1. Introduction
The increasing levels of CO
2 emissions have been widely and unanimously acknowledged as the main cause of anthropogenic climate change. In order to reduce the emissions of CO
2, several measures are being adopted by many countries. In this context, Carbon Capture and Utilization (CCU) is one of several approaches that have been proposed to decrease emissions [
1,
2,
3]. This work is focused on the utilization of CO
2 to transform it into dimethyl ether (DME) through a reaction with hydrogen obtained from renewable sources; DME, due to its chemical-physical properties, can be used as fuel in substitution of LPG (Liquified Petroleum Gas), maintaining the same transport and storage technologies. It can also be used as an additive to diesel fuel, granting better performances owing to its high cetane number; furthermore, in properly modified diesel engines, it can completely replace diesel fuel, giving rise to lower emissions of particulate, aromatic compounds, and sulfur [
4,
5,
6,
7,
8,
9,
10,
11,
12,
13]. Dimethyl ether is obtained from CO
2 through two subsequent reactions. The first one is the hydrogenation of CO
2 to methanol:
This reaction is then followed by the dehydration of methanol to DME:
Therefore, the global reaction is:
It has to be pointed out that, in the CO
2-to-DME process, CO
2 hydrogenation to methanol competes with the Reverse Water–Gas Shift (RWGS) reaction, which forms CO:
As regards the first reaction, the most widely proposed catalysts are based on Cu, the active phase of the catalyst, paired with ZnO, which acts as a promoter, and a third phase, usually Al
2O
3 or ZrO
2, which increases the stability of the system. Regarding the dehydration of methanol to DME, several solid acidic catalysts have been reported; particularly, zeolites [
14,
15] and γ-Al
2O
3 represent the most studied systems [
4,
5,
6]. γ-Al
2O
3, the focus of this work, is widely used as a methanol dehydration catalyst due to its low cost, compared to zeolites and other aluminosilicates, and due to the presence of Lewis acid sites of moderate strength, which lead to high DME selectivity; the presence of strong Brønsted sites, on the other hand, despite showing a higher activity in methanol dehydration, gives rise to a lower selectivity, due to the formation of hydrocarbons through the Methanol-To-Olefins (MTO) process and a potential deactivation of the catalyst due to coke formation [
10,
16,
17,
18]. For these reasons, γ-Al
2O
3 has been widely reported for the methanol-to-DME process [
19,
20,
21], as well as for the one-pot syngas-to-DME [
22,
23,
24] and CO
2-to-DME processes [
25,
26,
27].
The CO
2-to-DME process is often carried out using a one-step approach, in which the two reactions are performed simultaneously in the same reactor in the presence of both the redox and the acidic catalyst. In this context, the two catalytic systems are often combined by obtaining a physical mixture of the two phases; however, recently, the attention of several authors has been focused on bifunctional catalysts, which present an intimate contact between the redox and the acidic phases. These catalysts are usually synthesized by either coprecipitation or impregnation, dispersing the redox phase onto the surface of a support, which consists of the dehydrating acidic catalyst, such as zeolites [
4,
5] or disordered mesoporous aluminosilicates [
28]. A few cases of bifunctional catalysts consisting in a redox phase dispersed inside mesostructured γ-Al
2O
3 [
29,
30,
31,
32] or mesostructured silica [
33] have also been reported.
In this light, the present work focuses on developing bifunctional catalysts for a one-pot CO2-to-DME process, based on mesostructured γ-Al2O3 as the dehydration catalyst obtained by the EISA approach with two different templating agents (P123 and F127). The two acidic systems were used as supports for impregnation with a Cu/ZnO/ZrO2 redox catalyst, obtaining the nanocomposite bifunctional catalysts. Moving along this line, the final aim is to point out the effect of the close contact between the two catalytic phases in bifunctional catalysts obtained via impregnation compared to a physically mixed system, commonly proposed in the literature. Compared with physical mixtures, the deposition of the redox phase inside the pores of a mesostructured support to form nanocomposite catalysts should improve the dispersion and the surface area of Cu-based active sites, improving CO2 conversion. In this perspective, the attention was focused on mesostructured γ-Al2O3 instead of microporous materials since the larger size of mesopores should lead to a most efficient impregnation of the dehydration catalysts, allowing the deposition of the redox catalyst in the form of nanoparticles inside the pores, limiting their growth. The presence of the mesopore walls should also avoid sintering phenomena between the redox nanoparticles, rendering the catalyst stable and regenerable. Impregnation on zeolites, conversely, would lead to deposition of the redox phase only on the external surface of the support due to the small size of micropores.
2. Results and Discussion
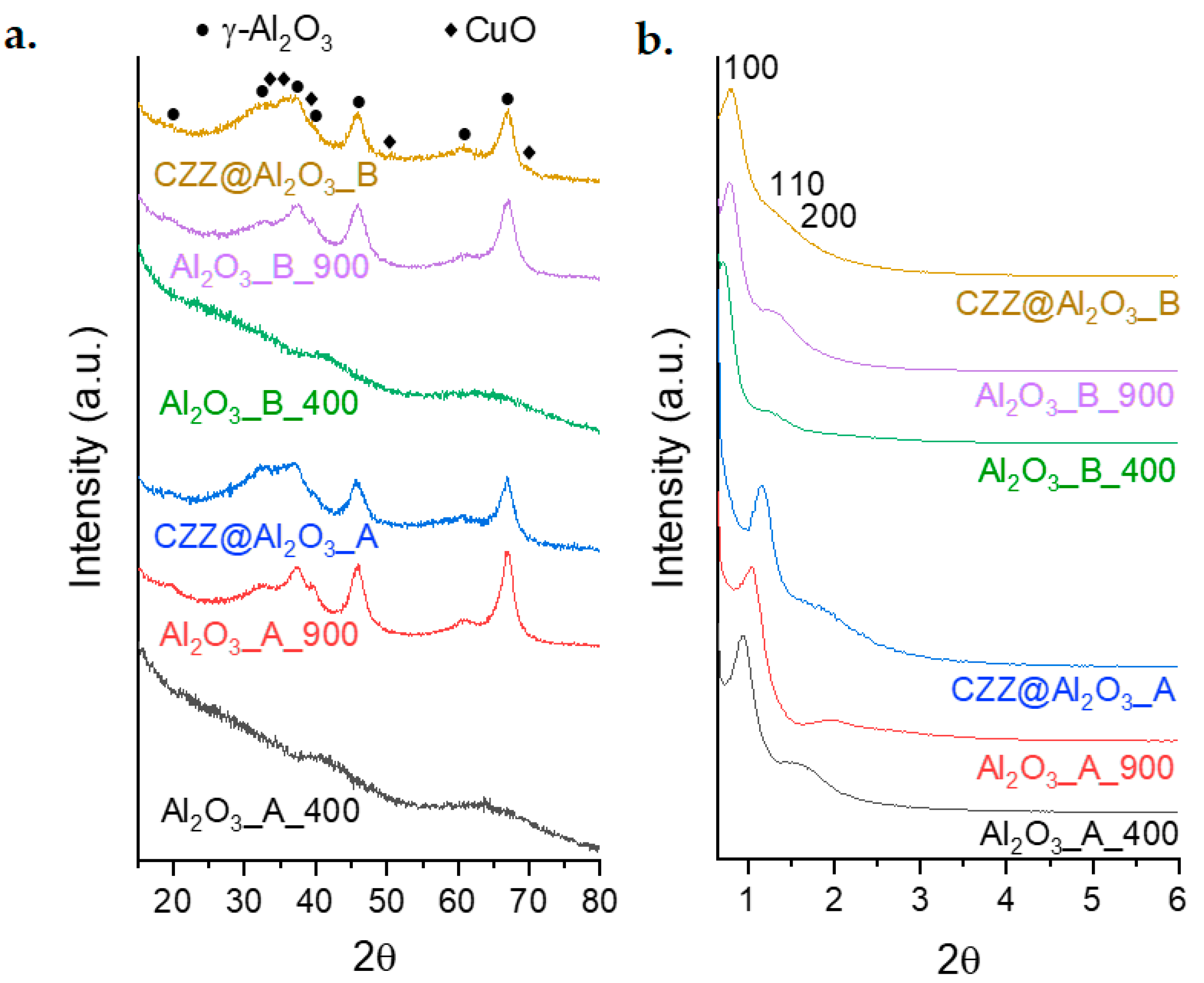
Wide Angle X-ray Diffraction (WA-XRD) measurements (
Figure 1a) point out the amorphous nature of both Al
2O
3 (A and B) thermally treated at 400 °C; after the 900 °C treatment, both samples show the formation of a cubic γ-Al
2O
3 phase (PDF card 00-047-1292) in the form of very small nanocrystals, as evidenced by the presence of broad diffraction peaks. As regards the composites, both the systems feature wide bands between 30° and 40°, the range where the main diffraction peaks of tenorite (CuO, PDF card 00-045-0937), zincite (ZnO, PDF Card 00-036-1451), and several crystalline phases of ZrO
2 are located; these signals overlap with those of γ-Al
2O
3, resulting in a single wide band located between 25° and 40°. No other signals attributable to the redox phase are observed, indicating that the redox phase is in the form of very small nanocrystals.
The Rietveld refinement (
Figure S1) performed on the pattern of Al
2O
3_A_900 evidences the presence of γ-Al
2O
3 with a mean crystallite size of 5.1(1) nm and a cell parameter of 7.879(4) Å, slightly smaller than the one reported in the literature (7.9448 Å, PDF card 00-047-1292). A similar result in terms of cell parameter (7.886 (4) Å) is obtained for Al
2O
3_B_900 (
Figure S2), while a slightly smaller mean crystallite size, namely 4.5(4) nm is observed. The refinement also highlights the presence of two broad bands at about 36° and 63°, ascribable to a small amount of amorphous alumina still present after the treatment at 900 °C.
Small-Angle X-ray Diffraction (SA-XRD) patterns (
Figure 1b) show the presence of a mesoporous order on both the supports obtained using P123 and F127 as templating agents, as well as on their corresponding composites. The mesostructure shows a hexagonal arrangement (p6 mm) indicated by the presence of a main peak located at values of 2θ between 0.6° and 1.5° and a shoulder located at higher values. A significant difference in terms of the main peak position (Δ2θ ≈ 0.3°) and, consequently, cell parameters can be observed between the systems obtained with the two Pluronics (
Table 1). As expected, a shift towards higher values of 2θ is observed for both samples (A and B) by increasing the temperature to 900 °C, indicating that the formation of a crystalline phase is associated with a decrease in the cell parameter of the mesoporous structure. Moreover, SA-XRD measurements point out that the mesostructure is maintained after the impregnation with the active phase, even if a further decrease in the mesostructure cell parameter is observed due to the functionalization process of the supports with the redox phase. Specifically, a
0 ranges from 10.8 nm for Al
2O
3_A_400 to 9.3 nm for Al
2O
3_A_900 and reaches 8.8 nm for CZZ@Al
2O
3_A; on the other hand, it decreases from 14.4 nm for Al
2O
3_B_400 to 13.1 nm for Al
2O
3_B_900, and to 12.7 nm for CZZ@Al
2O
3_B.
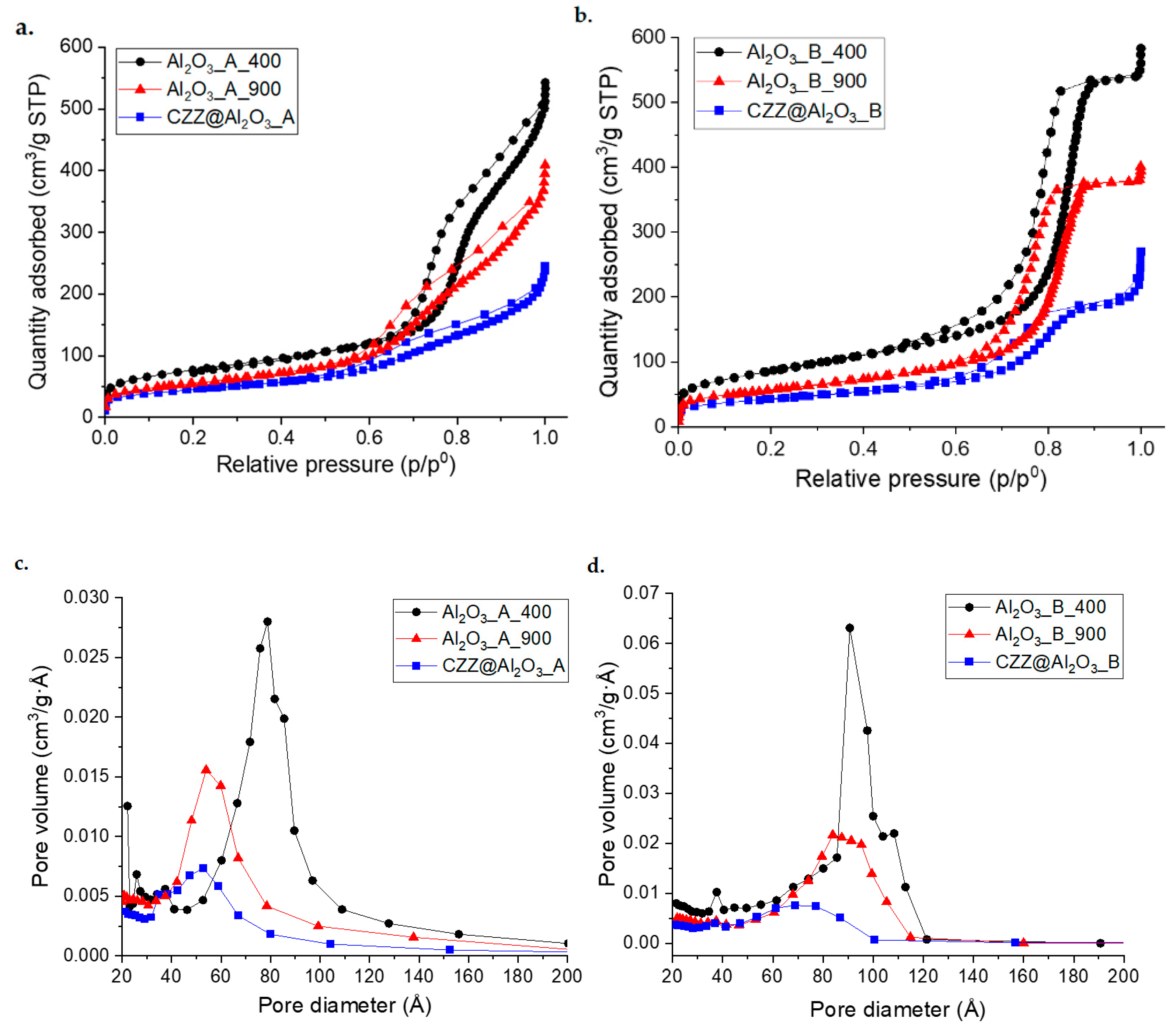
Nitrogen physisorption highlights the mesoporous nature of all samples, indicated by the presence of a capillary condensation branch in the isotherms. The series of samples synthesized using F127 as templating agent clearly shows type IV isotherms with a steep capillary condensation and parallel hysteresis branches, followed by a
plateau (
Figure 2b); on the other hand, the systems obtained with P123 present less steep capillary condensation curves and do not show a final
plateau (
Figure 2a), suggesting a lower degree of porous order and a possible macroporous contribution with inter-particle capillary condensation [
34]. As expected, for both samples, it can be observed how the thermal treatment at 900 °C causes a drop in the values of adsorbed nitrogen, indicating a partial collapse of the mesostructure, associated with a decrease in terms of both surface area (from 274 to 197 m
2/g for Al
2O
3_A and from 309 to 194 m
2/g for Al
2O
3_B) and pore volume (from 0.76 to 0.57 cm
3/g for Al
2O
3_A and from 0.83 to 0.57 cm
3/g for Al
2O
3_B). Al
2O
3_A_900 also shows a capillary condensation branch much less steep than that of Al
2O
3_A_400, indicating a loss of mesoporous order after the treatment. As expected, a decrease in porous order, together with a further lowering of the curves for both samples, is also observed after the functionalization process, associated with a decrease in terms of surface area and pore volume (
Table 1). For Al
2O
3_A and Al
2O
3_B treated at 400 °C, the pore size distribution (PSD) curves, determined with the BJH method (
Figure 2c,d), show similar shapes but a different mean size value, in agreement with the different lengths of the external chains of the triblock copolymers. For both A and B series, a gradual decrease in the mean values of pore diameter and a corresponding widening of the pore size distributions (more noticeable for the B series) were observed in association with both the thermal treatment at 900 °C and the functionalization process. Namely, for the samples obtained with P123, the mean pore diameter decreases from 7.8 nm (Al
2O
3_A_400) to 5.6 nm (Al
2O
3_A_900) up to 5.0 nm (CZZ@Al
2O
3_A); for the samples obtained using F127 the pore diameter decreases from 9.3 nm (Al
2O
3_B_400) to 8.7 (Al
2O
3_B_900) up to 6.7 nm (CZZ@Al
2O
3_B).
Transmission Electron Microscopy (TEM) images evidence that Al
2O
3_A_400 and Al
2O
3_B_400 present an ordered mesoporous structure with a hexagonal arrangement (
Figure 3a,d); this structure was maintained both after the treatment at 900 °C (
Figure 3b,e) and the impregnation process (
Figure 3c,f). The ordered mesoporous structure in all samples is accompanied by the presence of spheroidal particles, clearly visible in the A series of samples (see arrows in
Figure 3a,b), probably responsible for the disorder contribution evidenced by SA-XRD data and nitrogen physisorption. The impregnation process followed by the thermal treatment did not give rise to the formation of large external particles of the active phase, suggesting its homogeneous dispersion inside the mesopores, despite the high weight loading. These data are in good agreement with WA-XRD data, featuring only very broad diffraction bands associated with the dispersed phase.
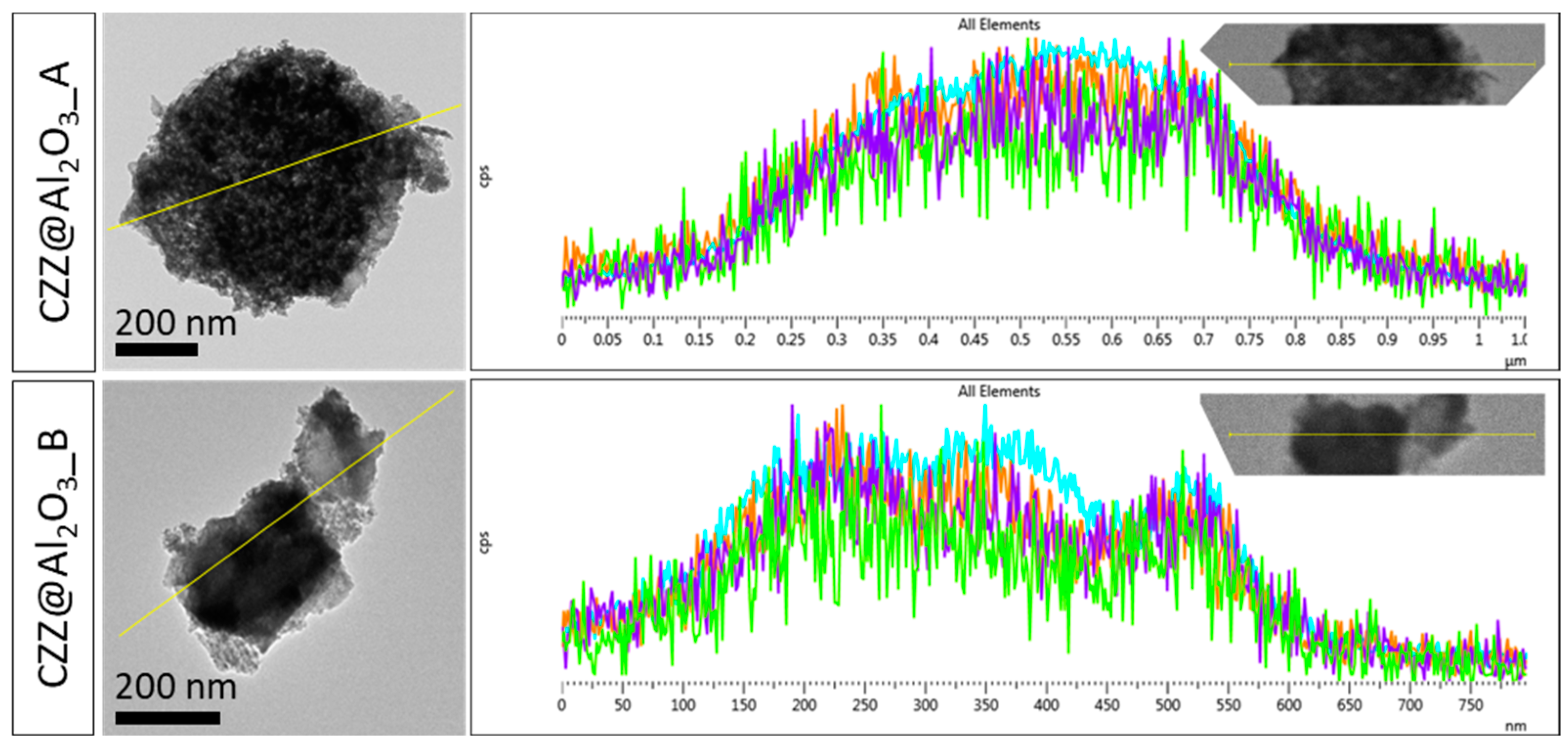
To further confirm the homogeneous nature of the dispersion of the redox phase throughout the support, Energy Dispersive X-ray (EDX) chemical mapping and linear profile analyses were performed. As can be seen from
Figure 4, the chemical mapping points out a homogeneous dispersion of the atomic species (Cu, Zn, and Zr) associated with the redox phase onto the support for both composites. Furthermore, the linear profile analysis (
Figure 5) allows highlighting how the composite obtained on the alumina sample synthesized with P123 (CZZ@Al
2O
3_A) shows a more homogeneous dispersion of the redox phase compared with CZZ@Al
2O
3_B.
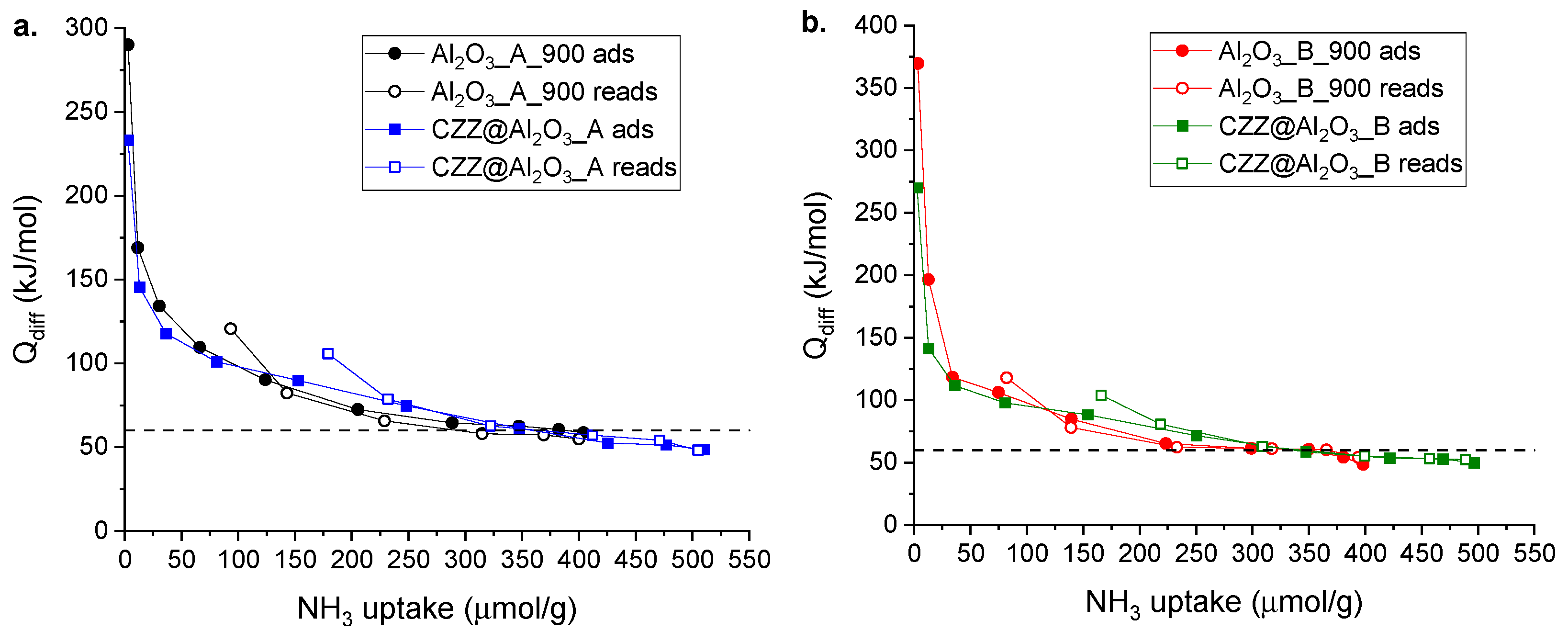
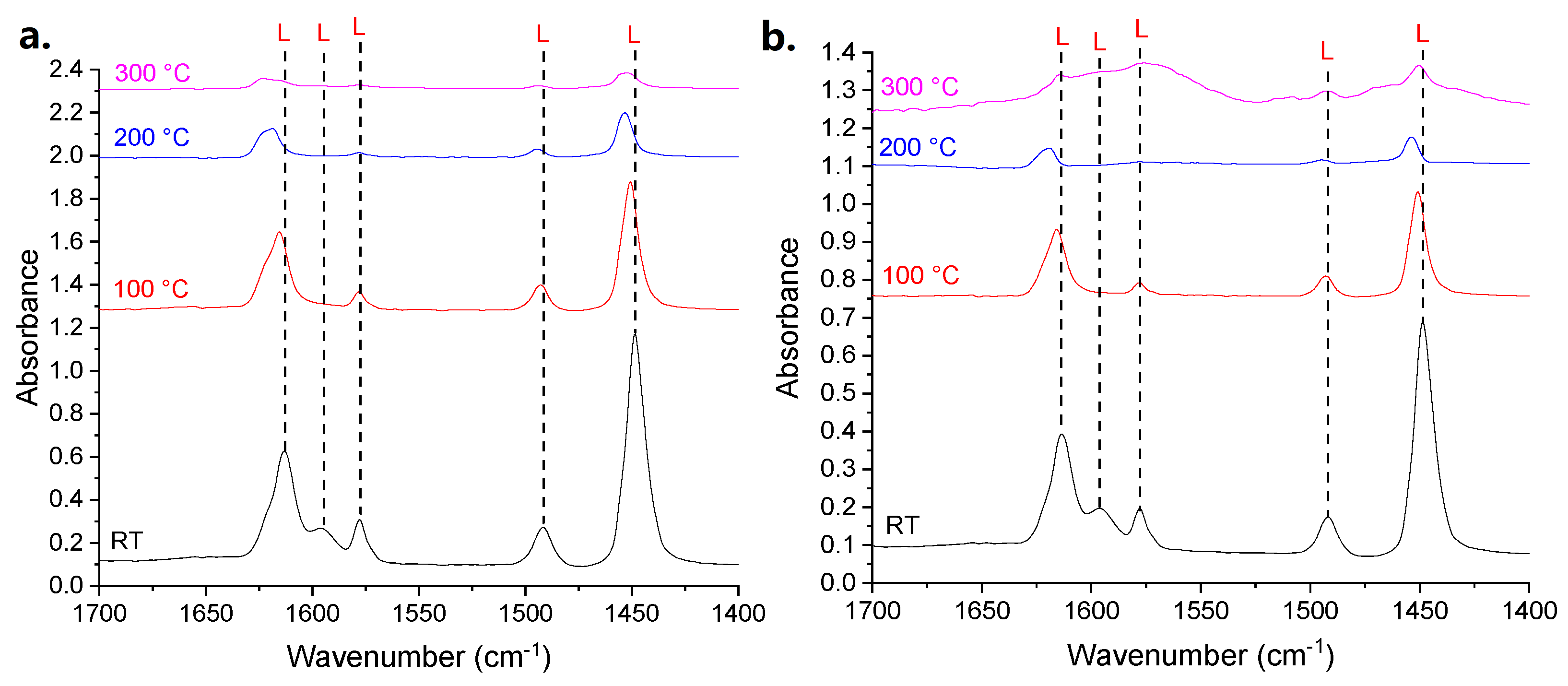
NH
3 adsorption microcalorimetry was used to characterize the surface acid properties of the γ-alumina dehydration catalysts and the results are shown in
Figure 6 and summarized in
Table 2 and
Figure S3. Both the Al
2O
3_A_900 and Al
2O
3_B_900 samples show very high initial values of Q
diff (>>150 kJ/mol, which is generally considered as the threshold value for acid sites of high strength) that indicate the presence of a small amount of very strong sites. However, an abrupt and continuous decrease in Q
diff with the increase in ammonia coverage is then observed, indicating a high heterogeneity of the surface from the energetic point of view. It can be seen from
Table 2 that sample Al
2O
3_B_900 shows slightly lower acidity than that of Al
2O
3_A_900 in terms of the amount of both total sites (n
tot) and sites irreversibly adsorbing ammonia (n
irr). The determined amounts of n
tot are in agreement with those reported in previous works [
35,
36]. The number of acid sites was also determined on the composite catalysts in order to assess the influence of the impregnation route on the acidic properties. As can be seen in
Table 2 and in
Figure S3, the total amount of acid sites showed by composite catalysts is similar to that determined on the corresponding supports. However, a significant difference can be seen in terms of sites on which ammonia is irreversibly adsorbed; indeed, composite catalysts show a higher amount of n
irr, reasonably due to the introduction of the redox phase into the support. The formation of these new strong sites might be attributed to the presence of ZrO
2, an acidic material, in the redox phase. This hypothesis is supported by literature data, in which a higher number of strong and medium acid sites is reported for ZrO
2, compared with γ-Al
2O
3 [
37,
38,
39,
40]. In the case of alumina supports, the nature of the acid sites was studied by Fourier-Transform Infrared Spectroscopy (FT-IR) of adsorbed pyridine (Py-FT-IR); this technique shows the presence of only Lewis sites (
Figure 7), according to previous FT-IR studies by other authors on the adsorption of ammonia [
41,
42] and pyridine [
42,
43,
44] on alumina. In agreement with NH
3-microcalorimetry results, Py-FT-IR points out a higher amount of acid sites for the sample obtained with Pluronic P123 (Al
2O
3_A_900); in this case, however, the difference between the two samples is more remarkable (
Table 3).
Catalytic Performance
The CZZ@Al
2O
3 composites were tested as catalysts for the CO
2 hydrogenation reaction in 35-h runs. Under the used experimental conditions, the main products were methanol, carbon monoxide, and DME, with water being coproduced in all reactions. Light hydrocarbons (methane, ethane, and propane) were also monitored during the reaction but were not detected.
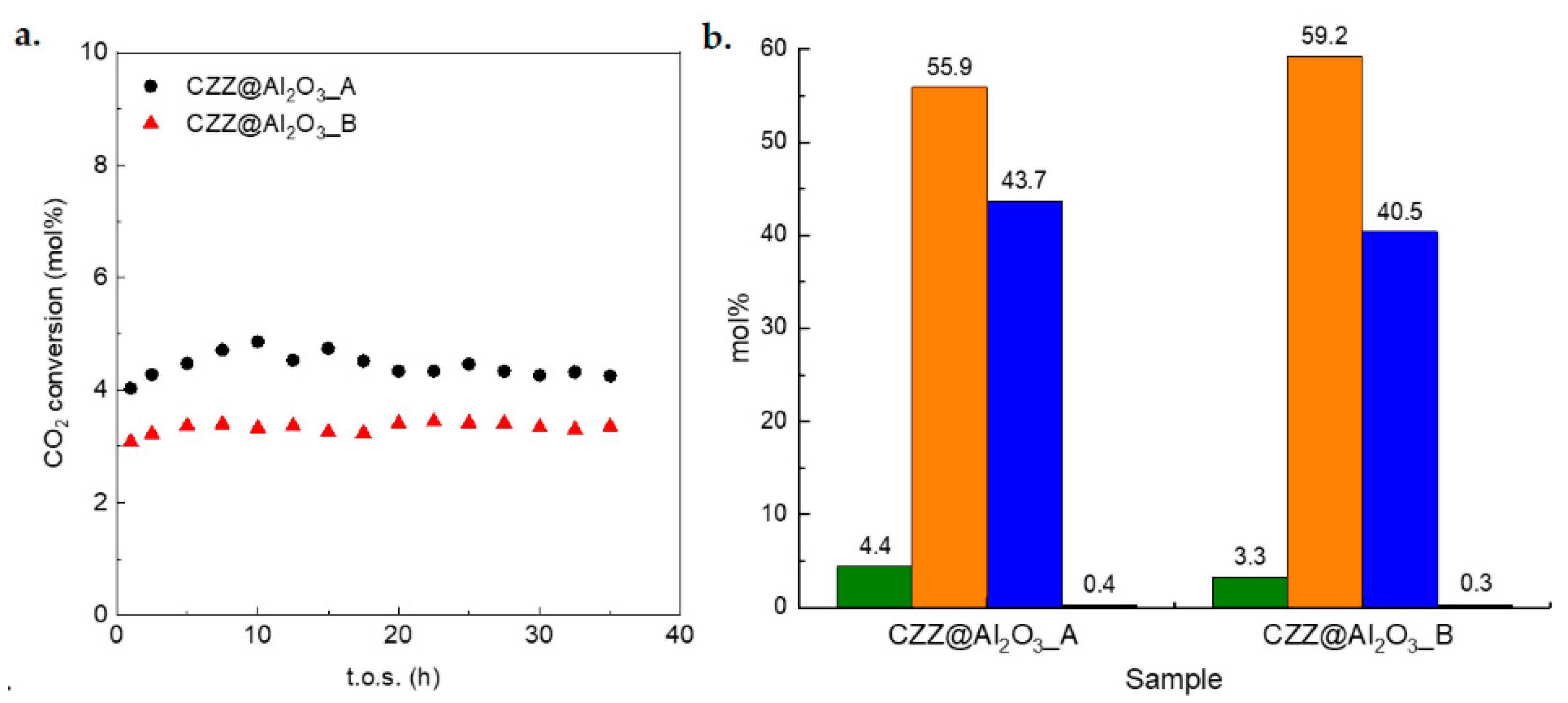
Figure 8a shows the CO
2 conversion as a function of time-on-stream (t.o.s.) for the two composites, which results in being stable within the investigated reaction time. Hence, the average values of CO
2 conversion and selectivity to methanol, DME, and CO were calculated and reported in
Figure 8b to compare their catalytic performances. Methanol and DME yields were also calculated and reported in
Table S1.
It can be observed that the CZZ@Al
2O
3_A catalyst shows a higher conversion (4.4 mol%) and methanol selectivity (44 mol%) compared to CZZ@Al
2O
3_B (
= 3.3 mol% and
= 40 mol%). Despite the same weight loading and the similar textural properties in terms of surface area and pore volume (
Table 1) of the two composites, the different CO
2 conversion could be ascribed to the different pore size and pore size distribution, which can lead to a different dispersion of the redox phase inside the mesochannels. Considering that the size of the mesopores of both the nanocomposites is significantly larger than that of the involved molecules, the difference in pore size should not affect the mass transfer phenomena, which can therefore be neglected. Noteworthy, despite the acidic character of the supports, neither of the two composites is able of dehydrating methanol, as indicated by the formation of small amounts of DME (0.3–0.4 mol%). This finding could be explained by considering that the accessibility of the acidic sites of the mesostructured γ-Al
2O
3 samples is hampered due to the deposition of the CZZ redox phase by the post-synthesis wet impregnation route, thus inhibiting the dehydration reaction. Considering the combined data of XRD, TEM, and nitrogen physisorption, it can be assumed that the CZZ phase is homogeneously dispersed inside the pores as a thin layer rather than as nanoparticles. By observing the NH
3-microcalorimetry data on the supports and on the composite catalysts, it can be assumed that the irreversible acid sites introduced after the functionalization with the redox phase are too strong to be active toward methanol dehydration. Indeed, in previous work [
45], it has been observed that Lewis acid sites with high strength show a lower activity towards methanol dehydration due to their deactivation caused by water adsorption. A small difference in terms of S
DME (0.1 mol%) can be observed between the two composite catalysts and can be reasonably attributed to the different amounts of acid sites determined on the two acidic supports. The small entity of this difference might be ascribed to the coverage of the acid sites mentioned above. Very low performances in terms of DME formation rate were also observed by Bonura et al. [
46] for H-ZSM-5 combined with Cu/ZnO/ZrO
2 by a coprecipitation method; the authors ascribed this behavior to an ion-exchange phenomenon between the zeolite and the cations of the redox phase, which caused the disappearance of the acid sites responsible for methanol dehydration. However, in the present case, ion-exchange phenomena can be excluded since γ-Al
2O
3 only features Lewis acid sites; therefore, the very low DME selectivity value of the composite catalysts can be ascribed to the coverage of the acid sites of the dehydration catalyst. To confirm this hypothesis, the dehydration activity of the most active acid support (Al
2O
3_A) in the CO
2-to-DME conversion was investigated by physically mixing it with the most efficient commercial CZA catalyst. The relative amounts of the two components were suitably chosen to have the same quantity of the Cu/ZnO hydrogenation phase as that loaded in the CZZ@Al
2O
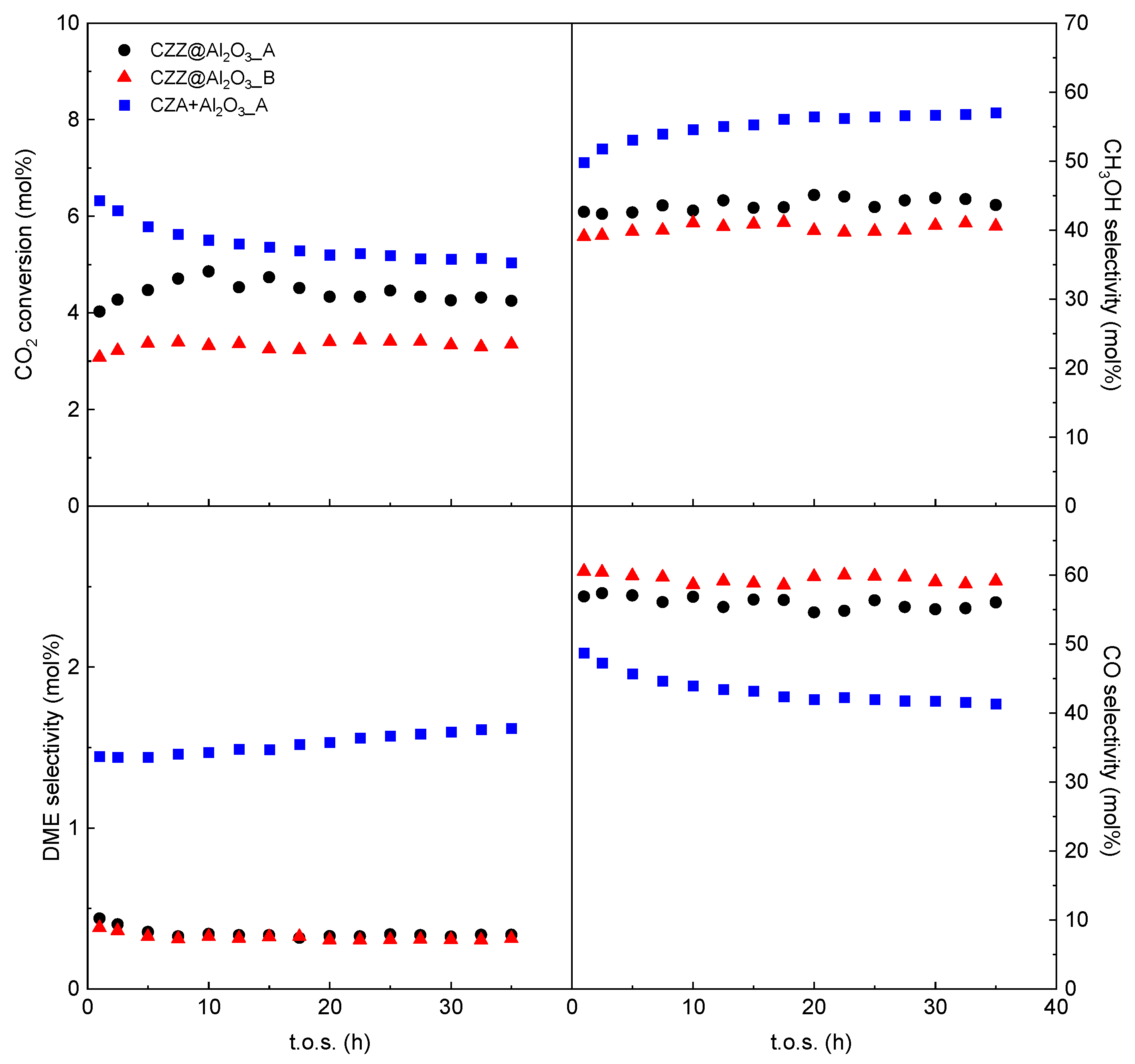
3 composites. The catalytic results are shown in
Figure 9 and compared to those obtained on the composite catalysts.
It is worthy of note that selectivity to DME is increased about five times compared to that of the composites, strongly supporting the assumption that the dehydrating activity of the acidic component in the composite catalysts is almost completely inhibited by the intimate contact of the two phases deriving by the dispersion of the redox phase into the mesochannels as nanolayer. A significant improvement in DME production rate has also been reported by other authors for a CZZ + H-ZSM-5 physical mixture, where the contact between the two phases is minimized, compared to a CZZ@H-ZSM-5 bifunctional catalyst obtained by coprecipitation [
46]. The slightly better performances in terms of CO
2 conversion of the CZA + Al
2O
3_A physical mixture in comparison with the composites (
Figure 9) could be ascribed to the different activity of the CZA redox phase and the CZZ one in the CO
2 hydrogenation reaction [
47]. In addition, the substantial difference in terms of methanol selectivity between the physical mixture CZA + Al
2O
3_A (
ca. 55 mol%) and the two composites (43.7 mol% for CZZ@Al
2O
3_A and 40.5 mol% for CZZ@Al
2O
3_B) can be almost exclusively ascribed to a different catalytic activity of the two redox phases (CZA vs. CZZ) and/or to their two different mixing approaches with the dehydration phase. Considering the higher DME selectivity of the physical mixture, a lower methanol selectivity would be expected; however, CH
3OH selectivity results in being higher, thus excluding an influence of the dehydration phase. It can be then assumed that the higher S
CH3OH for the physical mixture is caused by the lower activity of the CZA catalyst towards the RWGS reaction, which, on the other hand, seems to be predominant for the CZZ phase, causing a significantly higher S
CO for the composites.
Interestingly, at variance with the CZZ@Al
2O
3 catalysts, a slight deactivation with t.o.s. is observed for the physical mixture. Deactivation phenomena can be ascribed to the presence of water formed during the whole CO
2-to-DME process. According to the literature, water can be adsorbed on the metallic Cu sites of the redox catalyst, causing re-oxidation and sintering phenomena [
4], and on the Lewis acid sites of γ-Al
2O
3, which can be progressively deactivated by the formation of an acid-base adduct with water [
4,
10]. In the latter case, the amount of formed methanol that is dehydrated to DME should decrease; however, in the present study, an increase in both S
CH3OH and S
DME is observed, together with a decrease in S
CO. The obtained results suggest that deactivation is mainly related to the CZA catalyst, associated with a decreased contribution of RWGS.
Regarding the data reported in the literature for similar systems, a direct comparison between the systems developed and tested in this work and other γ-Al
2O
3-based catalysts proposed in the literature may be difficult due to the remarkable differences in terms of reactions conditions, such as GHSV, redox/acid catalysts ratio, pressure, temperature, and type of reactor. Nevertheless, regarding the CO
2-to-DME reaction performed on physical mixtures consisting of a redox catalyst mixed with γ-Al
2O
3, it appears that γ-Al
2O
3 does not show promising performances in terms of DME selectivity, if compared with other acidic catalysts such as zeolites, due to the deactivation of its Lewis acid sites caused by water adsorption. In this context, Naik et al. [
26] tested two different physical mixtures consisting of a redox catalyst (CuO/ZnO/Al
2O
3) mixed with γ-Al
2O
3 and H-ZSM-5, respectively. The physical mixture featuring γ-Al
2O
3 as a dehydration catalyst showed a very low DME yield (0.4%); this value is higher than those obtained by us (0.02% for CZZ@Al
2O
3_A; 0.08 for CZA + Al
2O
3_A), but it must be considered that this is most likely due to the significantly lower value of GHSV used by Naik et al. (3000 NmL/g·h) compared to ours (48,000 NmL/g·h). Concerning bifunctional catalysts, interesting results were obtained on non-mesoporous alumina by da Silva et al. [
25]. The authors compared two bifunctional catalysts obtained by impregnating γ-Al
2O
3 with a CuO/ZnO redox phase using either precipitation or a wet impregnation method. The catalyst obtained through precipitation did not show any dehydration activity; on the other hand, at the same temperature and pressure values of this work (250 °C, 30 bar), the impregnated system showed a DME selectivity of 35%. In addition, in this case, however, the significantly lower GHSV used by the authors (12,000 NmL/g·h) and the higher relative amount of acidic phase (6.9 wt% of redox phase loading) make a direct comparison in terms of DME selectivity difficult.
As for composite catalysts based on mesostructured γ-Al
2O
3, to the best of our knowledge, only four papers report on this type of system; however, three of them deal with the DME synthesis from syngas, and only one regards the CO
2-to-DME process [
31]. The Cu/γ-Al
2O
3 composite catalyst was tested at two different GHSV values, namely 2000 and 4000 NmL/g·h. The test carried out at 2000 NmL/g·h showed a DME selectivity of 12.6% but at GHSV = 4000 NmL/g·h, its value dropped to about 2%, indicating a strong influence of GHSV on product selectivity and thus explaining the lower selectivity values obtained on our samples using a much higher GHSV value (48,000 NmL/g·h).
3. Materials and Methods
3.1. Chemicals
The following chemicals were used as received without further purification: PEG20-PPG70-PEG20 (Pluronic P123) average Mn ~ 5800 (Aldrich Chemistry, St. Louis, MO, USA), PEG101-PPG56-PEG101 (Pluronic F127) (Sigma Life Science St. Louis, MO, USA), aluminum isopropoxide > 98% (Alfa Aesar, Kandel, Germany), nitric acid (HNO3) ≥ 65% (Honeywell Fluka, Muskegon, MI, USA), copper nitrate hemi-pentahydrate Cu(NO3)2·2.5H2O 98% (Alfa Aesar, Kandel, Germany), zinc nitrate hexahydrate Zn(NO3)2·6H2O 98% (Sigma-Aldrich, St. Louis, MO, USA), zirconium(IV) oxynitrate ZrO(NO3)2 99% (Aldrich Chemistry, St. Louis, MO, USA), absolute ethanol (Honeywell Fluka, Muskegon, MI, USA), copper-based methanol synthesis catalyst (CZA, Alfa Aesar, Kandel, Germany).
3.2. Synthesis of Mesostructured Supports
Two samples of Al
2O
3 were synthesized through an adapted EISA method [
48], using two different triblock copolymers, namely P123 (Al
2O
3_A) and F127 (Al
2O
3_B), in order to induce the formation of a mesostructure. Typically, 1.5 g of mesostructured Al
2O
3 can be obtained by dissolving 3 g of triblock copolymer (either P123 or F127) in 75 mL of absolute ethanol into a flask under continuous stirring at room temperature. Then, 4.8 mL of HNO
3 and 6.24 g of aluminum isopropoxide were added after 120 min, and the resulting solution was maintained under stirring at room temperature for one day. The mixture was then put into a Petri dish inside a controlled-humidity chamber (H ≈ 20%) on a heating plate at 70 °C in order to promote the self-assembly process by controlled evaporation. After two days, the gel was first treated at 400 °C in static air for 4 h with a 1 °C/min ramp (Al
2O
3_A_400; Al
2O
3_B_400) to induce the decomposition of the templating agent and then at 900 °C for 2 h with a 1 °C/min ramp (Al
2O
3_A_900; Al
2O
3_B_900) to convert the amorphous phase into γ-Al
2O
3 crystalline phase, which shows an acidic character [
4,
5,
10].
3.3. Synthesis of Mesostructured Composites
Al2O3_A_900 and Al2O3_B_900 were used as supports to be impregnated with CuO/ZnO/ZrO2-redox phase with a molar ratio of 2/1/1.3 via a post-synthesis impregnation route based on self-combustion. The two supports were impregnated with a weight load of 20 wt% of the active phase; it must be taken into account that the active phase of the redox catalyst is only composed of CuO/ZnO, so the weight of ZrO2 was not taken into the calculation of the weight load of redox catalyst. The total load, including ZrO2, reaches 33 wt%.
Typically, an aqueous solution of Cu(NO
3)
2·2.5H
2O, Zn(NO
3)
2·6H
2O, and ZrO(NO
3)
2 with a total metal ion concentration of 0.894 mol/L was prepared and mixed with a second solution of glycine, keeping a glycine/metal ions molar ratio of 1.44. 1 g of support, previously dried at 120 °C overnight, was then dispersed into 10 mL of the metal nitrates/glycine solution into a beaker under constant vigorous stirring up to almost complete evaporation of the solvent. The resulting gel was sonicated for 5 min and then submitted to direct treatment at 300 °C by putting it into a preheated oven in order to induce the self-combustion process between the nitrates (oxidizers) and glycine (reducing agent) [
49,
50,
51,
52,
53]. The catalysts discussed here are hereinafter labeled as CZZ@Al
2O
3_A and CZZ@Al
2O
3_B, where CZZ indicates the redox phase.
3.4. Characterization Techniques
Small-angle (SA-XRD, 2θ = 0.7–6°) X-ray diffraction patterns were recorded on a Seifert X3000 instrument (Seifert, Radevormwald, Germany) with a θ−θ geometry and a Cu anode; wide-angle (WA-XRD, 2θ = 15–80°) X-ray diffraction patterns were recorded using a PANalytical X’pert Pro (Malvern PANalytical, Malvern, UK) equipped with Cu Kα radiation (1.5418 Å). The lattice parameter was calculated using the equation
[
54,
55,
56]. Rietveld refinement was carried out on the XRD pattern of γ-Al
2O
3 using the software MAUD [
57]. LaB
6 from NIST was used as a standard reference for determining the instrumental parameters. The CIF structure used for the refinement was 1200015 from Crystallography Open Database [
58].
Textural analyses were performed on a Micromeritics ASAP 2020 system (Micromeritics, Norcross, GA, USA) by determining the nitrogen adsorption–desorption isotherms at −196 °C. Prior to the analyses, all samples were heated for 12 h under a vacuum at 250 °C (heating rate, 1 °C min−1). The Brunauer-Emmett-Teller (BET) specific Surface Area (SA) was calculated from the adsorption data in the P/P0 range 0.05–0.25. The total pore volume (Vp) was calculated at P/P0 = 0.9975, while the mean pore diameter (Dp) was determined by applying the Barrett-Joyner-Halenda (BJH) model to the desorption branch isotherm.
Transmission Electron Microscopy (TEM) micrographs and Energy Dispersive X-ray (EDX) analyses were obtained on a JEOL JEM 1400-PLUS microscope (JEOL, Akishima, Tokyo, Japan) operating at an accelerating voltage of 120 kV. Finely ground powders of the samples were first dispersed in ethanol and sonicated. The resulting suspensions were dropped onto 200 mesh carbon-coated copper grids.
A Tian-Calvet heat flow calorimeter (Setaram, Caluire-et-Cuire, France) equipped with a volumetric vacuum line was used for the microcalorimetric measurements. Samples (about 100 mg) were pretreated overnight at 300 °C under vacuum (1 Pa) prior to the successive introduction of small doses of the probe gas (ammonia). The equilibrium pressure relative to each adsorbed amount was measured by means of a differential pressure gauge (Datametrics, Bari, Italia) and the thermal effect recorded. The run was stopped at a final equilibrium pressure of ca. 133 Pa. The adsorption temperature was maintained at 80 °C in order to limit physisorption. After overnight outgassing at the same temperature, a second run was carried out. The adsorption and calorimetric isotherms were obtained from each adsorption run. The adsorption isotherms relate the amount of probe gas with the corresponding equilibrium pressure. The overall uptake of the probe gas on the solid was assessed from the first isotherm; the amount of the probe gas irreversibly adsorbed was calculated by subtracting the second isotherm, obtained after outgassing the sample, from the first one. The calorimetric isotherms relate the integral heat of adsorption with the corresponding equilibrium pressure. Combining the adsorption and calorimetric data, a plot of the differential heat of adsorption (Q
diff) as a function of the adsorbed amount was drawn, which gave information on the influence of the surface coverage on the energetics of the adsorption. Typically, the heat released during physisorption is two or three times as high as the condensation heat of the adsorbing molecule [
59], which, in the case of ammonia, is 20.2 kJ/mol at 80 °C [
60]. Accordingly, a Q
diff value of 60 kJ/mol (i.e., three times the NH
3 condensation heat) has been considered as the cut-off between specific and non-specific (physisorption) adsorbent/adsorbate interactions. Hence the fraction of ammonia uptake corresponding to differential heats below this value has been neglected in the assessment of the total acid site concentration.
To assess the nature of the acid sites, Fourier Transform Infrared spectroscopy (FT-IR) analyses were performed using pyridine as a probe molecule (Py-FT-IR). FT-IR spectra were acquired using a Nicolet iS50 spectrometer manufactured by Thermo Fisher Scientific, Waltham, MA, USA and equipped with a custom-made glass cell. The cell was evacuated (<1.3·10
−3 Pa) using a rotative and a turbomolecular pump. More detailed information on the experimental setup was provided in previous work [
45]. The FT-IR spectra were recorded between 1700 cm
−1 and 1400 cm
−1. Each sample was pressed into circular self-supported pellets with a diameter of 13 mm before the analysis. The obtained pellet was then inserted into the cell and heated at 250 °C (7.5 °C/min) for 1 h under high vacuum in order to completely desorb water molecules. The sample, kept under high vacuum, was subsequently moved to the measurement position, and its spectrum was acquired as a background. The cell was then saturated with pyridine, allowing it to reach a pressure of about 267 Pa with pyridine vapor and keeping these conditions for 10 min. The cell was subsequently evacuated at RT, and the spectrum was acquired. The analysis was then repeated by treating the sample at various temperatures (100 °C, 200 °C, 300 °C) under high vacuum. The area under the IR signals associated with each type of acid site was estimated to determine the number of acid sites still occupied by pyridine at each temperature. Specifically, the band at about 1455 cm
−1 was used to quantify the number of Lewis acid sites, using an Integrated Molar Extinction Coefficient (IMEC) of 2.22 cm/µmol; whereas the band at about 1545 cm
−1 was used for the Brønsted acid sites, with an IMEC of 1.67 cm/µmol [
61].
3.5. Catalytic Tests
The DME synthesis experiments were carried out in a customized Microactivity Effi (PID Eng&Tech, Madrid, Spain) bench-scale plant, employing a high-pressure fixed-bed stainless steel reactor (length 304.8 mm, inner diameter 9.1 mm). A porous plate (made of Hastelloy C, 20 μm) and quartz wool were used to support the catalytic bed inside the isothermal temperature zone of the reactor. For the catalytic tests on bifunctional composites, 0.25 g of catalyst were used. The test of Al2O3_A_900 as a dehydration catalyst was performed using a physical mixture made up of 0.05 g of the commercial Cu-based redox catalyst (CZA) and 0.2 g of the dehydration catalyst. The obtained catalytic systems were diluted with 3.2 g of α-Al2O3, a chemically inert material, in order to reach a total bed volume of ca. 3 cm3. As a result, keeping the inlet flow rate constant, the Gas Hourly Space Velocity (GHSV) was 48,000 Ncm3 gcat−1 h−1. To rule out possible effects of the inert material, a blank test was performed with α-Al2O3, highlighting the lack of any catalytic activity.
Before the catalytic tests, all fresh catalysts were reduced in situ in a stream of an H
2/N
2 mixture (H
2, 15 vol% in N
2). Composite catalysts were reduced at 300 °C, and the physical mixture was reduced at 250 °C; both reductions were carried out for 2 h under atmospheric pressure. Upon completion of the reduction process, the system was brought at 250 °C, and the reaction gas mixture containing H
2 and CO
2 (molar ratio of 3:1) and 10 vol% of N
2 (used as the internal standard for gas chromatographic analysis) was fed, and the pressure was allowed to reach 3.0 MPa. After allowing the system to reach the steady state in 1 h on stream, analysis was periodically performed within the run. Runs were carried out for at least 36 h. The reaction stream was analyzed by a 7890B (Agilent Technologies, Santa Clara, CA, USA) gas chromatograph equipped with a Flame-Ionized Detector (FID) for carbon-containing compounds and with a Thermal Conductivity Detector (TCD) for permanent gases. Two columns connected in series were used to identify the components of the outlet gas mixture. In particular, CO
2, methanol, DME, ethane, and propane were separated by an HP-PLOT Q (Agilent Technologies, Santa Clara, CA, USA) column (length 30 m, inner diameter 0.53 mm, film thickness 40 μm), while an HP-PLOT Molesieve (Agilent Technologies, Santa Clara, CA, USA) column (length 30 m, inner diameter 0.53 mm, film thickness 50 μm) was used for H
2, N
2, CH
4, and CO. To avoid condensation of condensable products, the connection lines between the plant gas outlet and gas chromatograph inlet were heated at 180 °C. CO
2 conversion (
XCO2), products selectivity (
SP, with P: CH
3OH, DME, or CO), and products yield (
YP, with P: CH
3OH or DME) were calculated as follows:
where
i,in and
i,out are the molar flow rates of the
i-th species in the feed or in the gas mixture exiting from the reactor, respectively, and
νi is the stoichiometric coefficient of the
i-th species in the corresponding balanced equation.
In order to assess the error associated with the catalytic tests, a catalytic run using commercial catalysts was performed three times, obtaining a relative standard deviation in the 2–5% range for both conversion and selectivity [
62].
4. Conclusions
In this work, two mesostructured γ-Al2O3 samples obtained with an EISA approach using two different templating agents (Pluronic P123 and F127) were synthesized, characterized, and used as supports to develop bifunctional composite catalysts for the CO2-to-DME process. The mesostructured supports were functionalized with a Cu/ZnO/ZrO2 redox phase using an impregnation method modified with a self-combustion reaction to obtain the composite catalysts, which were tested for the CO2-to-DME reaction. The main findings are reported below:
(i) the self-combustion impregnation was demonstrated to be an effective method to create a homogeneous dispersion of the redox phase in the form of nanoparticles on mesostructured supports (nanocomposites). The absence of diffraction peaks attributable to the redox phase and the preservation of the mesostructure in the composites suggest that the redox phase has been homogeneously dispersed inside the pores. This finding has also been confirmed by TEM micrographs, which pointed out the absence of particles outside the mesopores of the support.
(ii) both the composite catalysts show low values of DME selectivity (0.3–0.4 mol%), which have been ascribed to the coverage of the acid sites deriving from the homogeneous dispersion of the redox phase as a thin layer rather than in the form of nanoparticles. This assumption is confirmed by the test of Al2O3_A_900 in the form of a physical mixture with the commercial redox catalyst CZA (CZA + Al2O3_A), which shows a significantly higher DME selectivity (1.7 mol%) than the corresponding composite. This suggests that, in the case of a physical mixture, the minor contact between the two phases preserves the accessibility of the acid sites of γ-Al2O3 and, consequently, their dehydration activity.
In the perspective of developing new efficient catalysts for the one-pot CO2-to-DME process, future studies may focus on the optimization of the weight loading of the redox phase in bifunctional composite catalysts; a smaller amount of redox phase could indeed lead to a lower coverage degree of the support, allowing to design bifunctional composites with accessible acid sites. Furthermore, the accessibility of the acid sites could be improved by using different impregnation strategies to modify the size and morphology of the redox phase and, thus, its contact with the dehydration catalyst.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}