

Influence of the Valence of Iron on the NO Reduction by CO over Cu-Fe-Mordenite

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. X-ray Diffraction Studies and Chemical Composition
2.2. Morphology and Textural Studies
2.3. Surface Acidity
2.4. Adsorption of Probe Molecules
2.5. Reduction of NO via CO Oxidation
3. Materials and Methods
3.1. Samples Preparation
3.2. Characterization Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwak, J.H.; Tran, D.; Burton, S.D.; Szanyi, J.; Lee, J.H.; Peden, C.H.F. Effects of Hydrothermal Aging on NH3-SCR Reaction over Cu/Zeolites. J. Catal. 2012, 287, 203–209. [Google Scholar] [CrossRef]
- Chatterjee, D.; Deutschmann, O.; Warnatz, J. Detailed Surface Reaction Mechanism in a Three-Way Catalyst. Faraday Discuss. 2001, 119, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Peng, Q.; Jiaqiang, E.; Xie, B.; Wei, J.; Yin, R.; Fu, G. Mechanism, Performance and Modification Methods for NH3-SCR Catalysts: A Review. Fuel 2023, 331, 125885. [Google Scholar] [CrossRef]
- Gramigni, F.; Iacobone, U.; Nasello, N.D.; Selleri, T.; Usberti, N.; Nova, I. Review of Hydrocarbon Poisoning and Deactivation Effects on Cu-Zeolite, Fe-Zeolite, and Vanadium-Based Selective Catalytic Reduction Catalysts for NOx Removal from Lean Exhausts. Ind. Eng. Chem. Res. 2021, 60, 6403–6420. [Google Scholar] [CrossRef]
- Kubo, T.; Tominaga, H.; Kunugi, T. Oxidation of Carbon Monoxide over Transition Metal Ion-Zeolite Catalysts. Bull. Chem. Soc. Jpn. 1973, 46, 3549–3552. [Google Scholar] [CrossRef] [Green Version]
- Bulánek, R.; Wichterlová, B.; Sobalík, Z.; Tichý, J. Reducibility and Oxidation Activity of Cu Ions in Zeolites. Appl. Catal. B Environ. 2001, 31, 13–25. [Google Scholar] [CrossRef]
- Gao, F.; Zheng, Y.; Kukkadapu, R.K.; Wang, Y.L.; Walter, E.D.; Schwenzer, B.; Szanyi, J.; Peden, C.H.F. Iron Loading Effects in Fe/SSZ-13 NH3-SCR Catalysts: Nature of the Fe Ions and Structure–Function Relationships. ACS Catal. 2016, 6, 2939–2954. [Google Scholar] [CrossRef]
- Cheung, T.; Bhargava, S.K.; Hobday, M.; Foger, K. Adsorption of NO on Cu Exchanged Zeolites, an FTIR Study: Effects of Cu Levels, NO Pressure, and Catalyst Pretreatment. J. Catal. 1996, 158, 301–310. [Google Scholar] [CrossRef]
- Metkar, P.S.; Harold, M.P.; Balakotaiah, V. Selective Catalytic Reduction of NOx on Combined Fe- and Cu-Zeolite Monolithic Catalysts: Sequential and Dual Layer Configurations. Appl. Catal. B Environ. 2012, 111–112, 67–80. [Google Scholar] [CrossRef]
- Deka, U.; Lezcano-Gonzalez, I.; Weckhuysen, B.M.; Beale, A.M. Local Environment and Nature of Cu Active Sites in Zeolite-Based Catalysts for the Selective Catalytic Reduction of NOx. ACS Catal. 2013, 3, 413–427. [Google Scholar] [CrossRef]
- Sazama, P.; Moravkova, J.; Sklenak, S.; Vondrova, A.; Tabor, E.; Sadovska, G.; Pilar, R. Effect of the Nuclearity and Coordination of Cu and Fe Sites in β Zeolites on the Oxidation of Hydrocarbons. ACS Catal. 2020, 10, 3984–4002. [Google Scholar] [CrossRef]
- Bayliss, P.; Kolitsch, U.; Nickel, E.H.; Pring, A. Alunite Supergroup: Recommended Nomenclature. Mineral. Mag. 2010, 74, 919–927. [Google Scholar] [CrossRef]
- Zhukov, Y.M.; Shelyapina, M.G.; Efimov, A.Y.; Zhizhin, E.V.; Petranovskii, V. Recognition of Depth Composition Profiles of Copper-Exchanged Mordenites Applying Analytical Methods with Different Depth Vision. Mater. Chem. Phys. 2019, 236, 121787. [Google Scholar] [CrossRef]
- Bogdanov, D.S.; Novikov, R.G.; Pestsov, O.S.; Baranov, D.A.; Shelyapina, M.G.; Tsyganenko, A.A.; Kasatkin, I.A.; Kalganov, V.D.; Silyukov, O.I.; Petranovskii, V. Formation of admixed phase during microwave assisted Cu ion exchange in mordenite. Mater. Chem. Phys. 2021, 261, 124235. [Google Scholar] [CrossRef]
- Shelyapina, M.; Krylova; Zhukov, Y.; Zvereva; Rodriguez-Iznaga, I.; Petranovskii, V.; Fuentes-Moyado, S. Comprehensive Analysis of the Copper Exchange Implemented in Ammonia and Protonated Forms of Mordenite Using Microwave and Conventional Methods. Molecules 2019, 24, 4216. [Google Scholar] [CrossRef] [Green Version]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry and Use; John Wiley & Sons, Inc.: New York, NY, USA, 1974; p. 771. [Google Scholar]
- Efimov, A.Y.; Petranovsky, V.P.; Fedotov, M.A.; Khripun, M.K.; Myund, L.A. On the role of triethanolamine in the Charnell synthesis of zeolites. J. Struct. Chem. 1993, 34, 548–551. [Google Scholar] [CrossRef]
- Luo, W.; Yang, X.; Wang, Z.; Huang, W.; Chen, J.; Jiang, W.; Wang, L.; Cheng, X.; Deng, Y.; Zhao, D. Synthesis of ZSM-5 aggregates made of zeolite nanocrystals through a simple solvent-free method. Microporous Mesoporous Mater. 2017, 243, 112–118. [Google Scholar] [CrossRef]
- Matsukata, M.; Osaki, T.; Ogura, M.; Kikuchi, E. Crystallization behavior of zeolite beta during steam-assisted crystallization of dry gel. Microporous Mesoporous Mater. 2002, 56, 1–10. [Google Scholar] [CrossRef]
- Inagaki, S.; Nakatsuyama, K.; Saka, Y.; Kikuchi, E.; Kohara, S.; Matsukata, M. Elucidation of Medium-Range Structure in a Dry Gel-Forming *BEA-Type Zeolite. J. Phys. Chem. C. 2007, 111, 10285–10293. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Gurgul, J.; Łątka, K.; Sánchez-López, P.; Bogdanov, D.; Kotolevich, Y.; Petranovskii, V.; Fuentes, S. Mechanism of formation of framework Fe3+ in bimetallic Ag-Fe mordenites-Effective catalytic centers for deNOx reaction. Microporous Mesoporous Mater. 2019, 299, 109841. [Google Scholar] [CrossRef]
- Chen, L.; Janssens, T.V.W.; Skoglundh, M.; Grönbeck, H. Interpretation of NH3-TPD Profiles from Cu-CHA Using First-Principles Calculations. Top. Catal. 2018, 62, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Benaliouche, F.; Boucheffa, Y.; Ayrault, P.; Mignard, S.; Magnoux, P. NH3-TPD and FTIR Spectroscopy of Pyridine Adsorption Studies for Characterization of Ag- and Cu-Exchanged X Zeolites. Microporous Mesoporous Mater. 2008, 111, 80–88. [Google Scholar] [CrossRef]
- Davydov, A. The Nature of Oxide Surface Centers. In Molecular Spectroscopy of Oxide Catalyst Surfaces; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 27–179. [Google Scholar]
- Marie, O.; Massiani, P.; Thibault-Starzyk, F. Infrared evidence of a third brønsted site in mordenites. J. Phys. Chem. B 2004, 108, 5073–5081. [Google Scholar] [CrossRef]
- Makarova, M.A.; Wilson, A.E.; Van Liemt, B.J.; Mesters, C.M.; De Winter, A.M.; Williams, C. Quantification of Brønsted acidity in mordenites. J. Catal. 1997, 172, 170–177. [Google Scholar] [CrossRef]
- Jiang, M.; Karge, H.G. Investigation of acid properties of dealuminated H-mordenite zeolites by low-temperature diffuse reflectance FTIR. J. Chem. Soc. Faraday Trans. 1996, 92, 2641–2649. [Google Scholar] [CrossRef]
- Wu, P.; Komatsu, T.; Yashima, T. Isomorphous substitution of Fe3+ in the framework of aluminosilicate mordenite by hydrothermal synthesis. Microporous Mesoporous Mater. 1998, 20, 139–147. [Google Scholar] [CrossRef]
- Bevilacqua, M.; Busca, G. A study of the localization and accessibility of Brønsted and Lewis acid sites of H-mordenite through the FT-IR spectroscopy of adsorbed branched nitriles. Catal. Commun. 2002, 3, 497–502. [Google Scholar] [CrossRef]
- Antúnez-García, J.; Galván, D.H.; Petranovskii, V.; Murrieta-Rico, F.N.; Yocupicio-Gaxiola, R.I.; Shelyapina, M.G.; Fuentes-Moyado, S. Aluminum Distribution in Mordenite-Zeolite Framework: A New Outlook Based on Density Functional Theory Calculations. J. Solid State Chem. 2022, 306, 122725. [Google Scholar] [CrossRef]
- Sobalík, Z.; Tvarůžková, Z.; Vondrová, A.; Schwarze, M. Targeted Preparation of Fe-Zeolites with Iron Prevailing in Extraframework Cationic Positions. Stud. Surf. Sci. Catal. 2006, 162, 889–896. [Google Scholar]
- Sklenak, S.; Andrikopoulos, P.C.; Boekfa, B.; Jansang, B.; Nováková, J.; Benco, L.; Bucko, T.; Hafner, J.; Dědeček, J.; Sobalík, Z. N2O Decomposition over Fe-Zeolites: Structure of the Active Sites and the Origin of the Distinct Reactivity of Fe-Ferrierite, Fe-ZSM-5, and Fe-Beta. A Combined Periodic DFT and Multispectral Study. J. Catal. 2010, 272, 262–274. [Google Scholar] [CrossRef]
- Bols, M.L.; Hallaert, S.D.; Snyder, B.E.R.; Devos, J.; Plessers, D.; Rhoda, H.M.; Dusselier, M.; Schoonheydt, R.A.; Pierloot, K.; Solomon, E.I.; et al. Spectroscopic Identification of the α-Fe/α-O Active Site in Fe-CHA Zeolite for the Low-Temperature Activation of the Methane C–H Bond. J. Am. Chem. Soc. 2018, 140, 12021–12032. [Google Scholar] [CrossRef] [PubMed]
- Daouli, A.; Hessou, E.P.; Monnier, H.; Dziurla, M.-A.; Hasnaoui, A.; Maurin, G.; Badawi, M. Adsorption of NO, NO2 and H2O in Divalent Cation Faujasite Type Zeolites: A Density Functional Theory Screening Approach. Phys. Chem. Chem. Phys. 2022, 24, 15565–15578. [Google Scholar] [CrossRef] [PubMed]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.L.; Hallaert, S.D.; Böttger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Devos, J.; Bols, M.L.; Plessers, D.; Goethem, C.V.; Seo, J.W.; Hwang, S.-J.; Sels, B.F.; Dusselier, M. Synthesis–Structure–Activity Relations in Fe-CHA for C–H Activation: Control of Al Distribution by Interzeolite Conversion. Chem. Mater. 2019, 32, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Bols, M.L.; Snyder, B.E.R.; Rhoda, H.M.; Cnudde, P.; Fayad, G.; Schoonheydt, R.A.; Van Speybroeck, V.; Solomon, E.I.; Sels, B.F. Coordination and Activation of Nitrous Oxide by Iron Zeolites. Nat. Catal. 2021, 4, 332–340. [Google Scholar] [CrossRef]
- Mesilov, V.; Pon, L.; Dahlin, S.; Bergman, S.L.; Pettersson, L.J.; Bernasek, S.L. Computational Study of Noble Metal CHA Zeolites: NO Adsorption and Sulfur Resistance. J. Phys. Chem. C 2022, 126, 7022–7035. [Google Scholar] [CrossRef]
- Kostrov, V.V.; Lokhov, Y.; Morozov, L.N.; Davydov, A. Study of the state of transition-metal cations on catalyst surfaces by IR spectroscopy of adsorbed test molecules (CO, NO). 3. Copper-aluminum catalysts (CuO-Al2O3). Kinet. Catal. 1980, 21, 943–946. [Google Scholar]
- Adak, S.; Pal, R.S.; Khan, T.S.; Poddar, M.K.; Ahmad, M.S.; Prasad, V.V.; Haider, M.A.; Bal, R. Role of Interfacial Cu-Ions in Polycrystalline Cu-CeO2: In-Situ Raman, In-situ DRIFT and DFT Studies for Preferential Oxidation of CO in Presence of Excess H2. ChemistrySelect 2021, 6, 13051–13059. [Google Scholar] [CrossRef]
- Szanyi, J.; Kwak, J.H.; Zhu, H.; Peden, C.H.F. Characterization of Cu-SSZ-13 NH3 SCR Catalysts: An in Situ FTIR Study. Phys. Chem. Chem. Phys. 2013, 15, 2368. [Google Scholar] [CrossRef]
- Ramírez-Garza, R.E.; Rodríguez-Iznaga, I.; Simakov, A.; Farías, M.H.; Castillón-Barraza, F.F. Cu-Ag/Mordenite Catalysts for NO Reduction: Effect of Silver on Catalytic Activity and Hydrothermal Stability. Mater. Res. Bull. 2018, 97, 369–378. [Google Scholar] [CrossRef]
- Davydov, A. Study of Cation States by DRES and FTIR Spectroscopies of the Probe Molecules. In Molecular Spectroscopy of Oxide Catalyst Surfaces; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 181–275. [Google Scholar]
- Gao, F. Fe-Exchanged Small-Pore Zeolites as Ammonia Selective Catalytic Reduction (NH3-SCR) Catalysts. Catalysts 2020, 10, 1324. [Google Scholar] [CrossRef]
- Kuterasiński, Ł.; Podobiński, J.; Madej, E.; Smoliło-Utrata, M.; Rutkowska-Zbik, D.; Datka, J. Reduction and oxidation of cu species in Cu-faujasites studied by IR spectroscopy. Molecules 2020, 25, 4765. [Google Scholar] [CrossRef] [PubMed]
- Brandenberger, S.; Kröcher, O.; Tissler, A.; Althoff, R. The State of the Art in Selective Catalytic Reduction of NOxby Ammonia Using Metal-Exchanged Zeolite Catalysts. Catal. Rev. 2008, 50, 492–531. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
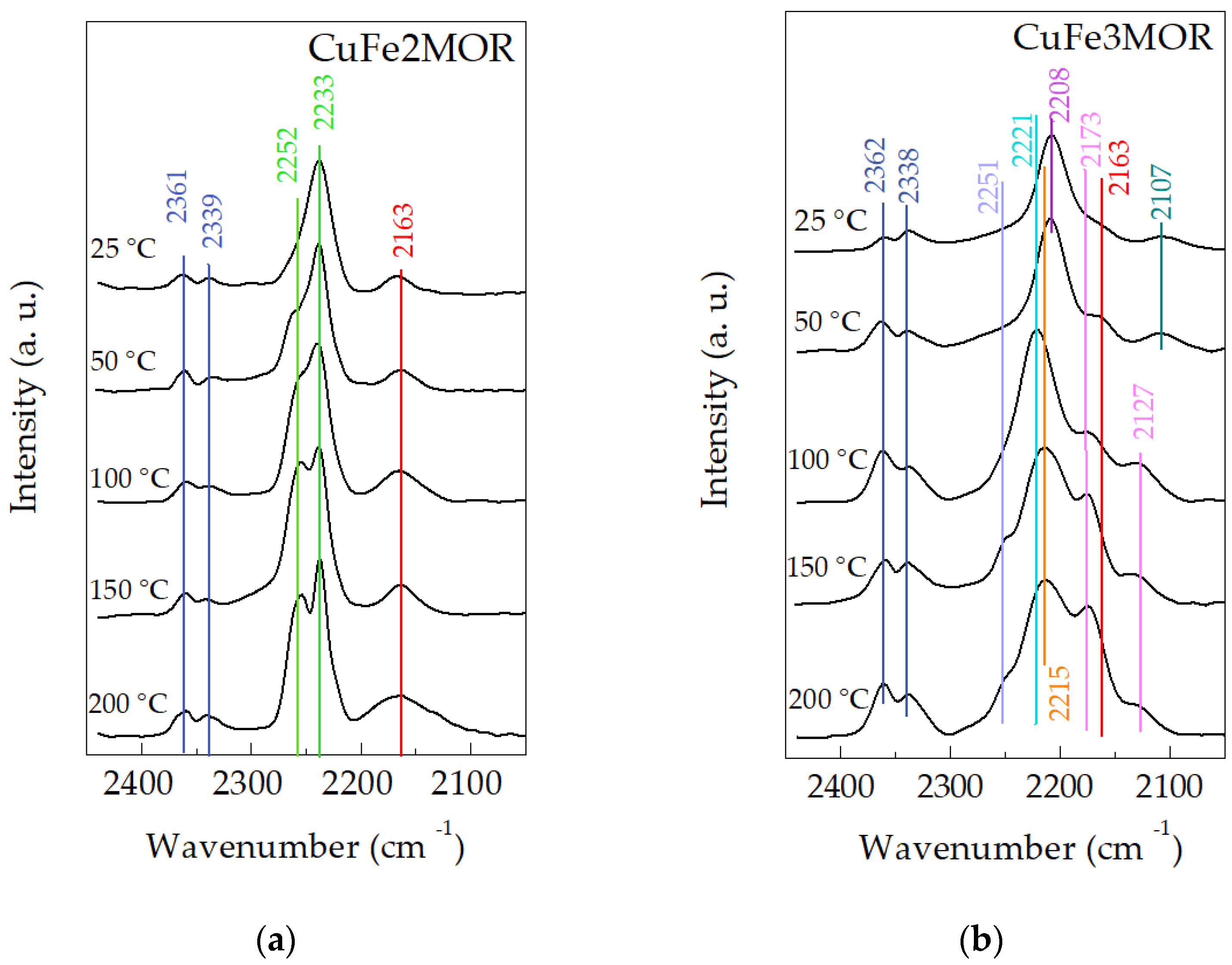{kind=link}
{kind=link}
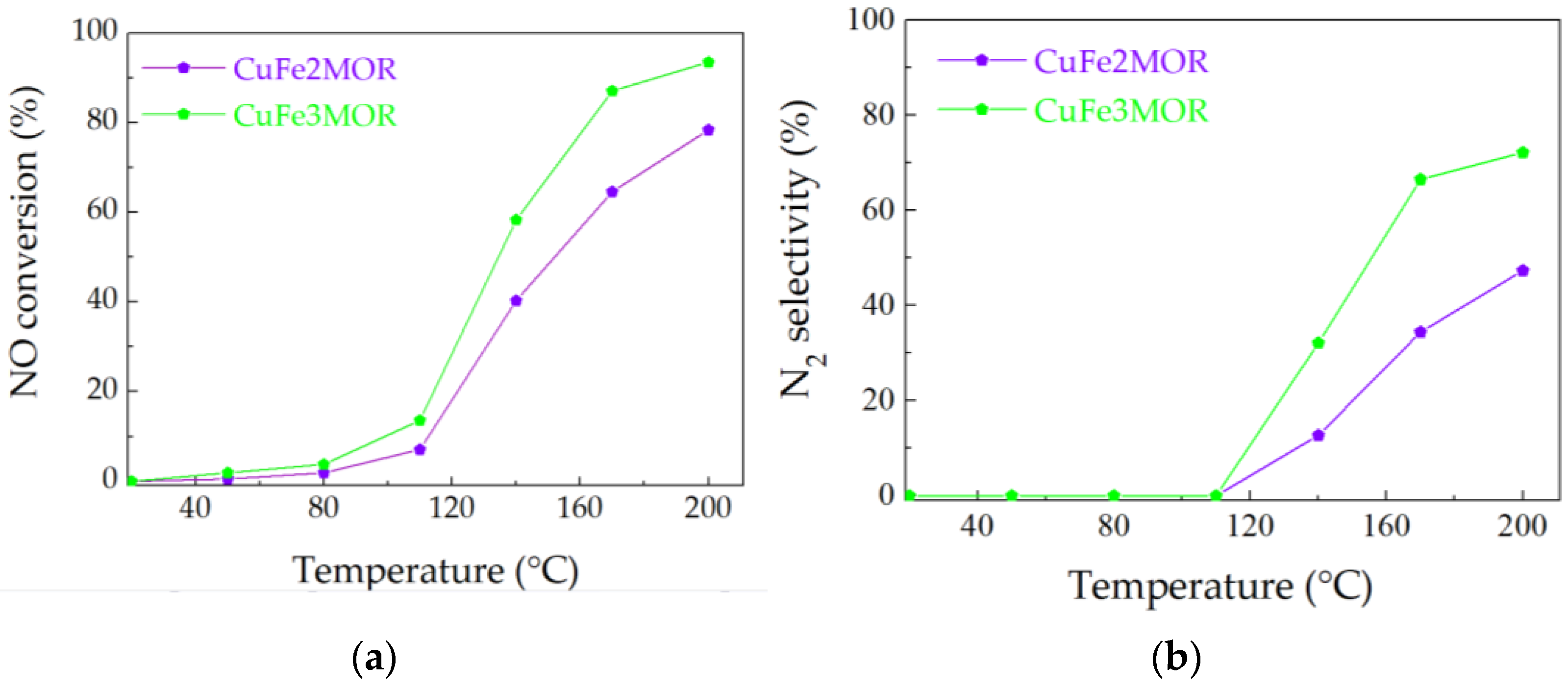{kind=link}
| Sample | a | b | c | K = a/b |
|---|---|---|---|---|
| NaMOR | 18.12 | 20.46 | 7.51 | 0.886 |
| CuMOR | 18.15 | 20.42 | 7.51 | 0.889 |
| Fe2MOR | 18.16 | 20.43 | 7.51 | 0.889 |
| CuFe2MOR | 18.11 | 20.41 | 7.51 | 0.887 |
| Fe3MOR | 18.14 | 20.36 | 7.50 | 0.891 |
| CuFe3MOR | 18.14 | 20.37 | 7.50 | 0.891 |
| Sample | Si | Al | Si/Al | Na | Cu | Fe | EIEM |
|---|---|---|---|---|---|---|---|
| NaMOR | 40.04 | 6.12 | 6.55 | 7.00 | - | - | 1.14 |
| CuMOR | 38.97 | 5.88 | 6.62 | 3.34 | 1.41 | - | 1.05 |
| Fe2MOR | 41.80 | 6.19 | 6.75 | 3.82 | - | 1.18 | 0.71 |
| CuFe2MOR | 41.10 | 6.00 | 6.85 | 3.82 | 0.85 | 0.62 | 0.74 |
| Fe3MOR | 35.40 | 4.10 | 8.63 | 3.82 | - | 3.26 | 1.20 |
| CuFe3MOR | 36.82 | 4.79 | 7.68 | 3.63 | 0.88 | 1.81 | 0.98 |
| Sample | SBET, m2∙g−1 | Vtotal, cm3∙g−1 | d, Å |
|---|---|---|---|
| NaMOR | 297 | 0.04 | 170 |
| CuMOR | 309 | 0.04 | 360 |
| Fe2MOR | 275 | 0.04 | 201 |
| CuFe2MOR | 313 | 0.05 | 379 |
| Fe3MOR | 288 | 0.05 | 171 |
| CuFe3MOR | 302 | 0.04 | 346 |
| Sample | Amount of Acid Cites | Total Acidity, mmol/g | ||
|---|---|---|---|---|
| Weak, % | Medium, % | Strong, % | ||
| NaMOR | 100.0 | 0.0 | 0.0 | 1439 |
| CuMOR | 70.7 | 16.1 | 13.2 | 2108 |
| Fe2MOR | 74.3 | 17.4 | 8.3 | 1732 |
| CuFe2MOR | 43.0 | 34.9 | 22.2 | 2991 |
| Fe3MOR | 64.4 | 25.8 | 9.9 | 1218 |
| CuFe3MOR | 66.9 | 12.7 | 20.4 | 1653 |
| Sample | Ion Exchange Solution | CCu+CFe in Solution |
|---|---|---|
| CuMOR | CuSO4 | 0.5 N of Cu2+ |
| Fe2MOR | FeSO4, H2SO4, until pH = 2 | 0.5 N of Fe2+ |
| CuFe2MOR | CuSO4, FeSO4, H2SO4 until pH = 2 | 0.5 N, Cu2+:Fe2+ = 1:1 |
| Fe3MOR | Fe2(SO4)3 | 0.5 N of Fe3+ |
| Cu3FeMOR | CuSO4, Fe2(SO4)3 | 0.5 N, Cu2+:Fe3+ = 1:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotolevich, Y.; Zepeda-Partida, T.; Yocupicio-Gaxiola, R.; Antúnez-Garcia, J.; Pelaez, L.; Avalos-Borja, M.; Vázquez-Salas, P.J.; Fuentes-Moyado, S.; Petranovskii, V. Influence of the Valence of Iron on the NO Reduction by CO over Cu-Fe-Mordenite. Catalysts 2023, 13, 484. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13030484
Kotolevich Y, Zepeda-Partida T, Yocupicio-Gaxiola R, Antúnez-Garcia J, Pelaez L, Avalos-Borja M, Vázquez-Salas PJ, Fuentes-Moyado S, Petranovskii V. Influence of the Valence of Iron on the NO Reduction by CO over Cu-Fe-Mordenite. Catalysts. 2023; 13(3):484. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13030484
Chicago/Turabian StyleKotolevich, Yulia, Trino Zepeda-Partida, Rosario Yocupicio-Gaxiola, Joel Antúnez-Garcia, Luis Pelaez, Miguel Avalos-Borja, Pedro Jovanni Vázquez-Salas, Sergio Fuentes-Moyado, and Vitalii Petranovskii. 2023. "Influence of the Valence of Iron on the NO Reduction by CO over Cu-Fe-Mordenite" Catalysts 13, no. 3: 484. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13030484