H2 Thermal Desorption Spectra on Pt(111): A Density Functional Theory and Kinetic Monte Carlo Simulation Study

Abstract
:1. Introduction
2. Results and Discussion
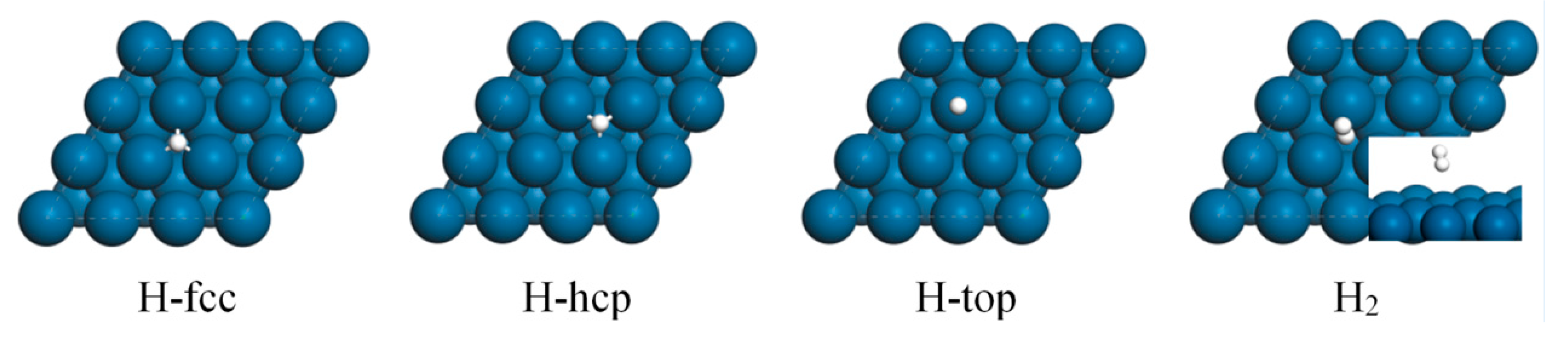
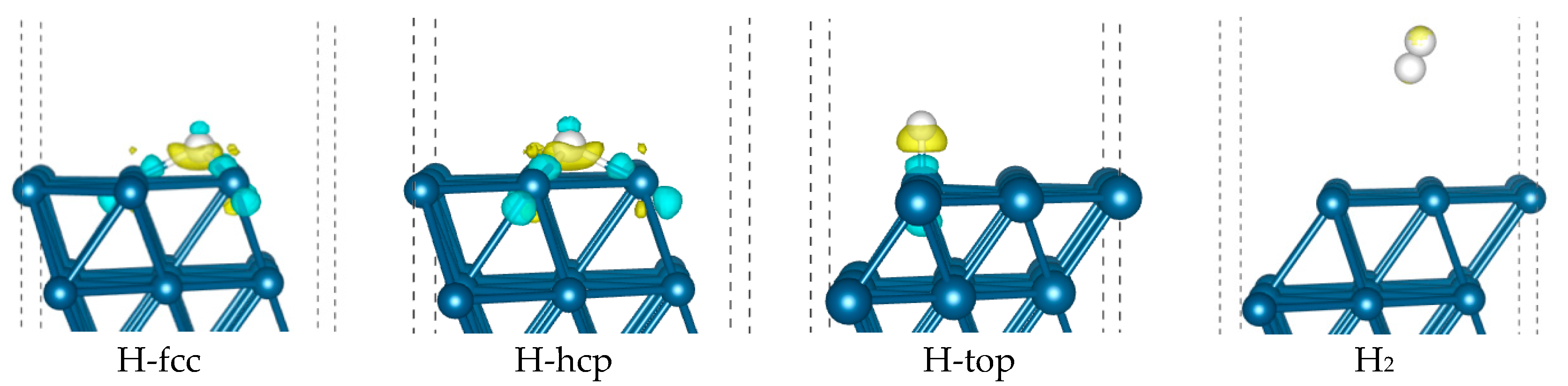
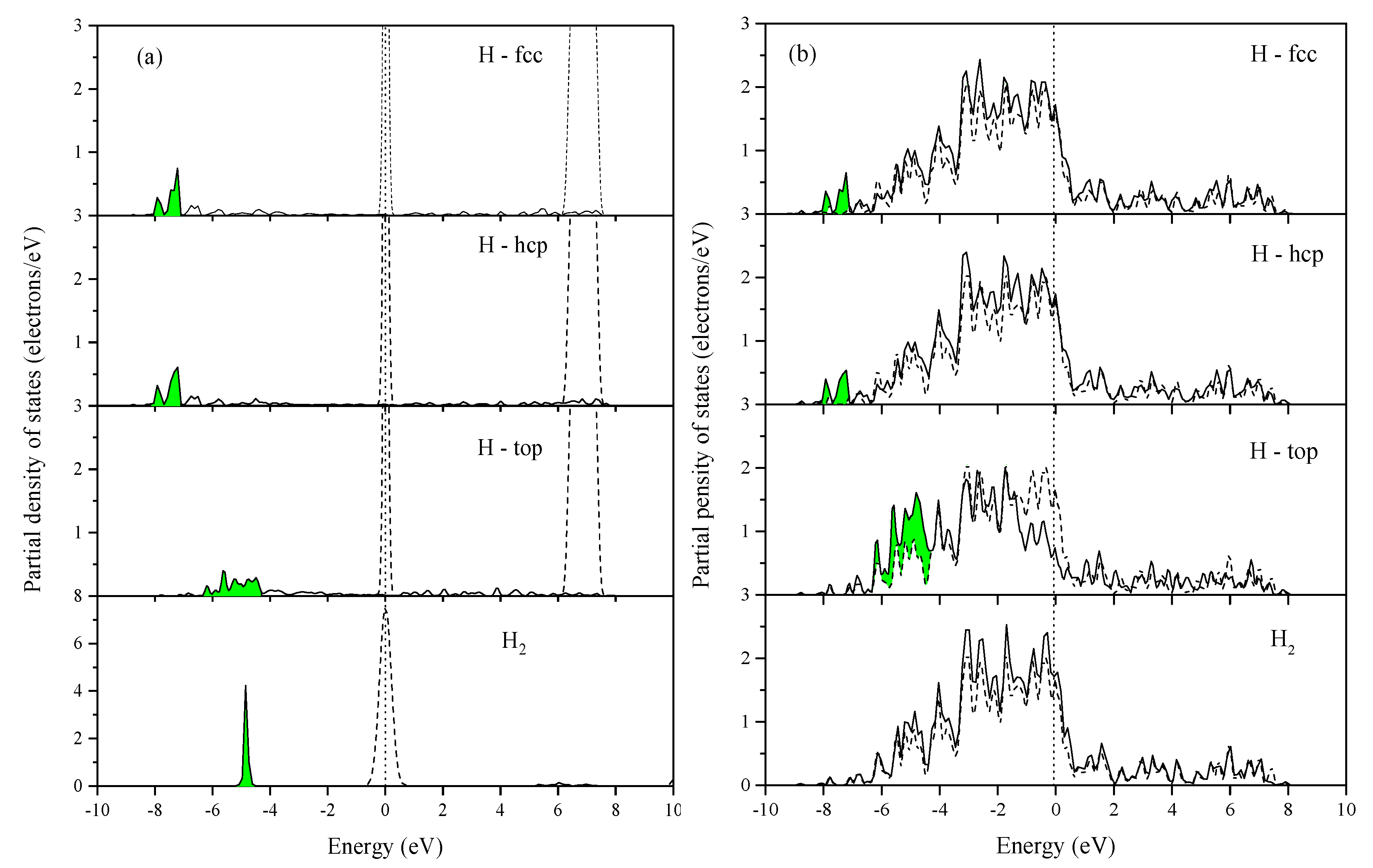
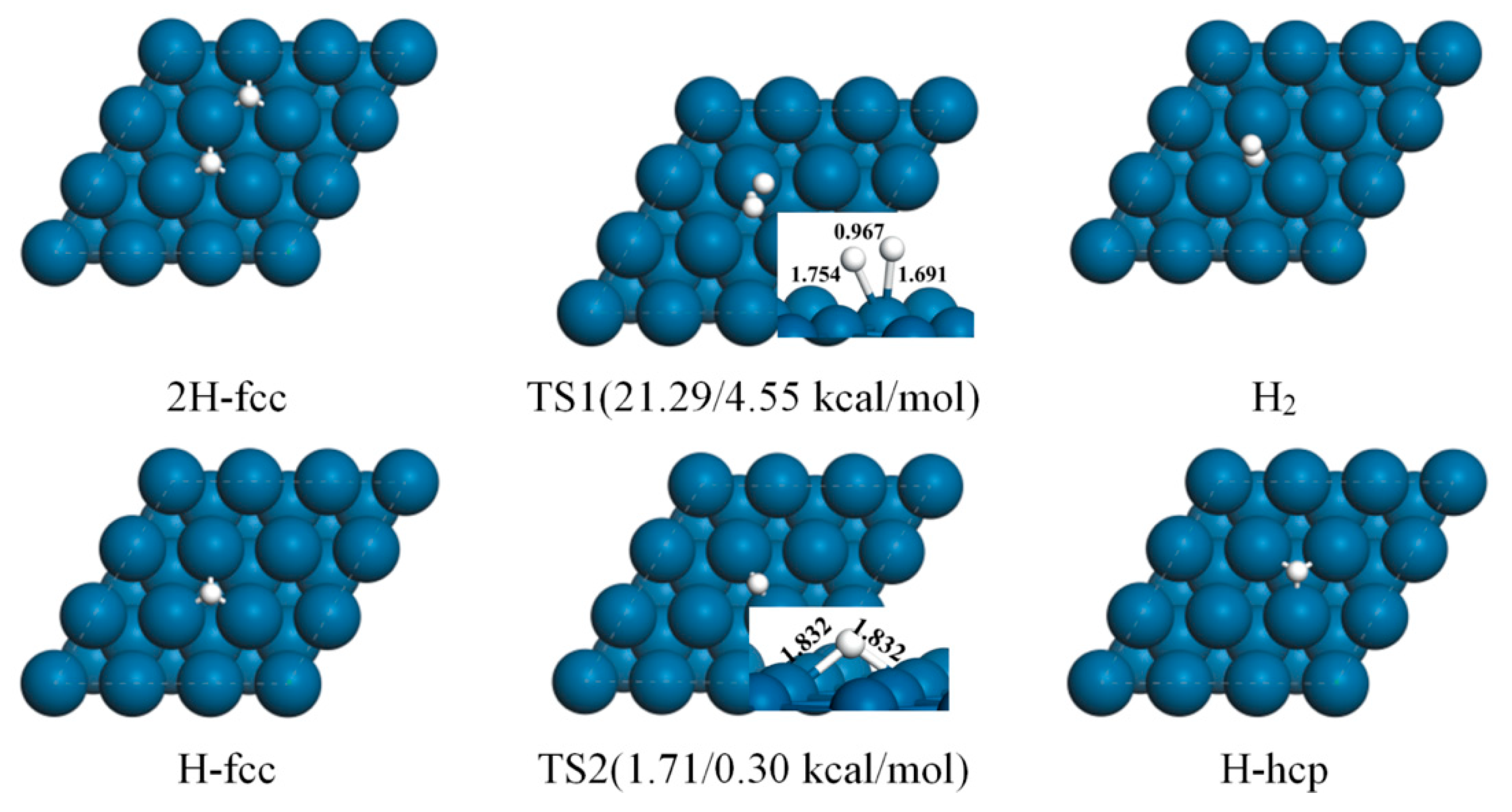
2.1. DFT Study of H2 and H on Pt(111)
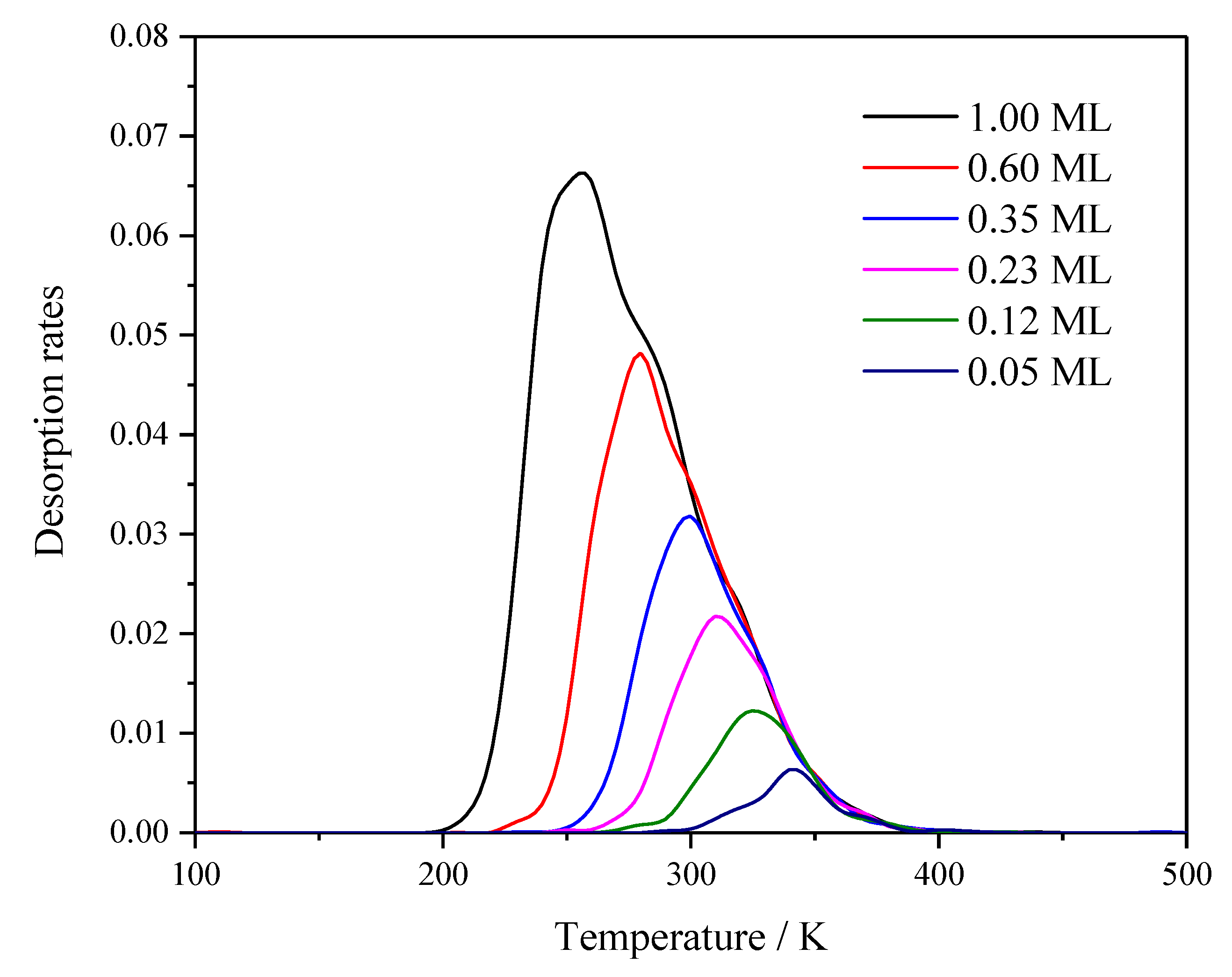
2.2. kMC Simulation of H2 TDS on Pt(111)
- (1)
- H adatom diffusion: H(*) + * → * + H(*)
- (2)
- H2 desorption: 2H(*) → H2(g) + 2*
- (3)
- H2 dissociation: H2(g) + 2* → 2H(*)
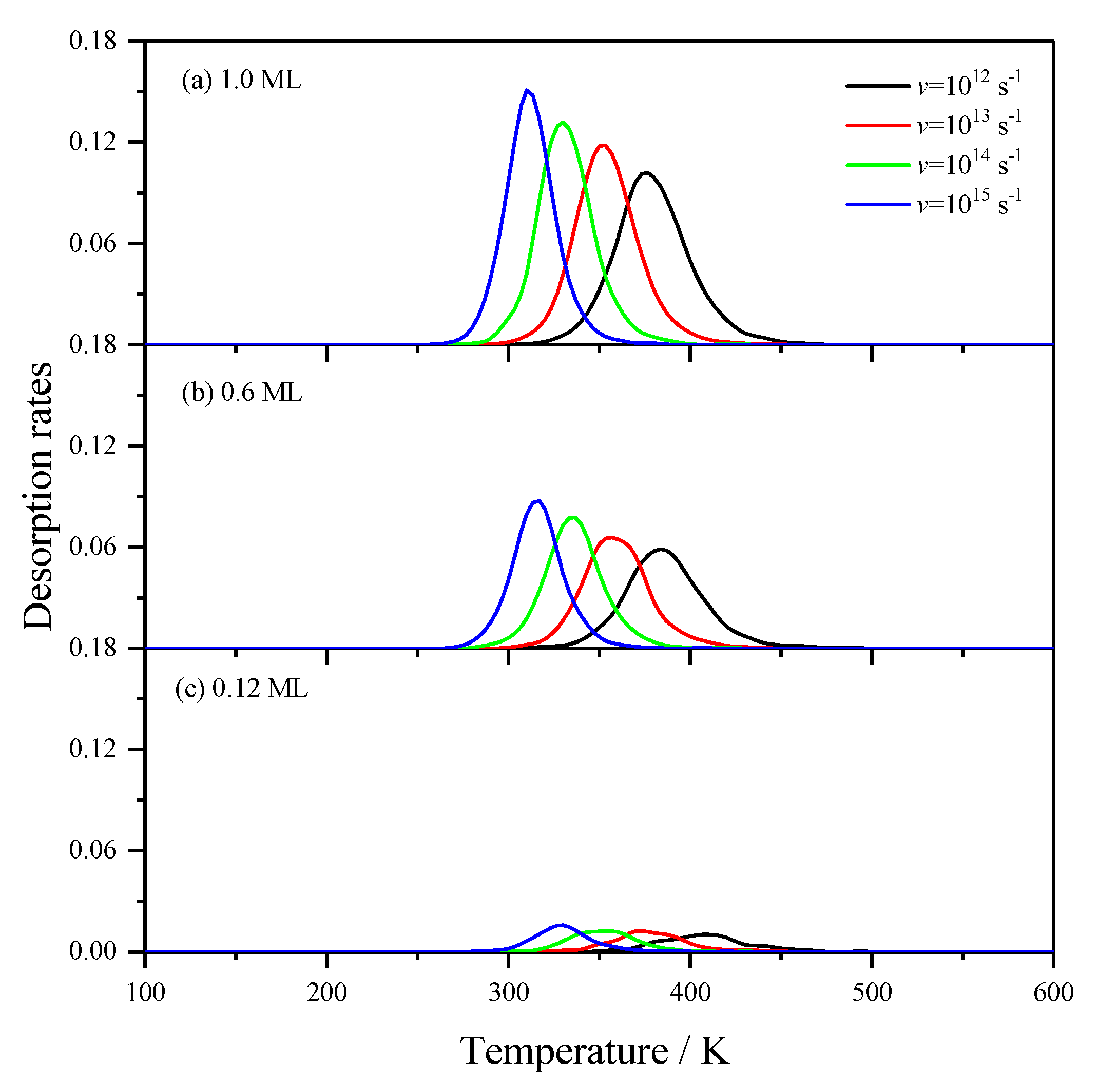
2.2.1. Effects of the Pre-Exponential Factor of H2 Desorption
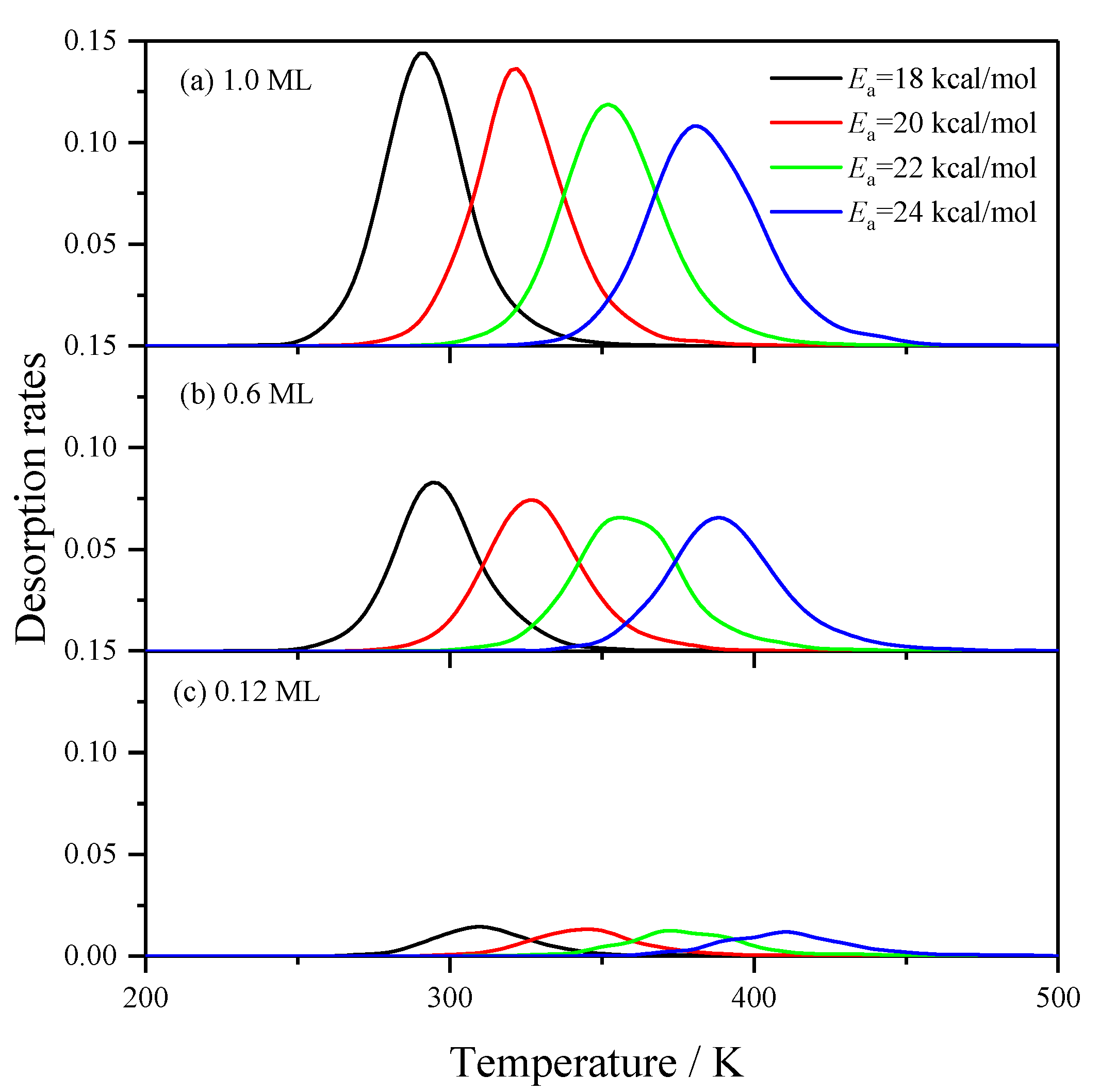
2.2.2. Effects of the Desorption Barrier of H2 Desorption
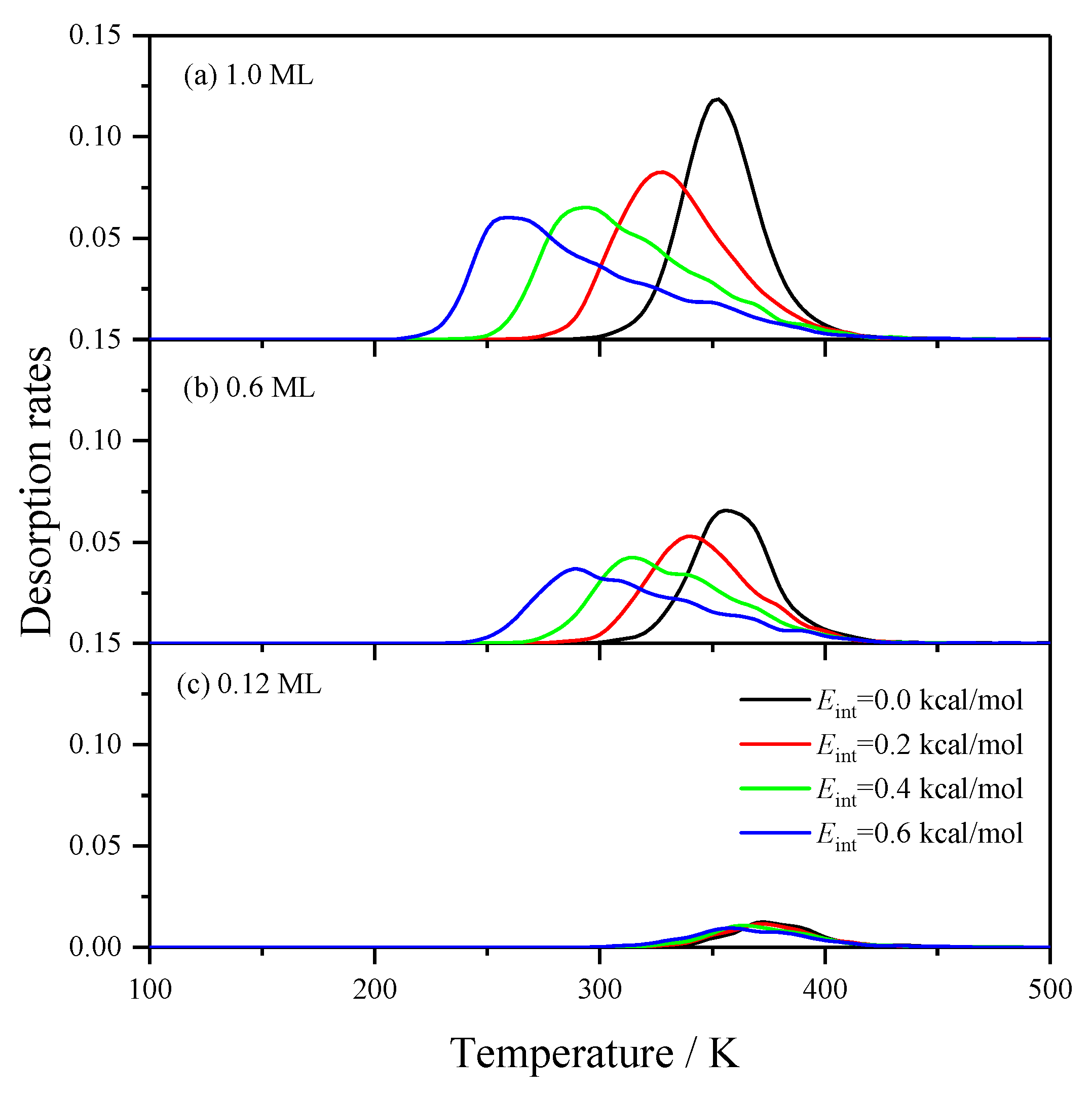
2.2.3. Effects of the Lateral Interactions among H(a)
2.3. kMC Simulation of H2 TDS on Pt(111) Based on DFT Calculation
2.3.1. Parameters Calculated by DFT Method
2.3.2. kMC Simulation Based on DFT Calculation
3. Model and Method
3.1. DFT Calculation Details
3.2. kMC Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Baxter, E.T.; Ha, M.A.; Cass, A.C.; Alexandrova, A.N.; Anderson, S.L. Ethylene Dehydrogenation on Pt4,7,8 Clusters on Al2O3: Strong Cluster Size Dependence Linked to Preferred Catalyst Morphologies. ACS Catal. 2017, 7, 3322–3335. [Google Scholar] [CrossRef]
- An, W.; Men, Y.; Wang, J.G. Comparative study on hydrogenation of propanal on Ni(111) and Cu(111) from density functional theory. Appl. Surf. Sci. 2017, 394, 333–339. [Google Scholar] [CrossRef]
- Liu, Y.L.; Liu, J.; Feng, G.; Yin, S.; Cen, W.L.; Liu, Y.J. Interface effects for the hydrogenation of CO2 on Pt4/γ-Al2O3. Appl. Surf. Sci. 2016, 386, 196–201. [Google Scholar] [CrossRef]
- Gonda, M.; Ohshima, M.A.; Kurokawa, H.; Miura, H. Toluene hydrogenation over Pd and Pt catalysts as a model hydrogen storage process using low grade hydrogen containing catalyst inhibitors. Int. J. Hydrogen Energy 2014, 39, 16339–16346. [Google Scholar] [CrossRef]
- Shen, W.; Wu, B.; Liao, F.; Jiang, B.; Shao, M. Optimizing the hydrogen evolution reaction by shrinking Pt amount in Pt-Ag/SiNW nanocomposites. Int. J. Hydrogen Energy 2017, 42, 15024–15030. [Google Scholar] [CrossRef]
- Verdinelli, V.; Germán, E.; Jasen, P.; González, E.; Marchetti, J.M. The influence of pre-adsorbed Pt on hydrogen adsorption on B2 FeTi(111). Int. J. Hydrogen Energy 2014, 39, 8621–8630. [Google Scholar] [CrossRef]
- Esan, D.A.; Ren, Y.; Feng, X.; Trenary, M. Adsorption and Hydrogenation of Acrolein on Ru(001). J. Phys. Chem. C 2017, 121, 4384–4392. [Google Scholar] [CrossRef]
- Julkapli, N.M.; Bagheri, S. Graphene supported heterogeneous catalysts: An overview. Int. J. Hydrogen Energy 2015, 40, 948–979. [Google Scholar] [CrossRef]
- Jung, H.; Park, K.T.; Gueye, M.N.; So, S.H.; Park, C.R. Bio-inspired graphene foam decorated with Pt nanoparticles for hydrogen storage at room temperature. Int. J. Hydrogen Energy 2016, 41, 5019–5027. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, Z.; Yang, C.; Wang, M.; Ma, X. Hydrogen generation from water molecule with Pt7 clusters. Int. J. Hydrogen Energy 2017, 42, 4032–4039. [Google Scholar] [CrossRef]
- Avanesian, T.; Gusmão, G.S.; Christopher, P. Mechanism of CO2 reduction by H2 on Ru(0001) and general selectivity descriptors for late-transition metal catalysts. J. Catal. 2016, 343, 86–96. [Google Scholar] [CrossRef]
- Clay, J.P.; Greeley, J.P.; Ribeiro, F.H.; Nicholas Delgass, W.; Schneider, W.F. DFT comparison of intrinsic WGS kinetics over Pd and Pt. J. Catal. 2014, 320, 106–117. [Google Scholar] [CrossRef]
- Liu, D.; Li, G.F.; Yang, F.F.; Wang, H.; Han, J.Y.; Zhu, X.L.; Ge, Q.F. Competition and Cooperation of Hydrogenation and Deoxygenation Reactions during Hydrodeoxygenation of Phenol on Pt(111). J. Phys. Chem. C 2017, 121, 12249–12260. [Google Scholar] [CrossRef]
- Stamatakis, M.; Chen, Y.; Vlachos, D.G. First-Principles-Based Kinetic Monte Carlo Simulation of the Structure Sensitivity of the Water–Gas Shift Reaction on Platinum Surfaces. J. Phys. Chem. C 2011, 115, 24750–24762. [Google Scholar] [CrossRef]
- Taylor, M.J.; Jiang, L.; Reichert, J.; Papageorgiou, A.C.; Beaumont, S.K.; Wilson, K.; Lee, A.F.; Barth, J.V.; Kyriakou, G. Catalytic Hydrogenation and Hydrodeoxygenation of Furfural over Pt(111): A Model System for the Rational Design and Operation of Practical Biomass Conversion Catalysts. J. Phys. Chem. C 2017, 121, 8490–8497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, J.M.; Skavdahl, I.J.; McEwen, J.S.; Schneider, W.F. First-principles reaction site model for coverage-sensitive surface reactions: Pt(111)–O temperature programmed desorption. Surf. Sci. 2014, 622, L1–L6. [Google Scholar] [CrossRef]
- Gudmundsdottir, S.; Skulason, E.; Weststrate, K.J.; Juurlink, L.; Jonsson, H. Hydrogen adsorption and desorption at the Pt(110)-(1×2) surface: Experimental and theoretical study. Phys. Chem. Chem. Phys. 2013, 15, 6323–6332. [Google Scholar] [CrossRef] [PubMed]
- Hanh, T.T.T.; Takimoto, Y.; Sugino, O. First-principles thermodynamic description of hydrogen electroadsorption on the Pt(111) surface. Surf. Sci. 2014, 625, 104–111. [Google Scholar] [CrossRef]
- Shi, Q.; Sun, R. Adsorption manners of hydrogen on Pt(100), (110) and (111) surfaces at high coverage. Comput. Theor. Chem. 2017, 1106, 43–49. [Google Scholar] [CrossRef]
- Karlberg, G.S.; Jaramillo, T.F.; Skulason, E.; Rossmeisl, J.; Bligaard, T.; Norskov, J.K. Cyclic voltammograms for H on Pt(111) and Pt(100) from first principles. Phys. Rev. Lett. 2007, 99, 126101. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.S.; Ma, Y.S.; Zhang, Y.L.; Teng, B.T.; Jiang, Z.Q.; Huang, W.X. Revisiting H/Pt(111) by a combined experimental study of the H-D exchange reaction and first-principles calculations. Sci. China Chem. 2011, 54, 745–755. [Google Scholar] [CrossRef]
- Lu, K.E.; Rye, R.R. Flash desorption and equilibration of H2 and D2 on single crystal surfaces of platinum. Surf. Sci. 1974, 45, 677–695. [Google Scholar] [CrossRef]
- Christmann, K.; Ertl, G. Interaction of hydrogen with Pt(111): The role of atomic steps. Surf. Sci. 1976, 60, 365–384. [Google Scholar] [CrossRef]
- Baró, A.M.; Ibach, H.; Bruchmann, H.D. Vibrational modes of hydrogen adsorbed on Pt(111): Adsorption site and excitation mechanism. Surf. Sci. 1979, 88, 384–398. [Google Scholar] [CrossRef]
- Koeleman, B.J.; de Zwart, S.T.; Boers, A.L.; Poelsema, B.; Verheij, L.K. Information on adsorbate positions from low-energy recoil scattering: Adsorption of hydrogen on Pt. Phys. Rev. Lett. 1986, 56, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.A.; Kroes, G.J.; Baerends, E.J. Atomic and molecular hydrogen interacting with Pt(111). J. Chem. Phys. 1999, 111, 11155–11163. [Google Scholar] [CrossRef] [Green Version]
- Watson, G.W.; Wells, R.P.K.; Willock, D.J.; Hutchings, G.J. A Comparison of the Adsorption and Diffusion of Hydrogen on the {111} Surfaces of Ni, Pd, and Pt from Density Functional Theory Calculations. J. Phys. Chem. B 2001, 105, 4889–4894. [Google Scholar] [CrossRef]
- Papoian, G.; Nørskov, J.K.; Hoffmann, R. A Comparative Theoretical Study of the Hydrogen, Methyl, and Ethyl Chemisorption on the Pt(111) Surface. J. Am. Chem. Soc. 2000, 122, 4129–4144. [Google Scholar] [CrossRef]
- Mei, D.; Hansen, E.W.; Neurock, M. Ethylene Hydrogenation over Bimetallic Pd/Au(111) Surfaces: Application of Quantum Chemical Results and Dynamic Monte Carlo Simulation. J. Phys. Chem. B 2003, 107, 798–810. [Google Scholar] [CrossRef]
- Mei, D.; Du, J.; Neurock, M. First-Principles-Based Kinetic Monte Carlo Simulation of Nitric Oxide Reduction over Platinum Nanoparticles under Lean-Burn Conditions. Ind. Eng. Chem. Res. 2010, 49, 10364–10373. [Google Scholar] [CrossRef] [Green Version]
- Van Bavel, A.P.; Hopstaken, M.J.P.; Curulla, D.; Niemantsverdriet, J.W.; Lukkien, J.J.; Hilbers, P.A.J. Quantification of lateral repulsion between coadsorbed CO and N on Rh(100) using temperature-programmed desorption, low-energy electron diffraction, and Monte Carlo simulations. J. Chem. Phys. 2003, 119, 524–532. [Google Scholar] [CrossRef]
- Teng, B.T.; Huang, W.X.; Wu, F.M.; Lan, Y.Z.; Cao, D.B. A density functional theory study of the CH2I2 reaction on Ag(111): Thermodynamics, kinetics, and electronic structures. J. Chem. Phys. 2010, 132, 024715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Teng, B.T.; Wen, X.D.; Zhao, Y.; Chen, Q.P.; Zhao, L.H.; Luo, M.F. Superoxide and Peroxide Species on CeO2(111), and Their Oxidation Roles. J. Phys. Chem. C 2012, 116, 15986–15991. [Google Scholar] [CrossRef]
- Teng, B.T.; Lang, J.J.; Wen, X.D.; Zhang, C.; Fan, M.H.; Harris, H.G. O2 Adsorption and Oxidative Activity on Gold-Based Catalysts with and without a Ceria Support. J. Phys. Chem. C 2013, 117, 18986–18993. [Google Scholar] [CrossRef]
- Boekfa, B.; Treesukol, P.; Injongkol, Y.; Maihom, T.; Maitarad, P.; Limtrakul, J. The Activation of Methane on Ru, Rh, and Pd Decorated Carbon Nanotube and Boron Nitride Nanotube: A DFT Study. Catalysts 2018, 8, 190. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, L.; Yan, W.; Li, J.; Liu, C.; Lu, X. Theoretical Study of the Mechanism for CO2 Hydrogenation to Methanol Catalyzed by trans-RuH2(CO)(dpa). Catalysts 2018, 8, 244. [Google Scholar] [CrossRef]
- Chen, S.; Luo, J.; Bu, S. Morphology transition of Ag ultrathin films on Pt(111): Kinetic Monte Carlo simulation. Appl. Surf. Sci. 2014, 301, 289–292. [Google Scholar] [CrossRef]
- Prats, H.; Álvarez, L.; Illas, F.; Sayós, R. Kinetic Monte Carlo simulations of the water gas shift reaction on Cu(111) from density functional theory based calculations. J. Catal. 2016, 333, 217–226. [Google Scholar] [CrossRef]
- Exner, K.S.; Heß, F.; Over, H.; Seitsonen, A.P. Combined experiment and theory approach in surface chemistry: Stairway to heaven? Surf. Sci. 2015, 640, 165–180. [Google Scholar] [CrossRef]
- Hong, Q.; Liu, Z. Mechanism of CO2 hydrogenation over Cu/ZrO2(212) interface from first-principles kinetics Monte Carlo simulations. Surf. Sci. 2010, 604, 1869–1876. [Google Scholar] [CrossRef]
- Farkas, A.; Hess, F.; Over, H. Experiment-Based Kinetic Monte Carlo Simulations: CO Oxidation over RuO2(110). J. Phys. Chem. C 2012, 116, 581–591. [Google Scholar] [CrossRef]
- Hermse, C.G.M.; Frechard, F.; van Bavel, A.P.; Lukkien, J.J.; Niemantsverdriet, J.W.; van Santen, R.A.; Jansen, A.P.J. Combining density-functional calculations with kinetic models: NO/Rh(111). J. Chem. Phys. 2003, 118, 7081–7089. [Google Scholar] [CrossRef] [Green Version]
- Van Bavel, A.P.; Hermse, C.G.M.; Hopstaken, M.J.P.; Jansen, A.P.J.; Lukkien, J.J.; Hilbers, P.A.J.; Niemantsverdriet, J.W. Quantifying lateral adsorbate interactions by kinetic Monte-Carlo simulations and density-functional theory: NO dissociation on Rh(100). Phys. Chem. Chem. Phys. 2004, 6, 1830. [Google Scholar] [CrossRef]
- Källén, G.; Wahnström, G. Quantum treatment of H adsorbed on a Pt(111) surface. Phys. Rev. B 2001, 65, 033406. [Google Scholar] [CrossRef]
- Muscat, J.P. Role of multi-adatom interactions in the formation of ordered structures on metal surfaces: Application to H/Fe(110). Surf. Sci. 1984, 139, 491–504. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal amorphous semiconductor transition in germanium. Phys. Rev. B Condens. Matter 1994, 49, 14251. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J. Special points for Brillouin-zone integrations. Phys. Rev. B Condens. Matter 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Jonsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transition. In CLassical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1997; pp. 385–404. [Google Scholar]
- Ramalho, J.P.P.; Gomes, J.R.B.; Illas, F. Accounting for van der Waals interactions between adsorbates and surfaces in density functional theory based calculations: Selected examples. RSC Adv. 2013, 3, 13085–13100. [Google Scholar] [CrossRef]
- Prats, H.; Gamallo, P.; Sayos, R.; Illas, F. Unexpectedly large impact of van der Waals interactions on the description of heterogeneously catalyzed reactions: The water gas shift reaction on Cu(321) as a case example. Phys. Chem. Chem. Phys. 2016, 18, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Albao, M.A.; Padama, A.A.B. CO adsorption on W(100) during temperature-programmed desorption: A combined density functional theory and kinetic Monte Carlo study. Appl. Surf. Sci. 2017, 396, 1282–1288. [Google Scholar] [CrossRef]
- WELCOME to the CARLOS Project. Available online: https://carlos.win.tue.nl.
- Núñez, M.; Vlachos, D.G. Steady state likelihood ratio sensitivity analysis for stiff kinetic Monte Carlo simulations. J. Chem. Phys. 2015, 142, 044108. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Plaisance, C.P.; Reuter, K. Assessment of mean-field microkinetic models for CO methanation on stepped metal surfaces using accelerated kinetic Monte Carlo. J. Chem. Phys. 2017, 147, 152705. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Teng, B.; Wu, F.; Fang, Y. Fe nanostructures stabilized by long-range interactions on Cu(111): Kinetic Monte Carlo simulations. New J. Phys. 2008, 10, 023033. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eads/kcal/mol | ZPVE Corrected Eads/kcal/mol | dPt-H/Å | dH-H/Å | Bader Charge/e | ||
|---|---|---|---|---|---|---|
| H | Pt | |||||
| H-fcc | −8.86 | −9.12 | 1.874 | −0.100 | −0.027 | |
| 1.872 | 0.059 | |||||
| 1.875 | −0.019 | |||||
| H-hcp | −7.47 | −7.89 | 1.879 | −0.057 | −0.062 | |
| 1.866 | 0.036 | |||||
| 1.878 | −0.004 | |||||
| H-top | −8.35 | −7.35 | 1.572 | −0.035 | 0.029 | |
| H2-fcc | −0.09 | 0.13 | 3.789 | 0.748 | ||
| 3.712 | 0.025 | |||||
| 3.536 | −0.032 | |||||
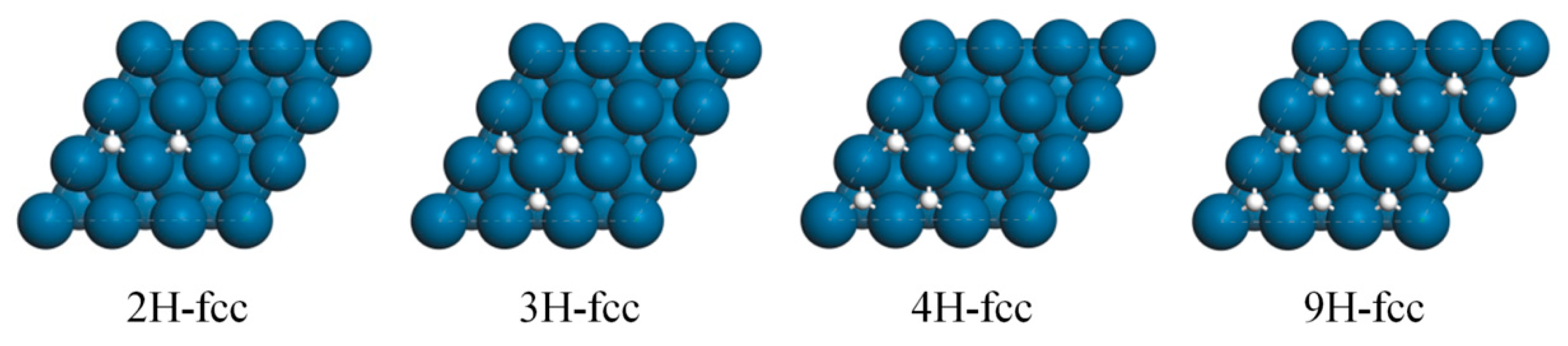
| Eads/kcal/mol | Eav/kcal/mol | Eint/kcal/mol | m | |
|---|---|---|---|---|
| 1H-fcc | −8.86 | −8.86 | - | 0 |
| 2H-fcc | −16.93 | −8.46 | 0.39 | 1 |
| 3H-fcc | −24.49 | −8.16 | 0.35 | 2 |
| 4H-fcc | −31.41 | −7.86 | 0.40 | 2.5 |
| 9H-fcc | −61.55 | −6.85 | 0.34 | 6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.; Wang, F.; Zhang, Y.; Zhao, L.; Teng, B.; Fan, M.; Liu, X. H2 Thermal Desorption Spectra on Pt(111): A Density Functional Theory and Kinetic Monte Carlo Simulation Study. Catalysts 2018, 8, 450. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8100450
Yu C, Wang F, Zhang Y, Zhao L, Teng B, Fan M, Liu X. H2 Thermal Desorption Spectra on Pt(111): A Density Functional Theory and Kinetic Monte Carlo Simulation Study. Catalysts. 2018; 8(10):450. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8100450
Chicago/Turabian StyleYu, Caoming, Fang Wang, Yunlei Zhang, Leihong Zhao, Botao Teng, Maohong Fan, and Xiaona Liu. 2018. "H2 Thermal Desorption Spectra on Pt(111): A Density Functional Theory and Kinetic Monte Carlo Simulation Study" Catalysts 8, no. 10: 450. https://0-doi-org.brum.beds.ac.uk/10.3390/catal8100450