]
- Cao, J.W.; Zhang, J.Y.; Dong, X.A.; Fu, H.L.; Zhang, X.; Lv, X.; Li, Y.H.; Jiang, G.M. Defective Borate-decorated Polymer Carbon Nitride: Enhanced Photocatalytic NO Removal, Synergy Effect and Reaction Pathway. Appl. Catal. B-Environ. 2019, 249, 266–274. [Google Scholar] [CrossRef]
- Ren, H.T.; Qi, F.; Labidi, A.; Zhao, J.J.; Wang, H.; Xin, Y.; Luo, J.M.; Wang, C.Y. Chemically bonded carbon quantum dots/Bi2WO6 S-scheme heterojunction for boosted photocatalytic antibiotic degradation: Interfacial engineering and mechanism insight. Appl. Catal. B-Environ. 2023, 330, 122587. [Google Scholar] [CrossRef]
- Wang, C.; Fu, M.; Cao, J.; Wu, X.L.; Hu, X.L.; Dong, F. BaWO4/g-C3N4 Heterostructure with Excellent Bifunctional Photocatalytic Performance. Chem. Eng. J. 2020, 385, 123833. [Google Scholar] [CrossRef]
- Tan, Y.W.; Wei, S.P.; Liu, X.Y.; Pan, B.Y.; Liu, S.; Wu, J.; Fu, M.; Jia, Y.M.; He, Y.Z. Neodymium oxide (Nd2O3) coupled tubular g-C3N4, an efficient dual-function catalyst for photocatalytic hydrogen production and NO removal. Sci. Total Environ. 2021, 773, 145583. [Google Scholar] [CrossRef]
- Li, J.L.; Zhang, Q.; Zou, Y.Z.; Cao, Y.H.; Cui, W.; Dong, F.; Zhou, Y. Ti3C2 MXene modified g-C3N4 with enhanced visible-light photocatalytic performance for NO purification. J. Colloid. Interf. Sci. 2020, 575, 443–451. [Google Scholar] [CrossRef]
- Ren, Y.Y.; Li, Y.; Wu, X.Y.; Wang, J.L.; Zhang, G.K. S-scheme Sb2WO6/g-C3N4 Photocatalysts with Enhanced Visible-light-induced Photocatalytic NO Oxidation Performance. Chin. J. Catal. 2021, 42, 69–77. [Google Scholar] [CrossRef]
- Cui, W.; Li, J.Y.; Dong, F.; Sun, Y.J.; Jiang, G.; Cen, W.; Lee, S.C.; Wu, Z.B. Highly Efficient Performance and Conversion Pathway of Photocatalytic NO Oxidation on SrO-Clusters@Amorphous Carbon Nitride. Environ. Sci. Technol. 2017, 51, 10682–10690. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.D.; Frost, R.L.; Kloprogge, J.T.; Duong, L. Infrared spectroscopy of goethite dehydroxylation: III. FT-IR microscopy of in situ study of the thermal transformation of goethite to hematite. Spectrochim. Acta A. 2002, 58, 443–451. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Xu, X.; Labidi, A.; Ren, H.; Allam, A.A.; Rady, A.; Huang, Y.; Wei, S.; Padervand, M.; Ghasemi, S.; et al. Cyano/Hydroxyl Groups Co-Functionalized g-C3N4 for Photocatalytic NO Removal: A Synergistic Strategy towards Inhibition of Toxic Intermediate NO2. Catalysts 2023, 13, 1433. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13111433
Wang H, Xu X, Labidi A, Ren H, Allam AA, Rady A, Huang Y, Wei S, Padervand M, Ghasemi S, et al. Cyano/Hydroxyl Groups Co-Functionalized g-C3N4 for Photocatalytic NO Removal: A Synergistic Strategy towards Inhibition of Toxic Intermediate NO2. Catalysts. 2023; 13(11):1433. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13111433
Chicago/Turabian StyleWang, Hui, Xiaoqian Xu, Abdelkader Labidi, Haitao Ren, Ahmed A. Allam, Ahmed Rady, Yu Huang, Shuwei Wei, Mohsen Padervand, Shahnaz Ghasemi, and et al. 2023. "Cyano/Hydroxyl Groups Co-Functionalized g-C3N4 for Photocatalytic NO Removal: A Synergistic Strategy towards Inhibition of Toxic Intermediate NO2" Catalysts 13, no. 11: 1433. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13111433