Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides



1
Department of Chemistry and Research Institute of Physics and Chemistry, Chonbuk National University, Jeonju 54896, Korea
2
Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science, Daejeon 34141, Korea
3
Department of Chemistry, Chonnam National University, Gwangju 61186, Korea
4
Department of Chemistry Education, Chungbuk National University, Cheongju 28644, Korea
*
Authors to whom correspondence should be addressed.
Catalysts 2019, 9(4), 311; https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040311
Submission received: 4 March 2019
/
Revised: 23 March 2019
/
Accepted: 27 March 2019
/
Published: 29 March 2019
(This article belongs to the Special Issue Recent Developments on Catalysis by Metalloporphyrins and Analogues)
Abstract
:Cationic tin(IV) porphyrins with tetracarbonyl cobaltates were synthesized, exhibiting bifunctional catalytic reactivity. The Lewis acidic tin-porphyrin center activated epoxides; concurrently, cobalt carbonyl anions efficiently opened epoxides and delivered carbonyl moieties. Thus, a series of β-lactones with a high synthetic value were obtained. This catalytic system showed excellent efficiency exceeding a turnover number of one thousand with a broad substrate scope. In addition, the presented tin porphyrin-based catalyst exhibited exclusive chemoselectivity to terminal epoxides over internal ones. The selective carbonylation of di-epoxides demonstrated the usefulness of these catalysts in the synthesis of complex molecular structures.
1. Introduction
Porphyrins and metalloporphyrins are found in many living organisms and play an essential role in sustaining life [1,2,3,4]. Incorporation of various metals into the porphyrin, contraction or expansion of the ring, and desymmetrization or extension of the π-system can induce intriguing properties [5,6,7,8,9,10,11,12,13,14]. Thus, many natural and nonnatural porphyrins and their derivatives have been widely used in versatile applications such as medicine, materials, catalysis, supramolecular chemistry, and biomimetic models [15,16,17,18,19,20,21].
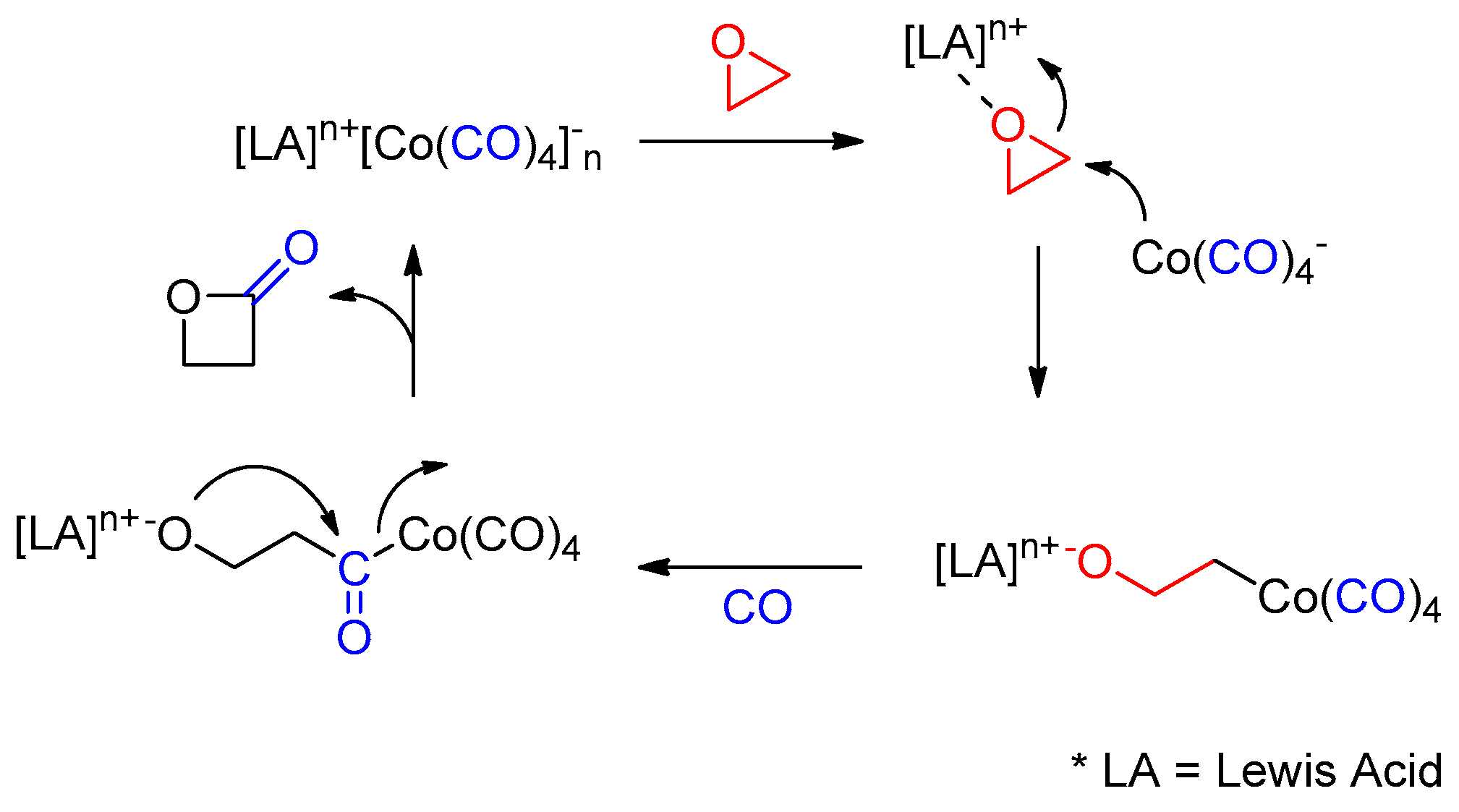
In the area of catalysis, numerous porphyrins, especially transition metal-based metalloporphyrins, promote many chemical transformations with high efficiency and reactivity; however, catalytic evaluation of main-group metal and semi-metal porphyrins has not received much attention despite the distinctive physical and chemical properties of such porphyrins [22,23,24,25,26]. Among those complexes, tin(IV) porphyrins have attracted our attention. Since the first synthesis of tin(IV) porphyrin in 1948 by Rothemund, a wide range of tin-based porphyrins have been developed [27,28,29,30,31,32,33]. As catalysts, they mediate hydrolysis [34], acetylation [30,31], alcoholysis [32], and epoxide ring-opening reactions [26], as well as polymerizations [26,28]. In general, the cationic porphyrin units with a +2 charge are balanced with non- or weakly coordinating anions such as ClO4−, BF4−, TfO−, Br−, F−, Cl−, and OH−. In a continuous effort to expand tin-based catalytic systems, we investigated novel tin complexes with a noninnocent tetracarbonyl cobaltate anion, which produced β-lactones through the selective mono carbonylation of epoxides. The direct conversion of epoxides to the corresponding β-lactones is a clean, one carbon homologation method with a 100% atom-economy [35]. A list of catalytic systems has been developed based on the combination of Lewis acid and metal carbonyl for the carbonylative ring expansion of epoxides (Scheme 1) [36,37,38,39]. Compared to the previous examples, our catalytic system consisting of Sn(IV)-centered porphyrins exhibited comparable reactivity with a turnover number (TON) of over 1000 and exclusive selectivity to β-lactones, without any side products or double carbonylations. In addition, highly selective carbonylation of terminal epoxide over internal epoxide allowed chemoselective lactone formation. The details of our findings are presented herein.
2. Results and Discussion
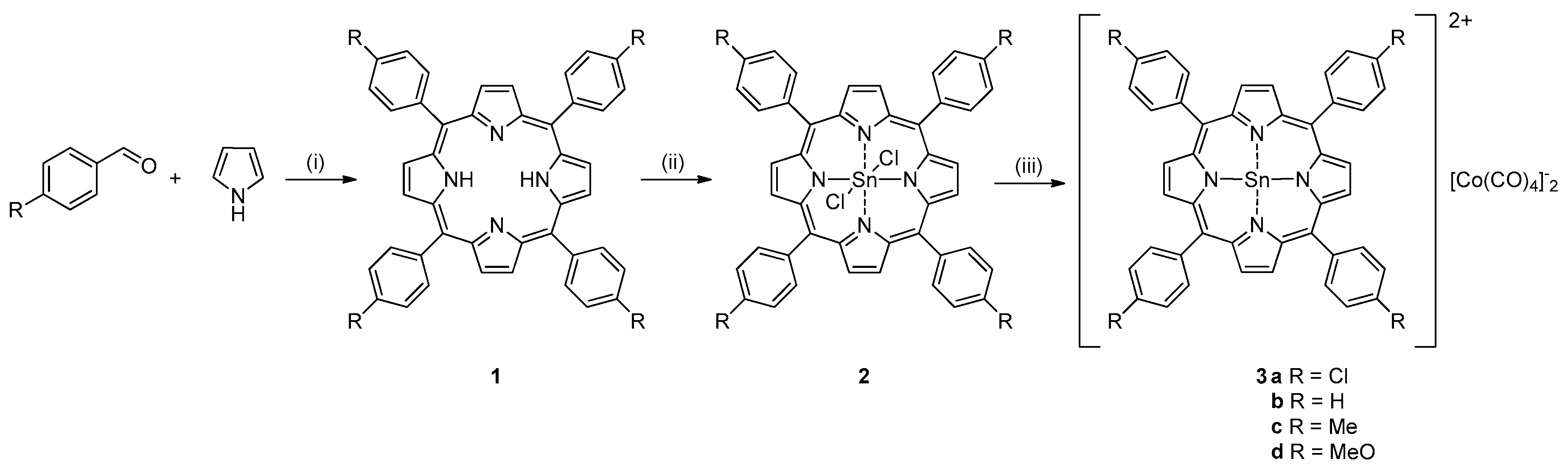
The syntheses of Sn(IV) porphyrins are outlined in Scheme 2. The conventional condensation reaction between benzaldehyde and pyrrole produced the porphyrins [40]. Subsequent reaction of porphyrins with Sn(II) chloride produced Sn(IV) porphyrin dichloride [41]. The X-ray crystal structure of 2a shows a chloride anion tightly bound to the Sn center (Figure S1). Finally, sodium tetracarbonyl cobaltate, NaCo(CO)4 reacted with Sn(IV) porphyrin dichloride precursors and produced the desired catalysts (3a–3d).
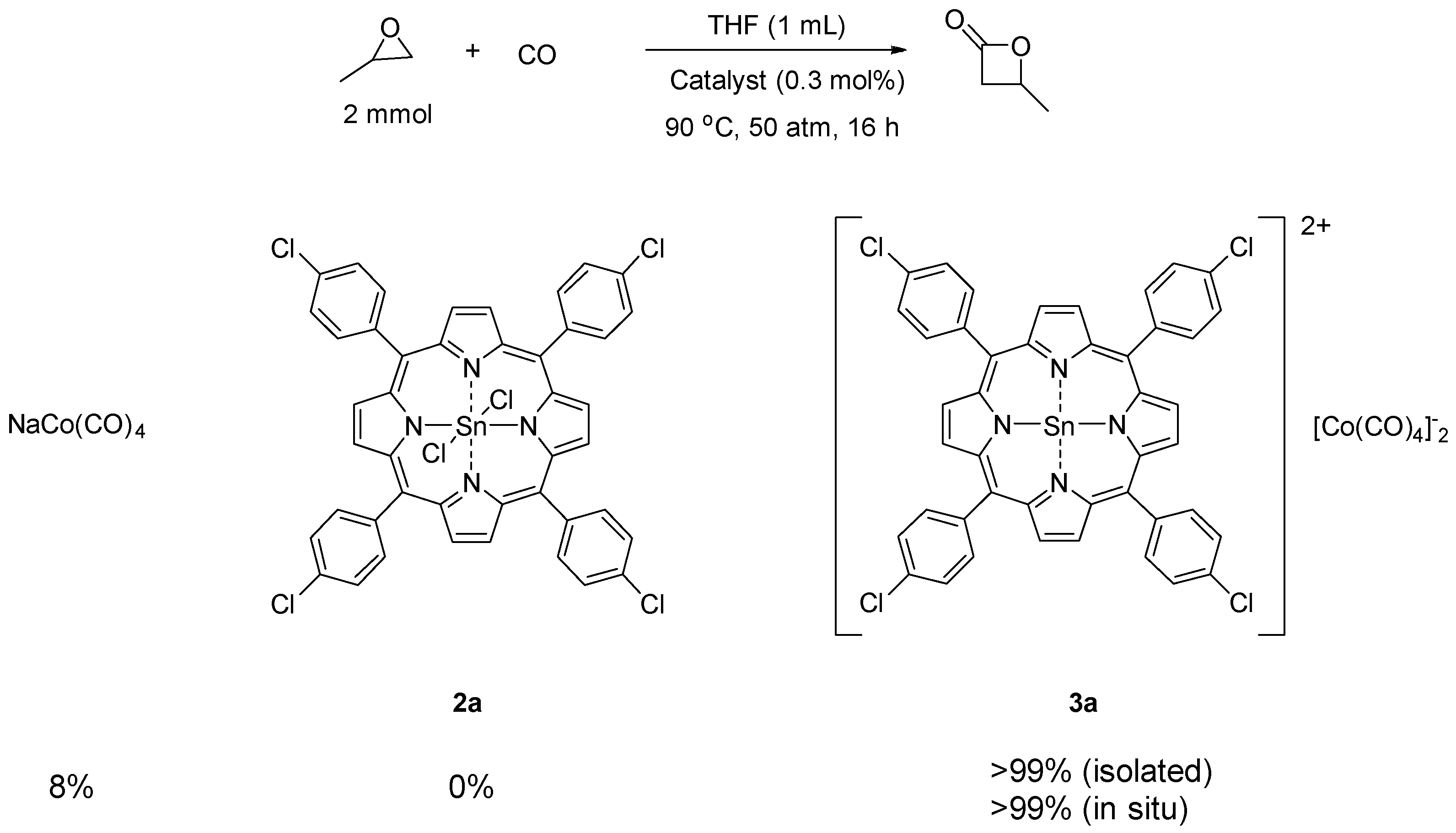
First, catalyst 3a (0.3 mol %) with electron withdrawing chlorine substituents was tested for carbonylation of propylene oxide (4a, 2 mmol) with carbon monoxide (50 atm) in tetrahydrofuran (THF, 1 mL) at 90 °C. After 16 h, the desired β-butyrolactone was formed quantitatively (Scheme 3). To elucidate the reactive catalytic species, we tested NaCo(CO)4 and Sn(IV) porphyrin dichloride 2a independently. Complex 2a with the tightly bound chloride anions did not promote carbonylation, and epoxide 4a remained unreacted. Interestingly, NaCo(CO)4 was solely active under the same CO pressure, but the efficiency was relatively low (8%). The complex 3a prepared in situ from the mixture of NaCo(CO)4 (0.6 mol %) and 2a (0.3 mol %) showed the same reactivity as isolated 3a. These results imply that the reaction between NaCo(CO)4 and 2a produces a more reactive Lewis acidic species, which is not exactly identified by analytical methods. However, currently, the formation of NaCl indicates that the Sn(IV) porphyrin cationic complex with an empty coordination site is the active catalytic species as a Lewis acid for epoxide activation.
Further optimizations were undertaken (Table 1), and the influence of the electronic nature of the catalyst was investigated. A shift to electron rich porphyrins by substitution of Cl (3a, entry 1) with H (3b, entry 2), methyl (3c, entry 3), and methoxy (3d, entry 4) groups exhibited a gradual decrease in catalytic turnover rates. The diminishing Lewis acid character was directly indicated by the catalytic efficiency. Additionally, the effect of reaction media was significant. THF and 2-methyl THF exhibited the same quantitative conversion with 0.3 mol % catalyst loading (entries 1 and 5), but another ethereal solvent 1,4-dioxane gave a lower conversion (entry 6). Another coordinating polar solvent, dimethyl formamide (DMF, entry 7) [42] and a protic polar solvent, methanol (MeOH, entry 8) showed complete catalyst inhibition. In noncoordinating and nonpolar solvents, 3a showed poor performance (entries 9–11). In their study with Cr-porphyrin catalysts, Coates and coworkers noted that the coordination of solvents such as THF is very important for high efficiency [43]. The trans-addition of THF to a metal facilitates the alkoxide departure from the metal center allowing the alkoxide to attack the carbonyl group and form β-lactone (Scheme 1). We believe that a similar solvent effect is valid in the Sn-porphyrin system as well, thus the use of a noncoordinating solvent could not promote a β-lactone formation step.
Next, the variations in temperature and carbon monoxide pressure were evaluated. The catalytic efficiency of 3a is sensitive to both temperature and CO pressure. Decreasing the reaction temperature to 70 °C gave 61% conversion under otherwise identical conditions (entry 12). At 50 °C, the conversion dropped to 20% (entry 13). The change in pressure to 30 atm maintained good reactivity with 95% conversion to β-lactone (entry 14), but a notable loss of conversion was observed at 10 atm of CO (63%, entry 15).
When the catalyst loading was decreased, higher temperature or pressure was required to maintain good β-lactone production. With 0.2 mol %, a longer reaction, 24 h, was needed to reach complete conversion (entries 16 and 17). When 0.1 mol % was used, changing the time and pressure to 36 h and 70 atm of CO, respectively, could achieve a TON of 1000 (entry 19). With the use of 0.05 mol %, 3a exhibited 63% conversion at 110 °C, with a TON of 1260 [44].
The direct carbonylation of epoxides provided a series of β-lactones with high efficiencies (Table 2). A substituent on the terminal epoxides did not hamper the carbonylation performance of 3a. Variations in the chain length (5a–c) and size (5d and e) of substituents showed no influence and quantitative β-lactones were obtained. Interestingly, the aromatic substituent exhibited positional sensitivity. Styrene oxide, surprisingly, did not react with catalyst 3a at all (5f); however, adding a methylene bridge restored its reactivity (5g). Various kinds of coordinating functional groups were introduced that could behave as potential catalyst inhibitors. Epoxides with alkene (5h), ether (5i), and nitrile (5j) groups were smoothly converted to the corresponding β-lactones with a slight increase in catalyst loadings. Moreover, ester and amide functionalities were well tolerated (5k–m). Dual carbonylations of di-epoxide were also successful (5n).
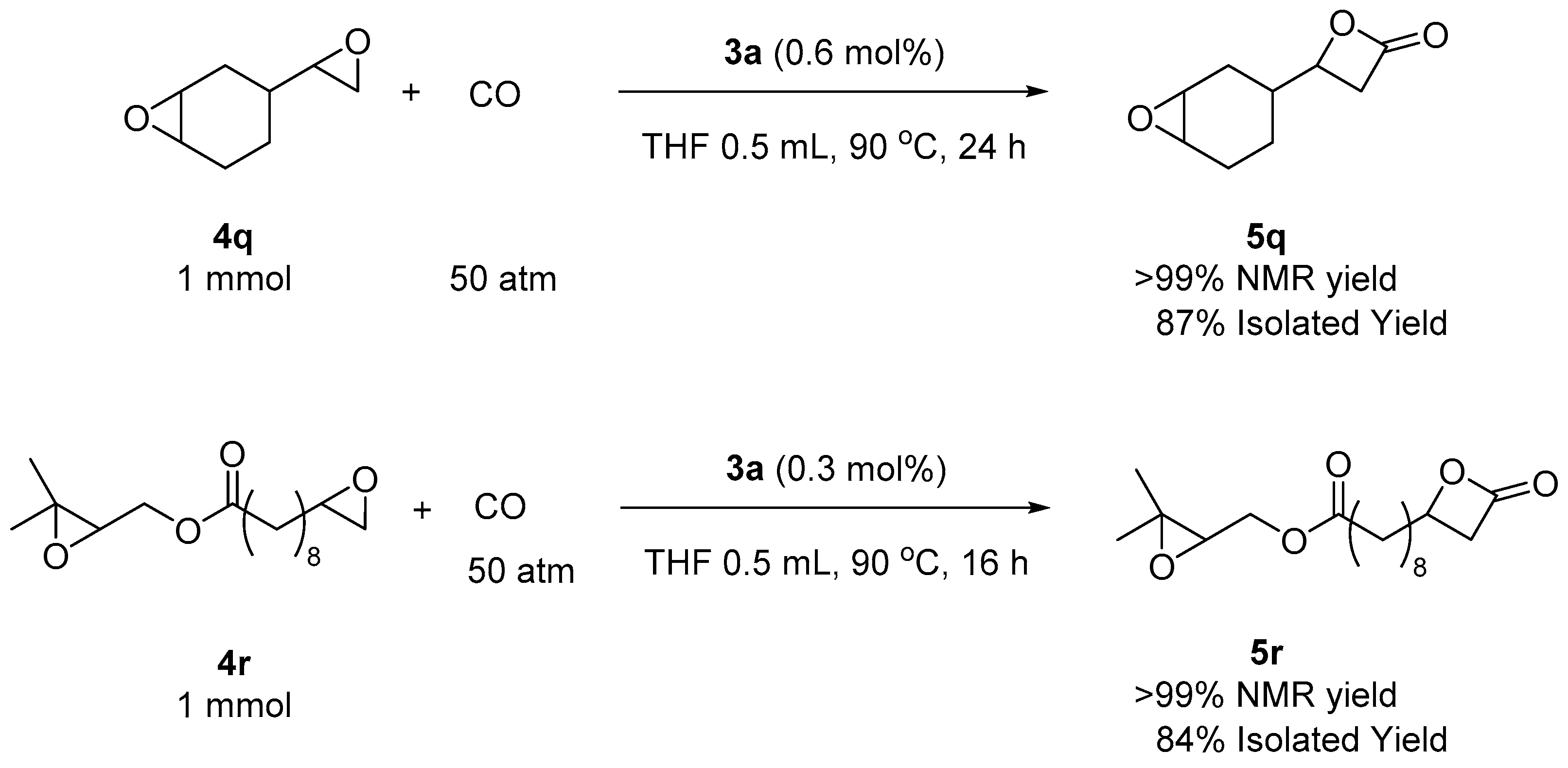
Next, sterically more congested epoxides with 1,1- and 1,2-substitution were investigated. 1,2-Epoxy-2-methylpropane (4o) gave only 27% of NMR yield under a high amount of catalyst loading. In the case of 1,2-substitution, such as with cyclohexene oxide (4p), carbon monoxide insertion did not occur and the starting material remained intact. This dramatic reactivity dependence on substitution patterns allowed for chemoselective carbonylation (Scheme 4). The di-epoxide (4q) has two distinctive epoxides: one has a single substitution and the other epoxide is internal and di-substituted. Catalyst 3a successfully discriminated the difference and only the terminal epoxide was converted to the corresponding β-lactone (5q). The other di-epoxide 4r also exhibited the same terminal selectivity to produce 5r with both epoxide and β-lactone. The use of these products in selective polymerization is currently under investigation.
3. Materials and Methods
3.1. General Considerations
All manipulations for air and moisture sensitive compounds were carried out under dry nitrogen using either a glove box (MBRAUN UNIlab, Garching, Germany) or standard Schlenk line technique. Carbonylations of epoxides were carried out in stainless steel reactors (Uto Engineering, Namyangju, Korea). Flash column chromatography was performed on silica gel 60 (230–400 mesh) (ZEOCHEM, Ruti, Switzerland). 1H and 13C NMR spectra were recorded on a Bruker Avance III HD Fourier transform 400 MHz NMR spectrometer (Billerica, MA, USA). Chemical shifts are reported in parts per million relative to residual solvent (CDCl3, 1H δ 7.26 ppm, and 13C δ 77.0 ppm), and coupling (J) constants are expressed in hertz (Hz). The multiplicities of 1H NMR are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, and m = multiplet. Infrared (IR) spectra were recorded on an ABB FT-IR spectrometer MB3000 (Zurich, Switzerland) and are reported in wavenumbers (cm−1). High resolution mass spectra were obtained from the Korea Basic Science Institute (Daegu, Korea) on a JEOL JMS 700 high resolution spectrometer (Tokyo, Japan). Carbon monoxide (99.999%) was purchased from Korea specialty gas (Iksan, Korea) and used without further purification. Unless otherwise stated, all reagents were purchased from Sigma-Aldrich (Seoul, Korea), Alfa Aesar (Seoul, Korea), or Acros Organics (Seoul, Korea) and used as received. 1,4-Dioxane, tetrahydrofuran, 2-methyltetrahydrofuran, toluene, o-xylene, and hexane were dried using activated 3 Å molecular sieves and degassed by repeated freeze-pump-thaw cycles before use. All epoxides were stirred for a few days over CaH2, vacuum distilled, and degassed by freeze-pump-thaw prior to use. NaCo(CO)4 [45], 5,6-epoxyhexanenitrile, 3,4-epoxybutyl butyrate [46], cyclohexyl oxirane [47], 2-(tert-butyl)oxirane [48], and 2-benzyl oxirane [49] were synthesized following previously reported methods.
3.2. Syntheses of Substrates
Synthesis of Ethyl 9-(oxiran-2-yl)nonanoate (4l). First, 10-Undecenoyl chloride (2.6 mL, 12 mmol) was mixed with ethanol (5.0 mL). The mixture was stirred at room temperature for 2 h. Removal of excess ethanol and vacuum drying gave a colorless liquid of ethyl undec-10-enoate which was directly used for the epoxidation using a modified previous report [37]. The ethyl undec-10-enoate was dissolved in 100 mL of CH2Cl2 and cooled at 0 °C. m-Chloroperoxybenzoic acid (70–75% purity, Acros Organics) (3.7 g, 15 mmol) was gradually added to the solution. The resulting mixture was stirred at 0 °C for 2 h and then at room temperature for 14 h. The excess peroxide was quenched by the addition of a 10% solution of NaHSO3 at 0 °C. A saturated solution of NaHCO3 was added until gas evolution ceased. Then, the aqueous layer was extracted with CH2Cl2. The separated organic layer was washed with NaHCO3 and dried with anhydrous MgSO4. The removal of solvent in the rotary evaporator and subsequent vacuum distillation gave the epoxide product 4l (0.80 g, 72%). The NMR data is identical to reported that reported previously [50]. 1H NMR (400 MHz, CDCl3) δ 4.11 (q, J = 7.2 Hz, 2H), 2.88–2. 91 (m, 1H), 2.73 (dd, J = 5.1, 4.0 Hz, 1H), 2.45 (dd, J = 5.1, 2.8 Hz, 1H), 2.27 (t, J = 7.8 Hz, 2H), 1.66–1.55 (m, 2H), 1.56–1.48 (m, 2H), 1.49–1.37 (m, 2H), 1.29–1.35 (m, 8H), 1.24 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.8, 60.1, 52.3, 47.1, 34.3, 32.4, 29.3, 29.3, 29.1, 29.0, 25.9, 24.9, 14.2.
Synthesis of N,N-Diethyl-10,11-epoxyundecylamide (4m). Diethyl amine (1.0 mL, 10.0 mmol) and triethyl amine (2.8 mL, 20.0 mmol) were mixed in CH2Cl2 (100 mL) at 0 °C. Then, 10-Undecenoyl chloride (2.6 mL, 12 mmol) was added dropwise. The mixture was stirred at room temperature for 14 h. A saturated solution of NaHCO3 was added and then extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4 and filtered. The solvent was removed using a rotary evaporator and the product was purified by flash column chromatography using hexane/ethyl acetate as the eluent. The resulting N,N-diethylunde-10-enamide (1.7 g, 7.1 mmol) was dissolved in 100 mL of CH2Cl2 and cooled at 0 °C. m-Chloroperoxybenzoic acid (70–75% purity, Acros Organics) (2.8 g, 9.2 mmol) was gradually added to the solution. The resulting mixture was stirred at 0 °C for 2 h and then at room temperature for 14 h. The excess peroxide was quenched by addition of a 10% solution of NaHSO3 at 0 °C. A saturated solution of NaHCO3 was added until gas evolution ceased. Then, the aqueous layer was extracted with CH2Cl2. The separated organic layer was washed with NaHCO3 and dried over anhydrous MgSO4. The removal of solvent in the rotary evaporator and subsequent vacuum distillation gave the epoxide product 4m (1.0 g, 56%). 1H NMR (400 MHz, CDCl3) δ 3.35 (q, J = 7.2 Hz, 2H), 3.28 (q, J = 7.2 Hz, 2H), 2.86–2.90 (m, 1H), 2.73 (dd, J = 5.0, 4.0 Hz, 1H), 2.44 (dd, J = 5.0, 2.7 Hz, 1H), 2.34–2.18 (m, 2H), 1.68–1.58 (m, 2H), 1.56–1.47 (m, 2H), 1.47–1.37 (m, 2H), 1.37–1.26 (m, 8H), 1.15 (t, J = 7.2 Hz, 3H), 1.09 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.2, 52.3, 47.1, 41.9, 39.9, 33.1, 32.4, 29.4, 29.4, 29.3 (2C), 25.9, 25.4, 14.4, 13.1; IR (ATR) 1639 (amide) cm−1; HRMS m/z (ESI) calcd for C15H29NO2 [M+] 255.2198, found: 255.2200.
Synthesis of 3-(Oxiran-2-yl)-7-oxabicyclo[4.1.0]heptane (4q). First, 4-Vinylcyclohex-1-ene (3.0 g, 28 mmol) was dissolved in 100 mL of CH2Cl2 and cooled at 0 °C. m-Chloroperoxybenzoic acid (70–75% purity, Acros Organics) (12.0 g, 72.0 mmol) was gradually added to the solution. The resulting mixture was stirred at 0 °C for 2 h and then at room temperature for 14 h. The excess peroxide was quenched by the addition of a 10% solution of NaHSO3 at 0 °C. A saturated solution of NaHCO3 was added until gas evolution ceased. Then, the aqueous layer was extracted with CH2Cl2. The separated organic layer was washed with NaHCO3 and dried over anhydrous MgSO4. The removal of solvent in the rotary evaporator and subsequent vacuum distillation gave the epoxide product 4q (2.8 g, 72%) as the mixture of diastereomers. 1H NMR (400 MHz, CDCl3) δ 3.24–3.09 (m, 2H), 2.77–2.60 (m, 2H), 2.51–2.46 (m, 1H), 2.26–2.09 (m, 1H), 2.08–1.92 (m, 1H), 1.90–1.80 (m, 1H), 1.78–1.64 (m, 1H), 1.63–1.45 (m, 1H), 1.43–1.22 (m, 1H), 1.22–1.02 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 55.9, 55.65, 55.63, 52.5, 52.4, 52.3, 52.2, 51.6, 51.5, 50.9, 50.8, 46.2, 46.1, 45.6, 45.4, 35.8, 353, 32.7, 31.9, 27.5, 27.3, 27.0, 25.8, 24.4, 24.1, 23.6, 23.3, 22.9, 22.7, 21.4, 20.2; IR (ATR) 2927, 1434, 257, 929, 925, 856, 790 cm−1. HRMS m/z (ESI) calcd for C15H29NO2 [M+] 255.2198, found: 255.2200.
Synthesis of (3,3-Dimethyloxiran-2-yl)methyl 9-(oxiran-2-yl)nonanoate (4r). A 50 mL round-bottom flask was charged with 3-methylbut-2-en-1-ol (1.4 g, 16 mmol), toluene (15 mL), and triethyl amine (2.5 mL) at 0 °C. At 0 °C, 10-undecenoyl chloride (3.2 g, 16 mmol) was added dropwise. The mixture was stirred at 0 °C for 2 h and then at room temperature for 12 h. The toluene was evaporated after filtration. The resulting liquid was dissolved in 100 mL of CH2Cl2. After cooling at 0 °C, m-Chloroperoxybenzoic acid (70–75% purity, Acros Organics) (9.5 g, 41 mmol) was gradually added to the solution. The resulting mixture was stirred at 0 °C for 2 h and then at room temperature for 14 h. The excess peroxide was quenched by the addition of a 10% solution of NaHSO3 at 0 °C. The saturated solution of NaHCO3 was added until gas evolution ceased. Then, the aqueous layer was extracted with CH2Cl2. The separated organic layer was washed with NaHCO3 and dried over anhydrous MgSO4. The removal of solvent in the rotary evaporator and subsequent flash column chromatography gave the epoxide product 4r (3.5 g, 77%). 1H NMR (400 MHz, CDCl3) δ 4.32 (dd, J = 12.1, 4.3 Hz, 1H), 4.01 (dd, J = 12.1, 6.9 Hz, 1H), 2.98 (dd, J = 6.9, 4.3 Hz, 1H), 2.93–2.84 (m, 1H), 2.74 (dd, J = 4.9, 4.1 Hz, 1H), 2.45 (dd, J = 5.0, 2.7 Hz, 1H), 2.34 (t, J = 7.5 Hz, 2H), 1.71–1.58 (m, 2H), 1.513–1.50 (m, 2H), 1.47–1.41 (m, 2H), 1.36–1.27 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 173.7, 63.2, 60.5, 58.1, 52.3, 47.1, 34.1, 32.4, 29.3, 29.2, 29.1, 29.0, 25.9, 24.8, 24.5, 18.9; IR (ATR) 1737 (ester) cm−1. HRMS m/z (ESI) calcd for C8H12O2 [M+] 140.0837, found: 140.0834.
3.3. Catalyst Synthesis
General procedure for synthesis of 5,10,15,20-tetrakis(4-chlorophenyl)porphyrin dichlorotin(IV) (2a): 5,10,15,20-tetrakis(4-chlorophenyl)porphyrin (1.00 g, 1.33 mmol) synthesized from pyrrole and 4-chlorobenzaldehyde by a reported procedure was dissolved in 200 mL of pyridine, and tin(II) chloride dihydrate (0.805 g, 3.57 mmol) was added, after which the mixture was refluxed for 3 h. The product was precipitated by adding excess water. The precipitate was filter-washed sequentially with water, 1.0 M hydrochloric acid solution, and then again with water. The collected product was dried to give 5,10,15,20-tetrakis(4-chlorophenyl)porphyrin dichlorotin(IV) 2a (1.00 g, 80%) as a purple solid. An X-ray quality crystal was obtained from THF/benzene. 1H NMR (400 MHz, CDCl3) δ 9.22 (s, 8H, 4JH−Sn = 15.6 Hz), 8.25 (d, J = 8.3 Hz, 8H), 7.84 (d, J = 8.3 Hz, 8H); 13C NMR (100 MHz, CDCl3) δ 146.2 (8C), 138.7 (4C), 135.8 (8C), 135.4 (4C), 132.7 (8C), 127.5 (8C), 120.0 (4C).
5,10,15,20-Tetraphenylporphyrin dichlorotin(IV) TPPSnCl2 (2b) [51] was prepared from 5,10,15, 20-tetraphenylporphyrin (1.00 g, 1.64 mmol) and tin(II) chloride dihydrate (0.993 g, 4.40 mmol) following the general procedure (1.12 g, 85%). 1H NMR (400 MHz, CDCl3) δ 9.21 (s, 8H, 4JH−Sn = 15.6 Hz), 8.43–8.23 (m, 8H), 7.9–7.72 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 146.3 (8C), 140.5 (4C), 134.9 (8C), 132.7 (8C), 128.5 (4C), 127.1 (8C), 121.2 (4C).
5,10,15,20-tetrakis(p-tolyl)porphyrin dichlorotin(IV) MeTPPSnCl2 (2c) was prepared from 5,10,15,20-tetrakis(p-tolyl)porphyrin (1.00 g, 1.49 mmol) and tin(II) chloride dihydrate (0.902 g, 4.00 mmol) following the procedure of 2a (1.0 g, 78%). 1H NMR (400 MHz, CDCl3) δ 9.22 (s, 8H, 4JH−Sn = 15.6 Hz), 8.20 (d, J = 7.9 Hz, 8H), 7.62 (d, J = 7.7 Hz, 8H), 2.74 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 146.4 (8C), 138.3 (4C), 137.7 (4 ), 134.9 (8 ), 132.6 (8C), 127.8 (8 ), 121.2 (4C), 21.6 (4C).
5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin dichlorotin(IV) MeOTPPSnCl2 (2d) was prepared from 5,10,15,20-tetrakis(4-methoxyphenyl)porphyrin (1.00 g, 1.36 mmol) and tin(II) chloride dihydrate (0.821 g, 3.64 mmol) following a similar procedure to ClTPPSnCl2 (0.90 g, 72%). 1H NMR (400 MHz, CDCl3) δ 9.23 (s, 8H, 4JH−Sn = 15.6 Hz), 8.24 (d, J = 8.6 Hz, 8H), 7.36 (d, J = 8.6 Hz, 8H), 4.13 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 159.9 (4C), 146.6 (8C), 136.1 (8C), 133.0 (4C), 132.6 (8C), 120. 9 (4C), 112.6 (8C), 55.6 (4C).
Synthesis of Bimetallic Catalysts: [(ClTPP)Sn] [Co(CO)4]2 (3a). ClTPPSnCl2 (0.20 g, 0.21 mmol) and NaCo(CO)4 (81 mg, 0.42 mmol) were mixed in THF (20 mL) in a nitrogen atmosphere. The solution was stirred at room temperature, wrapped with aluminum foil for 48 h, and then allowed to settle. The resulting mixture was filtered and the solid was washed with additional THF. The filtrate was concentrated to 7–8 mL and was layered with hexane. The solid was filtered and washed with hexane and vacuum dried. Purple-greenish solid was obtained (0.18 g, 64%).
Similarly, the bimetallic catalysts [(TPP)Sn] [Co(CO)4]2 (3b), [(MeTPP)Sn] [Co(CO)4]2 (3c), and [(MeOTPP)Sn] [Co(CO)4]2 (3d) were synthesized in the same way as 3a from TPPSnCl2 (2b), MeTPPSnCl2 (2c), and MeOTPPSnCl2 (2d), respectively.
3.4. General Procedure for the Carbonylation of Epoxides
In a nitrogen glove box at room temperature, the appropriate amount of catalyst (3a) was added into a stainless-steel reactor. The solvent THF was added, followed by the addition of epoxide. The reactor was sealed properly and removed from the glove box. Then, the reactor was immediately pressurized to 50 atm pressure and stirred at 90 °C in a preheated heat bath. After the indicated time, the reactor was cooled at room temperature and with ice to a temperature of 0 °C. The excess CO gas was slowly vented. The lactone formed was determined by 1H NMR spectroscopy. The mixture was filtered through a short column packed with silica gel using diethyl ether. The lactones were further purified by flash column chromatography using hexane/ethyl acetate (4/1) as the eluent.
β-Butyrolactone (5a) [52]: The compound 5a was obtained as a colorless liquid by following a general procedure and only the 1H-NMR yield (99%) was recorded due to its low boiling point (71 °C). 1H NMR (400 MHz, CDCl3) δ 4.75–4.70 (m, 1H), 3.59 (dd, J = 16.3, 5.6 Hz, 1H), 3.09 (dd, J = 16.3, 4.0 Hz, 1H), 1.60 (d, J = 6.0, 3H).
β-Valerorolactone (5b) [53]: The compound 5b was obtained by using 4b (144 mg, 2.00 mmol), 3a (8.2 mg, 0.3 mol %), and CO (50 atm) in THF (1.0 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (168 mg, 84%).1H NMR (400 MHz, CDCl3) δ 4.49–4.43 (m, 1H), 3.49 (dd, J = 16.3, 5.7 Hz, 1H), 3.06 (dd, J = 16.3, 4.2 Hz, 1H), 1.94–184 (m, 1H), 1.84–1.73 (m, 1H), 1.01 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.3, 72.3, 42.3, 27.7, 8.9.
β-Heptanolactone (5c) [54]: The compound 5c was obtained by using 4c (200 mg, 2.00 mmol), 3a (8.2 mg, 0.3 mol %), and CO (50 atm) in THF (1.0 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (223 mg, 87%). 1H NMR (400 MHz, CDCl3) δ 4.53–4.47 (m, 1H), 3.50 (dd, J = 16.3, 5.8 Hz, 1H), 3.05 (dd, J = 16.3, 4.3 Hz, 1H), 1. 90–1.83 (m, 1H), 1.81–1.64 (m, 1H), 1.37 (m, 4H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.4, 71.3, 42.8, 34.3, 26.9, 22.2, 13.8.
4-Cyclohexyl-2-propiolactone (5d) [55]: The compound 5d was obtained by using 4d (126 mg, 1.00 mmol), 3a (4.1 mg, 0.3 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (146 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 4.20–4.17 (m, 1H), 3.42 (dd, J = 16.3, 5.8 Hz, 1H), 3.10 (dd, J = 16.3, 4.3 Hz, 1H), 1.98–1.91 (m, 1H), 1.87–1.51 (m, 5H), 1.36–1.10 (m, 3H), 1.07–0.92 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.6, 74. 9, 41.9, 41.0, 28.1, 27.0, 25. 9, 25.3, 25.1.
4-(tert-Butyl)oxetan-2-one (5e) [56]: The compound 5e was obtained by using 4e (100 mg, 1.00 mmol), 3a (4.1 mg, 0.3 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (114 mg, 89%). 1H NMR (400 MHz, CDCl3) δ 4.25–4.22 (m, 1H), 3.31 (dd, J = 16.5, 6.0 Hz, 1H), 3.14 (dd, J = 16.5, 4.5 Hz, 1H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 168.3, 77.9, 38.1, 32.8, 23.95.
4-Benzyloxetan-2-one (5g) [57]: The compound 5g was obtained by using 4g (134 mg, 1.00 mmol), 3a (4.1 mg, 0.3 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. White solid (155 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 7.31–7.18 (m, 3H), 7.18–7.12 (m, 2H), 4.67–4.63 (m, 1H), 3.40 (dd, J = 16.4, 5.7 Hz, 1H), 3.17–2.97 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 167.7, 134.9, 129.2 (2C), 128.8 (2C), 127.3, 70.8, 42.4, 40.4.
4-(But-3-enyl)-2-propiolactone (5h) [35]: The compound 5h was obtained by using 4h (196 mg, 2.00 mmol), 3a (10.8 mg, 0.4 mol %), and CO (50 atm) in THF (1.0 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (232 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 5.85–5.74 (m, 1H), 5.10–4.99 (m, 2H), 4.55–4.49 (m, 1H), 3.51 (dd, J = 16.3, 5.8 Hz, 1H), 3.08 (dd, J = 16.3, 4.3 Hz, 1H), 2.31–2.09 (m, 2H), 2.01–1.93 (m, 1H), 1.89–1.80 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 168.1, 136.3, 116.0, 70.6, 42.8, 33.8, 29.1.
4-(Methoxymethyl)-2-propiolactone (5i) [37]: The compound 5i was obtained by using 4i (176 mg, 2.00 mmol), 3a (13.5 mg, 0.5 mol %), and CO (50 atm) in THF (1.0 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (214 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 4.66–4.62 (m, 1H), 3.73 (dd, J = 11.7, 2.9 Hz, 1H), 3.63 (dd, J = 11.7, 4.5 Hz, 1H), 3.51–3.34 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 167.6, 71.8, 69.3, 59.5, 39.4.
4-(4-oxooxetan-2-yl)butanenitrile (5j): The compound 5j was obtained by using 4j (222 mg, 2.00 mmol), 3a (16.4 mg, 0.6 mol %), and CO (50 atm) in THF (1.0 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (248 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 4.57–4.49 (m, 1H), 3.56 (dd, J = 16.4, 5.8 Hz, 1H), 3.10 (dd, J = 16.4, 4.3 Hz, 1H), 2.45–2.41 (m, 2H), 2.03–1.70 (m, 4H); 13C NMR (100 MHz CDCl3) δ 167.5, 118.9, 70.0, 43.0, 33.3, 21.2, 16.6; IR (ATR) 1820 (lactone), 2248 (nitrile) cm−1; HRMS m/z (FAB) calcd for C7H9O2 [M+H]+ 140.0712, found: 140.0714.
4-(2-Butyroxyethyl)-2-propiolactone (5k) [37]: The compound 5k was obtained by using 4k (158 mg, 1.00 mmol), 3a (4.1 mg, 0.3 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (174 mg, 94%). 1H NMR (400 MHz, CDCl3) δ 4.69–4.57 (m, 1H), 4.32–4.14 (m, 2H), 3.59 (dd, J = 16.4, 5.8 Hz, 1H), 3.17 (dd, J = 16.4, 4.3 Hz, 1H), 2.29 (t, J = 7.4 Hz, 2H), 2.25–2.07 (m, 2H), 1.70–1.60 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.3, 167.5, 68.4, 59.9, 43.3, 36.0, 33.0, 18.3, 13.6.
Ethyl 9-(4-oxooxetan-2-yl)nonanoate (5l): The compound 5l was obtained by using 4l (228 mg, 1.00 mmol), 3a (4.1 mg, 0.3 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (246 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 4.51–4.45 (m, 1H), 4.09 (q, J = 7.2 Hz, 2H), 3.48 (dd, J = 16.3, 5.8 Hz, 1H), 3.03 (dd, J = 16.3, 4.3 Hz, 1H), 2.26 (t, J = 7.5 Hz, 2H), 1.88–1.79 (m, 1H), 1.78–1.66 (m, 1H), 1.62–1.55 (m, 2H), 1.47–1.25 (m, 10H), 1.22 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.6, 168.2, 71.1, 60.0, 42.7, 34.5, 34.1, 29.0, 28.9 (2C), 28. 8, 24.74, 24.71, 14.1; IR (ATR) 1822 (lactone), 1731 (ester) cm−1; HRMS m/z (FAB) calcd for C14H24O4 [M+H]+ 257.1755, found: 257.1755.
N,N-Diethyl 9-(4-oxooxetan-2-yl)nonamide (5m): The compound 5m was obtained by using 4m (255 mg, 1.00 mmol), 3a (13.5 mg, 1.0 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (269 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 4.46–4.41 (m, 1H), 3.43 (dd, J = 16.3, 5.8 Hz, 1H), 3.27 (q, J = 7.2 Hz, 2H), 3.27 (q, J = 7.2 Hz, 2H), 2.97 (dd, J = 16.3, 4.3 Hz, 1H), 2.25–2.12 (m, 2H), 1.82–1.72 (m, 1H), 1.69–1.64 (m, 1H), 1.59–1.49 (m, 2H), 1.33–1.19 (m, 10 H), 1.08 (t, J = 7.2 Hz, 3H), 1.01 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.9, 168.2, 71.1, 42.6, 41.7, 39.7, 34.4, 32.8, 29.1, 29.0, 28.9, 28.8, 25.1, 24.6, 14.1, 12.9; IR (ATR) 1824 (lactone) cm−1, 1635 (amide) cm−1; HRMS m/z (ESI) calcd for C16H29NO3 [M+] 283.2147, found: 283.2150.
4,4′-(Butane-1,4-diyl)bis(oxetan-2-one) (5n): The compound 5n was obtained by using 4n (142 mg, 1.00 mmol), 3a (8.2 mg, 0.6 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. White solid (196 mg, 99%). 1H NMR (400 MHz, CDCl3) δ 4.54–4.48 (m, 2H), 3.53 (dd, J = 16.3, 5.8 Hz, 2H), 3.07 (dd, J = 16.3, 4.3 Hz, 2H), 1.94–1.73 (m, 4H), 1.66–1.37 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 168.0 (2C), 70. 9 (2C), 42.93, 42.91, 34.5, 34.4, 24.6, 24.6; IR (ATR) 1816 cm−1; HRMS m/z (FAB) calcd for C10H14O4 [M+H]+ 199.0972, found: 199.0972.
4,4-Dimethyloxetan-2-one (5o) [58]: The compound 5o was obtained by using 4o (72.11 mg, 1.00 mmol), 3a (13.5 mg, 1.0 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure (27% NMR yield). 1H NMR (400 MHz, CDCl3, crude NMR) δ 3.19 (s, 2H), 1.62 (s, 6H); IR (ATR) 1811 cm−1.
4-(7-Oxabicyclo[4.1.0]heptan-3-yl)oxetan-2-one (5q): The compound 5q was obtained by using 4q (140 mg, 1.00 mmol), 3a (8.2 mg, 0.6 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 24 h by following the general procedure. Colorless liquid (146 mg, 87%) as the mixture of diastereomers. 1H NMR (400 MHz, CDCl3) δ 4.25–4.16 (m, 1H), 3.50–3.36 (m, 1H), 3.29–3.00 (m, 3H), 2.40–2.17 (m, 1H), 2.16–1.99 (m, 1H), 1.98–1.70 (m, 2H), 1.68–1.51 (m, 1H), 1.50–1.36 (m, 1H), 1.34–1.18 (m, 1H), 1.14–0.97 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 167.8, 73.9, 73.8, 62.1, 62.06, 62.00, 61.4, 60.4, 41.1, 41.07, 41.03, 37.1, 34.5, 34.2, 26.5, 26.1, 24.2, 22.6, 22.5, 22.1, 1.8, 18.6; IR (ATR) 1816 cm−1; HRMS m/z (ESI) calcd for C9H12O3 [M+] 168.0786, found: 168.0788.
(3,3-Dimethyloxiran-2-yl)methyl 9-(4-oxooxetan-2-yl)nonanoate (5r): The compound 5r was obtained by using 4r (284 mg, 1.00 mmol), 3a (4.1 mg 0.3 mol %), and CO (50 atm) in THF (0.5 mL) at 90 °C for 16 h by following the general procedure. Colorless liquid (262 mg, 84%). 1H NMR (400 MHz) δ 4.52–4.47 (m, 1H), 4.33 (dd, J = 12.1, 4.2 Hz, 1H), 4.01 (dd, J = 12.1, 7.0 Hz, 1H), 3.50 (dd, J = 16.3, 5.8 Hz, 1H), 3.05 (dd, J = 16.3, 4.3 Hz, 1H), 2.98 (dd, J = 6.9, 4.2 Hz, 1H), 2.35 (t, J = 7.5 Hz, 2H), 1.90–1.81 (m, 1H), 1.79–1.68 (m, 1H), 1.65–1.59 (m, 2H), 1.34–1.30 (m, 16H); 13C NMR (100 MHz, CDCl3) δ 173.6, 168.3, 71.3, 63.3, 60.5, 58.1, 42. 9, 34.6, 34.0, 29.1, 29.0 (2C), 28. 9, 24.8, 24.8, 24.6, 18.9; IR (ATR) 1824 (lactone), 1734 (ester) cm−1; HRMS m/z (ESI) calcd for C17H28O5 [M+] 312.1937, found: 312.1939.
4. Conclusions
A new highly reactive Sn(IV) porphyrin-mediated epoxide carbonylation was developed, producing synthetically valuable β-lactones. The Lewis acidic Sn(IV)-centered porphyrin activated an epoxide, and cobalt tetracarbonyl anions delivered carbon monoxide with high efficiencies. In particular, TONs up to 1260 were achieved. Furthermore, a variation of the porphyrin structure revealed the importance of high Lewis acidity and a broad substrate scope was disclosed. This new catalyst also exhibited interesting chemical selectivity on epoxide substitution patterns; thus, the synthesis of molecules with both epoxide and β-lactone has been realized, and its further utilization is currently in progress.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/9/4/311/s1, Figure S1: ORTEP of 2a.
Author Contributions
Conceptualization, J.G.K.; experiment design, J.G.K. and M.H.P.; catalyst study, E.R.B.; X-ray crystallography, D.K.; writing-original draft preparation, E.R.B. and J.G.K.; writing-review and editing, J.G.K., S.L., and M.H.P.; supervision, S.L., M.H.P., and J.G.K.
Funding
This research was funded by Next Generation Carbon Upcycling Project (NRF-2017M1A2A2043113).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kadish, K.; Smith, K.M.; Guilard, R. The Porphyrin Handbook: Chlorophylls and Bilins: Biosynthesis, Synthesis and Degradation; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Reddy, K.R.; Jiang, J.; Krayer, M.; Harris, M.A.; Springer, J.W.; Yang, E.; Jiao, J.; Niedzwiedzki, D.M.; Pandithavidana, D.; Parkes-Loach, P.S.; et al. Palette of lipophilic bioconjugatable bacteriochlorins for construction of biohybrid light-harvesting architectures. Chem. Sci. 2013, 4, 2036–2053. [Google Scholar] [CrossRef]
- Croce, R.; van Amerongen, H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Bio. 2014, 10, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Feher, G.; Allen, J.P.; Okamura, M.Y.; Rees, D.C. Structure and function of bacterial photosynthetic reaction centres. Nature 1989, 339, 111–116. [Google Scholar] [CrossRef]
- Sessler, J.L.; Seidel, D. Synthetic Expanded Porphyrin Chemistry. Angew. Chem. Int. Ed. 2003, 42, 5134–5175. [Google Scholar] [CrossRef]
- Saito, S.; Osuka, A. Expanded porphyrins: Intriguing Structures, Electronic Properties, and Reactivities. Angew. Chem. Int. Ed. 2011, 50, 4342–4373. [Google Scholar] [CrossRef]
- Jiang, L.; Engle, J.T.; Sirk, L.; Hartley, C.S.; Ziegler, C.J.; Wang, H. Triphenylene-Fused Porphyrins. Org. Lett. 2011, 13, 3020–3023. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Furuta, H.; Yoza, K.; Igarashi, S.; Osuka, A. meso-Aryl-Substituted Expanded Porphyrins. J. Am. Chem. Soc. 2001, 123, 7190–7191. [Google Scholar] [CrossRef]
- Kobayashi, N.; Takeuchi, Y.; Matsuda, A. meso-Aryl Subporphyrins. Angew. Chem. Int. Ed. 2007, 46, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Szyszko, B.; Latos-Grażyński, L. Core Chemistry and Skeletal Rearrangements of Porphyrinoids and Metalloporphyrinoids. Chem. Soc. Rev. 2015, 44, 3588–3616. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.J.; Latos-Grażyński, L. Core Modified Porphyrins—A Macrocyclic Platform for Organometallic Chemistry. Coord. Chem. Rev. 2005, 249, 2510–2533. [Google Scholar] [CrossRef]
- Carvalho, C.M.B.; Brocksom, T.J.; de Oliveira, K.T. Tetrabenzoporphyrins: Synthetic Developments and Applications. Chem. Soc. Rev. 2013, 42, 3302–3317. [Google Scholar] [CrossRef]
- Roznyatovskiy, V.V.; Lee, C.-H.; Sessler, J.L. π-Extended Isomeric and Expanded Porphyrins. Chem. Soc. Rev. 2013, 42, 1921–1933. [Google Scholar] [CrossRef]
- Davis, N.K.S.; Thompson, A.L.; Anderson, H.L. A Porphyrin Fused to Four Anthracenes. J. Am. Chem. Soc. 2011, 133, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Z.; Zhang, X.; Wang, Y.; Tian, J.; Huang, Y.; Ma, Y.; Zhang, X.; Chen, Y. A Graphene Hybrid Material Covalently Functionalized with Porphyrin: Synthesis and Optical Limiting Property. Adv. Mater. 2009, 21, 1275–1279. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Wathier, M.; Grinstaff, M.W. Synthesis and Properties of Supramolecular Ionic Networks. J. Am. Chem. Soc. 2008, 130, 9648–9649. [Google Scholar] [CrossRef]
- Drain, C.M.; Varotto, A.; Radivojevic, I. Self-Organized Porphyrinic Materials. Chem. Rev. 2009, 109, 1630–1658. [Google Scholar] [CrossRef]
- Vlascici, D.; Cosma, E.F.; Pica, E.M.; Cosma, V.; Bizerea, O.; Mihailescu, G.; Olenic, L. Free Base Porphyrins as Ionophores for Heavy Metal Sensors. Sensors 2008, 8, 4995–5004. [Google Scholar] [CrossRef]
- O’Connor, A.E.; Gallagher, W.M.; Byrne, A.T. Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol. 2009, 85, 1053–1074. [Google Scholar] [CrossRef]
- Endo, M.; Fujitsuka, M.; Majima, T. Diastereochemically Controlled Porphyrin Dimer Formation on a DNA Duplex Scaffold. J. Org. Chem. 2008, 73, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Takanami, T.; Hayashi, M.; Iso, K.; Nakamoto, H.; Suda, K. Highly Regioselective [3,3] Rearrangement of Aliphatic Allyl Vinyl Ethers Catalyzed by a Metalloporphyrin Complex, Cr (TPP) Cl. Tetrahedron 2006, 62, 9467–9474. [Google Scholar] [CrossRef]
- Brothers, P.J. Organoelement Chemistry of Main-Group Porphyrin Complexes. Adv. Organometal. Chem. 2001, 48, 289–342. [Google Scholar]
- Brothers, P.J. Recent developments in the coordination chemistry of porphyrin complexes containing non-metallic and semi-metallic elements. J. Porphyrins Phthalocyanines 2002, 6, 259–267. [Google Scholar] [CrossRef]
- Enakieva, Y.Y.; Volostnykh, M.V.; Nefedov, S.E.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y.; Bessmertnykh-Lemeune, A.G.; Stern, C.; Guilard, R. Gallium(III) and Indium(III) Complexes with meso-Monophosphorylated Porphyrins: Synthesis and Structure. A First Example of Dimers Formed by the Self-Assembly of meso-Porphyrinylphosphonic Acid Monoester. Inorg. Chem. 2017, 56, 3055–3070. [Google Scholar] [CrossRef]
- Chatterjee, C.; Chisholm, M.H. The Influence of the Metal (Al, Cr, and Co) and the Substituents of the Porphyrin in Controlling the Reactions Involved in the Copolymerization of Propylene Oxide and Carbon Dioxide by Porphyrin Metal(III) Complexes. 1. Aluminum Chemistry. Inorg. Chem. 2011, 50, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.; Menotti, A.R. Porphyrin Studies. V. The Metal Complex Salts of α,β,γ,δ-Tetraphenylporphine. J. Am. Chem. Soc. 1948, 70, 1808–1812. [Google Scholar] [CrossRef] [PubMed]
- Rafiemanzelat, F.; Abdollahi, E.; Moghadam, M.; Mirkhani, V.; Tangestaninejad, S.; Mohammadpoor-Baltork, I. Application of Tin(IV) Porphyrin Complexes as Novel catalysts for the Synthesis of New Copolyurethanes with Cyclopeptide Moiety. J. Appl. Polym. Sci. 2012, 124, 638–646. [Google Scholar] [CrossRef]
- Kumar, A.A.; Giribabu, L.; Reddy, D.R.; Maiya, B.G. New Molecular Arrays Based on a Tin(IV) Porphyrin Scaffold. Inorg. Chem. 2001, 40, 6757–6766. [Google Scholar] [CrossRef]
- Tangestaninejad, S.; Habibi, M.H.; Mirkhani, V.; Moghadam, M. Rapid and Efficient Acetylation of Alcohols and Phenols with Acetic Anhydride using Tin(IV) Porphyrin as Catalyst. Synth. Commun. 2002, 32, 1337–1343. [Google Scholar] [CrossRef]
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpour-Baltork, I.; Shaibani, R. Rapid and Efficient Acetylation of Alcohols and Phenols with Acetic Anhydride Catalyzed by Electron-deficient Tin (IV) Porphyrin. J. Mol. Catal. A: Chem. 2004, 219, 73–78. [Google Scholar] [CrossRef]
- Gharaati, S.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Kosari, F. Highly Efficient and Selective Methoxymethylation of Alcohols and Phenols Catalyzed by High-valent Tin (IV) Porphyrin. Inorg. Chim. Acta 2010, 363, 1995–2000. [Google Scholar] [CrossRef]
- Arnold, D.P.; Blok, J. The coordination chemistry of tin porphyrin complexes. Coord. Chem. Rev. 2004, 248, 299–319. [Google Scholar] [CrossRef]
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Taghavi, S.A. High-valent tin(IV) porphyrin, SnIV(TPP)(BF4)2, as an efficient catalyst for the ring-opening of epoxides. Catal. Commun. 2007, 8, 2087–2095. [Google Scholar] [CrossRef]
- Lee, J.T.; Thomas, P.J.; Alper, H. Synthesis of β-Lactones by the Regioselective, Cobalt and Lewis Acid Catalyzed Carbonylation of Simple and Functionalized Epoxides. J. Org. Chem. 2001, 66, 5424–5426. [Google Scholar] [CrossRef]
- Schmidt, J.A.R.; Mahadevan, V.; Getzler, Y.D.Y.L.; Coates, G.W. A Readily Synthesized and Highly Active Epoxide Carbonylation Catalyst Based on a Chromium Porphyrin Framework: Expanding the Range of Available β-Lactones. Org. Lett. 2004, 6, 373–376. [Google Scholar] [CrossRef]
- Schmidt, J.A.R.; Lobkovsky, E.B.; Coates, G.W. Chromium(III) Octaethylporphyrinato Tetracarbonylcobaltate: A Highly Active, Selective, and Versatile Catalyst for Epoxide Carbonylation. J. Am. Chem. Soc. 2005, 127, 11426–11435. [Google Scholar] [CrossRef]
- Kramer, J.W.; Lobkovsky, E.B.; Coates, G.W. Practical β-Lactone Synthesis: Epoxide Carbonylation at 1 atm. Org. Lett. 2006, 8, 3709–3712. [Google Scholar] [CrossRef]
- Mahadevan, V.; Getzler, Y.D.Y.L.; Coates, G.W. [Lewis Acid]+[Co(CO)4]− Complexes: A Versatile Class of Catalysts for Carbonylative Ring Expansion of Epoxides and Aziridines. Angew. Chem. Int. Ed. 2002, 41, 2781–2784. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Goldmacher, J.; Assour, J.; Korsakoff, L. A Simplified Synthesis for meso-Tetraphenylporphine. J. Org. Chem 1967, 32, 476. [Google Scholar] [CrossRef]
- Slagt, V.F.; van Leeuwen, P.W.N.M.; Reek, J.N.H. Supramolecular Bidentate Phosphorus Ligands Based on Bis-zinc (II) and Bis-tin (IV) Porphyrin Building Blocks. Dalton Trans. 2007, 2302–2310. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Torres, R.; Alvarez, S. Coordinating ability of Anions and Solvents towards Transition Metals and Lanthanides. Dalton Trans. 2011, 40, 10742–10750. [Google Scholar] [CrossRef]
- Rowley, J.M.; Lobkovsky, E.B.; Coates, G.W. Catalytic Double Carbonylation of Epoxides to Succinic Anhydrides: Catalyst Discovery, Reaction Scope, and Mechanism. J. Am. Chem. Soc. 2007, 129, 4948–4960. [Google Scholar] [CrossRef] [PubMed]
- Coates and coworkers’ original development of Cr-porphyrin catalyst exhibited high TON up to 10000 in ref. 43. However, the use of propylene oxide was not presented.
- Edgell, W.F.; Lyford, J., IV. The Preparation of Sodium Cobalt Tetracarbonyl. Inorg. Chem. 1970, 9, 1932–1933. [Google Scholar] [CrossRef]
- Sharma, R.; Bulger, P.G.; McNevin, M.; Dormer, P.G.; Ball, R.G.; Streckfuss, E.; Cuff, J.F.; Yin, J.; Chen, C.-y. A Cascade Approach to Cyclic Aminonitrones: Reaction Discovery, Mechanism, and Scope. Org. Lett. 2009, 11, 3194–3197. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Croft, R.A.; Whittingham, W.G.; Bower, J.F. Modular Access to Substituted Azocanes via a Rhodium-Catalyzed Cycloaddition–Fragmentation Strategy. J. Am. Chem. Soc. 2015, 137, 8054–8057. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.R.; Jung, Y.; Coates, G.W. Meinwald-type Rearrangement of Monosubstituted Epoxides to Methyl Ketones using an [Al porphyrin]+[Co(CO)4]− catalyst. Org. Chem. Front. 2015, 2, 346–349. [Google Scholar] [CrossRef]
- Taber, D.F.; Paquette, C.M.; Gu, P.; Tian, W. Cyclohexanones by Rh-Mediated Intramolecular C–H Insertion. J. Org. Chem. 2013, 78, 9772–9780. [Google Scholar] [CrossRef] [PubMed]
- Hao, E.; Wang, Z.; Jiao, L.; Wang, S. “Click” Tetradentate Ligands. Dalton Trans. 2010, 39, 2660–2666. [Google Scholar] [CrossRef]
- Arnold, D.P.; Morrison, E.A.; Hanna, J.V. Tin(IV) Porphyrin Complexes—III. NMR Trans-influences in Tin(IV) Tetraphenylporphyrin Complexes. Polyhedron 1990, 9, 1331–1336. [Google Scholar] [CrossRef]
- Sigma-Aldrich, commercial product, Cas number 3068-88-0.
- Ganji, P.; Doyle, D.J.; Ibrahim, H. In Situ Generation of the Coates Catalyst: A Practical and Versatile Catalytic System for the Carbonylation of meso-Epoxides. Org. Lett. 2011, 13, 3142–3145. [Google Scholar] [CrossRef]
- Nelson, S.G.; Spencer, K.L. Sequential Acyl Halide−Aldehyde Cyclocondensation and Enzymatic Resolution as a Route to Enantiomerically Enriched β-Lactones. J. Org. Chem. 2000, 65, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Gnanadesikan, V.; Corey, E.J. Enantioselective β-Lactone Formation from Ketene and Aldehydes Catalyzed by a Chiral Oxazaborolidine. Org. Lett. 2006, 8, 4943–4945. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Ageishi, S.; Isobe, H.; Hayashi, Y.; Yamamoto, Y. Lipase Promoted Asymmetric Transesterification of 4-Alkyl-, 3-Alkyl-and 3, 4-Dialkyloxetan-2-ones with Ring-opening. J. Chem. Soc. Perkin Trans. 2000, 71–77. [Google Scholar] [CrossRef]
- Concellón, J.M.; Concellón, C. Aldol-type Reactions of Unmasked Iodoacetic Acid with Carbonyl Compounds Promoted by Samarium Diiodide: Efficient Synthesis of Carboxylic 3-Hydroxyacids and Their Derivatives. J. Org. Chem. 2006, 71, 4428–4432. [Google Scholar] [CrossRef] [PubMed]
- Martín, C.; Belderraín, T.R.; Pérez, P.J. Rediscovering Copper-based Catalysts for Intramolecular Carbon–Hydrogen Bond Functionalization by Carbene Insertion. Org. Biomol. Chem. 2009, 7, 4777–4781. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
General reaction mechanism of epoxide carbonylation to form β-lactones.

Scheme 2.
Synthesis of Tin(IV) porphyrin catalysts. (i) Propionic acid, reflux, 1 h. (ii) SnCl2, 2H2O, pyridine, reflux, 3 h. (iii) NaCo(CO)4, THF, N2, rt, 48 h.
Scheme 2.
Synthesis of Tin(IV) porphyrin catalysts. (i) Propionic acid, reflux, 1 h. (ii) SnCl2, 2H2O, pyridine, reflux, 3 h. (iii) NaCo(CO)4, THF, N2, rt, 48 h.

Scheme 3.
Identification of reactive catalytic species.

Scheme 4.
Chemoselective carbonylations of di-epoxides.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization results 1,2.

| Entry | Catalyst | mol % | Temp (oC) | CO (atm) | Solvent | Yield (%)3 | TON 4 |
|---|---|---|---|---|---|---|---|
| 1 | 3a | 0.3 | 90 | 50 | THF | >99 | 333 |
| 2 | 3b | 0.3 | 90 | 50 | THF | 94 | 313 |
| 3 | 3c | 0.3 | 90 | 50 | THF | 83 | 277 |
| 4 | 3d | 0.3 | 90 | 50 | THF | 57 | 190 |
| 5 | 3a | 0.3 | 90 | 50 | 2-Me-THF | >99 | 333 |
| 6 | 3a | 0.3 | 90 | 50 | 1,4-dioxane | 89 | 297 |
| 7 | 3a | 0.3 | 90 | 50 | DMF | 0 | - |
| 8 | 3a | 0.3 | 90 | 50 | MeOH | 0 | - |
| 9 | 3a | 0.3 | 90 | 50 | o-xylene | 35 | 117 |
| 10 | 3a | 0.3 | 90 | 50 | toluene | 13 | 43 |
| 11 | 3a | 0.3 | 90 | 50 | n-hexane | 3 | 10 |
| 12 | 3a | 0.3 | 70 | 50 | THF | 61 | 203 |
| 13 | 3a | 0.3 | 50 | 50 | THF | 20 | 67 |
| 14 | 3a | 0.3 | 90 | 30 | THF | 95 | 317 |
| 15 | 3a | 0.3 | 90 | 10 | THF | 63 | 210 |
| 16 | 3a | 0.2 | 90 | 50 | THF | 91 | 455 |
| 17 | 3a (24 h) | 0.2 | 90 | 50 | THF | >99 | 500 |
| 18 | 3a (24 h) | 0.1 | 90 | 50 | THF | 80 | 800 |
| 19 | 3a (36 h) | 0.1 | 90 | 70 | THF | >99 | 1000 |
| 21 | 3a (36 h) | 0.05 | 110 | 70 | THF | 63 | 1260 |
1 Unless otherwise indicated, the reaction was carried out for 16 h. 2 Single experiment was performed on each entry. 3 Yields were determined by 1H NMR. 4 TON (turnover number) = Moles of product formed/moles of catalyst.
Table 2.
Scope of substrates 1,2.

 |
1 Single experiment was performed on each entry.2 Yields were determined by 1H NMR. Numbers in parentheses are isolation yields. 3 Catalyst loading = 0.4 mol %; 4 Catalyst loading = 0.5 mol %; 5 Catalyst loading = 0.6 mol %; 6 Catalyst loading = 1 mol %.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Baral, E.R.; Kim, D.; Lee, S.; Park, M.H.; Kim, J.G. Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides. Catalysts 2019, 9, 311. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040311
AMA Style
Baral ER, Kim D, Lee S, Park MH, Kim JG. Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides. Catalysts. 2019; 9(4):311. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040311
Chicago/Turabian StyleBaral, Ek Raj, Dongwook Kim, Sunwoo Lee, Myung Hwan Park, and Jeung Gon Kim. 2019. "Tin(IV)-Porphyrin Tetracarbonyl Cobaltate: An Efficient Catalyst for the Carbonylation of Epoxides" Catalysts 9, no. 4: 311. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9040311
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.