Stereoselective Crystallization of Chiral 3,4-Dimethylphenyl Glycerol Ether Complicated by Plurality of Crystalline Modifications

 , ,
, ,
Abstract
:
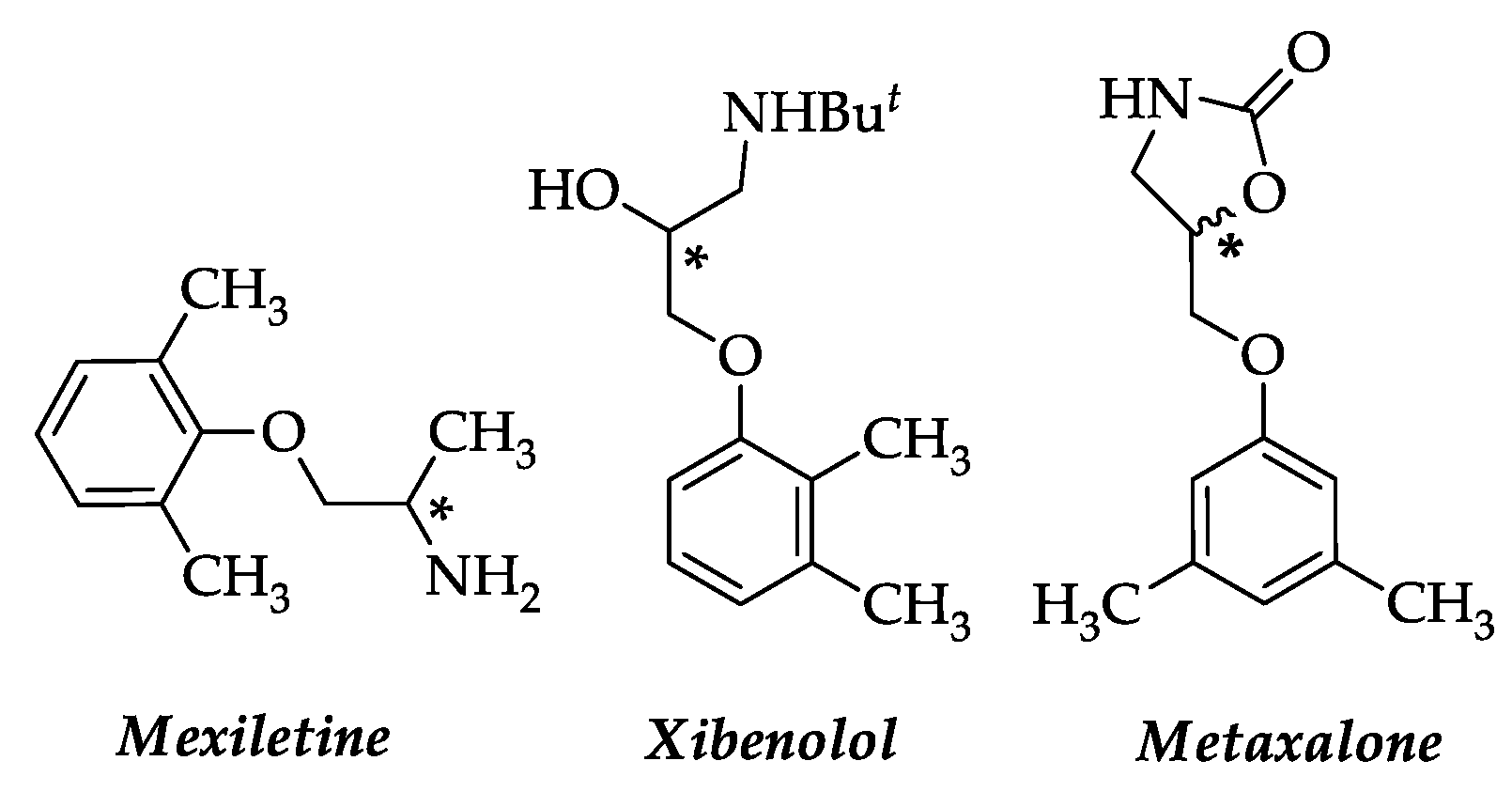
1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.2. Starting Materials
2.3. Synthesis and Samples Preparation
- (R)-3-(3,4-Dimethylphenoxy)propane-1,2-diol, (R)-1: mp 96–98 °C, [α]D20 –7.7 (c 1.0, EtOH), 99.9% ee (chiral HPLC analysis, tR 7.7 min). [Lit. [31] mp 96–98 °C, [α]D20 –7.7 (c 1.0, EtOH), 99.7% ee].
- (S)-3-(3,4-Dimethylphenoxy)propane-1,2-diol, (S)-1: mp 96–97 °C, [α]D20 +7.3 (c 1.0, EtOH), 99.2% ee (chiral HPLC analysis, tR 10.7 min). [Lit. [31] mp 96–97.5 °C, [α]D20 +6.7 (c 1.0, EtOH), 99.5% ee].
2.4. Single Crystal X-ray Analysis
2.5. Powder X-ray Diffraction Investigations
3. Results and Discussion
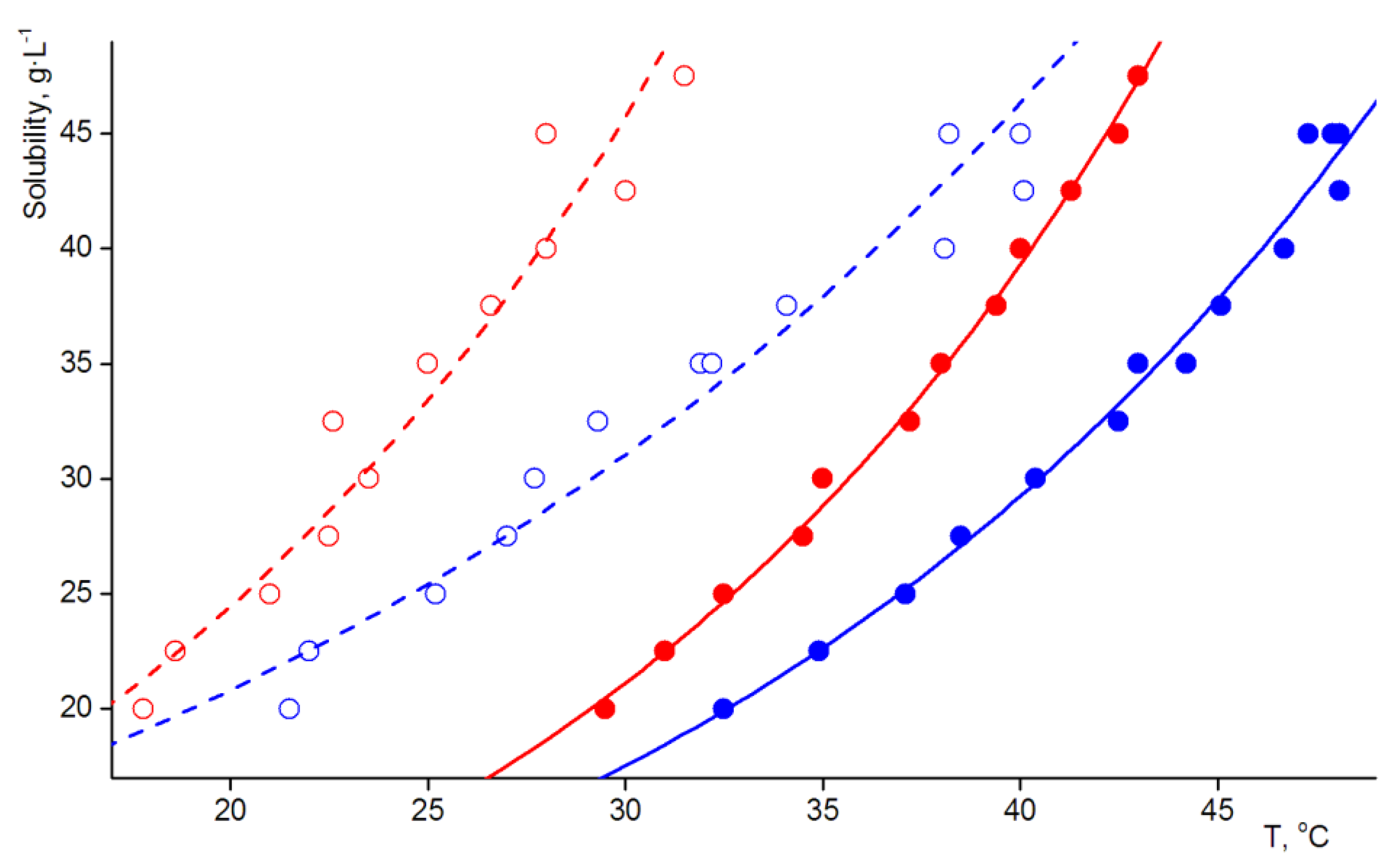
3.1. Solubility Test and Preliminary Entrainment Experiment
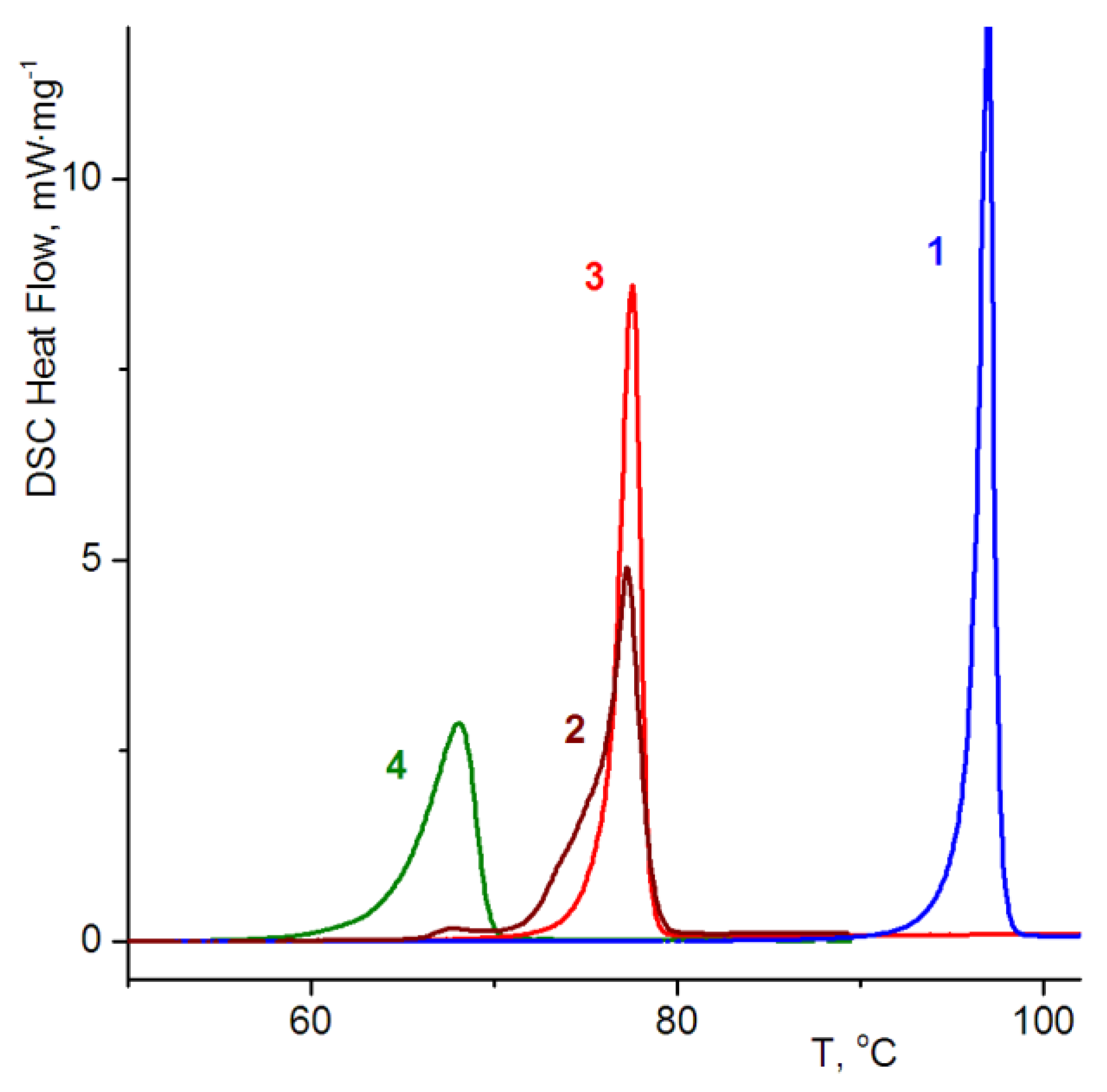
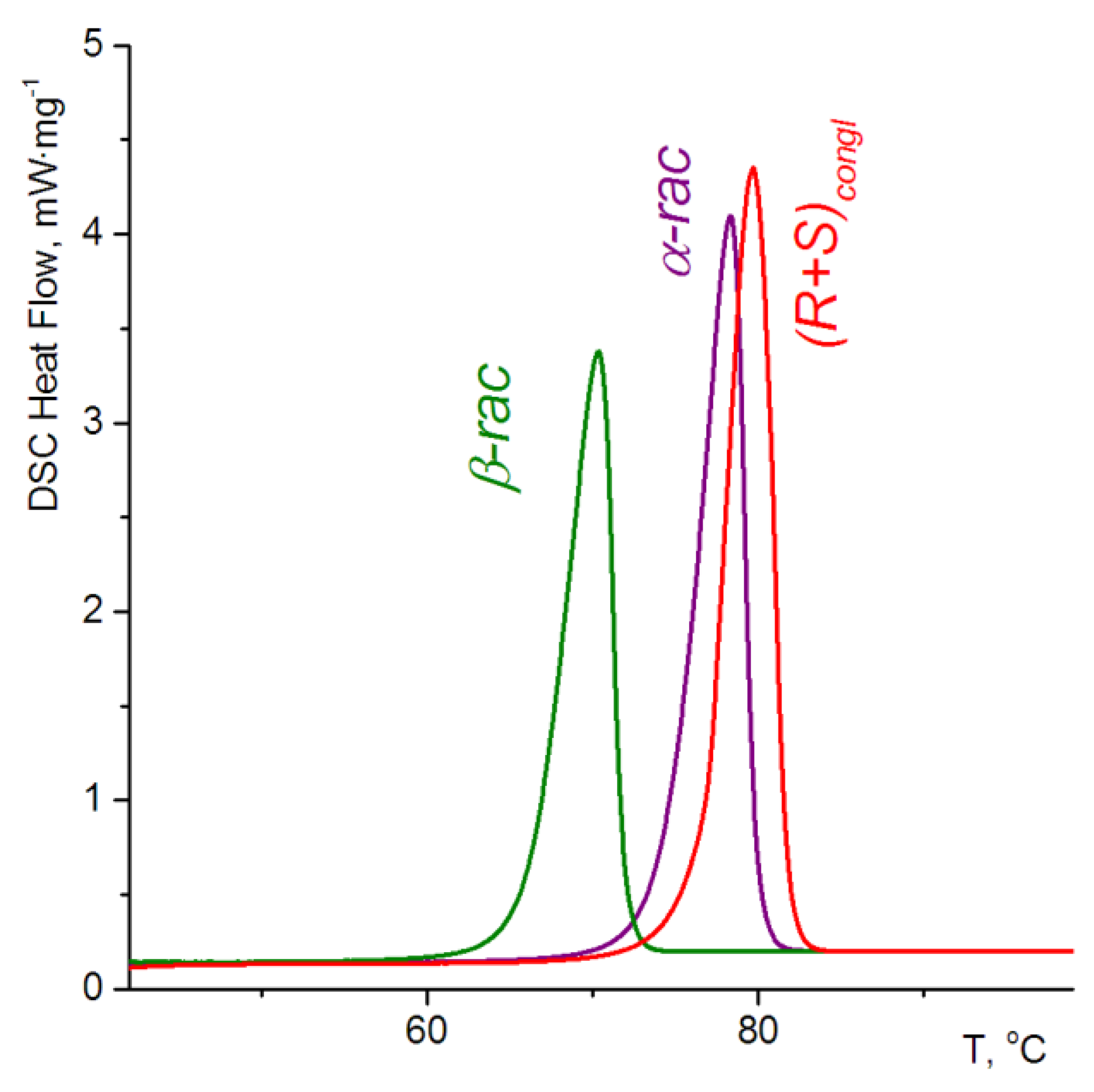
3.2. Thermochemical Investigations
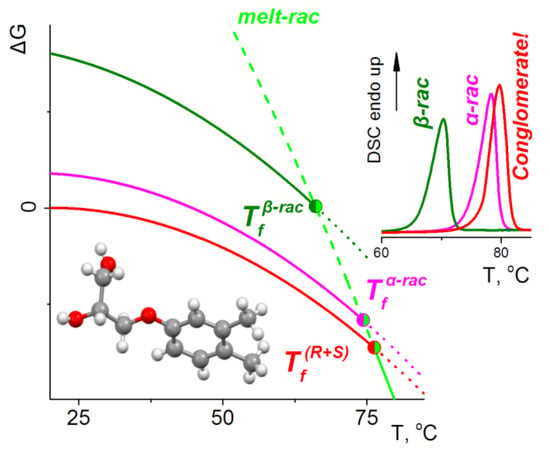
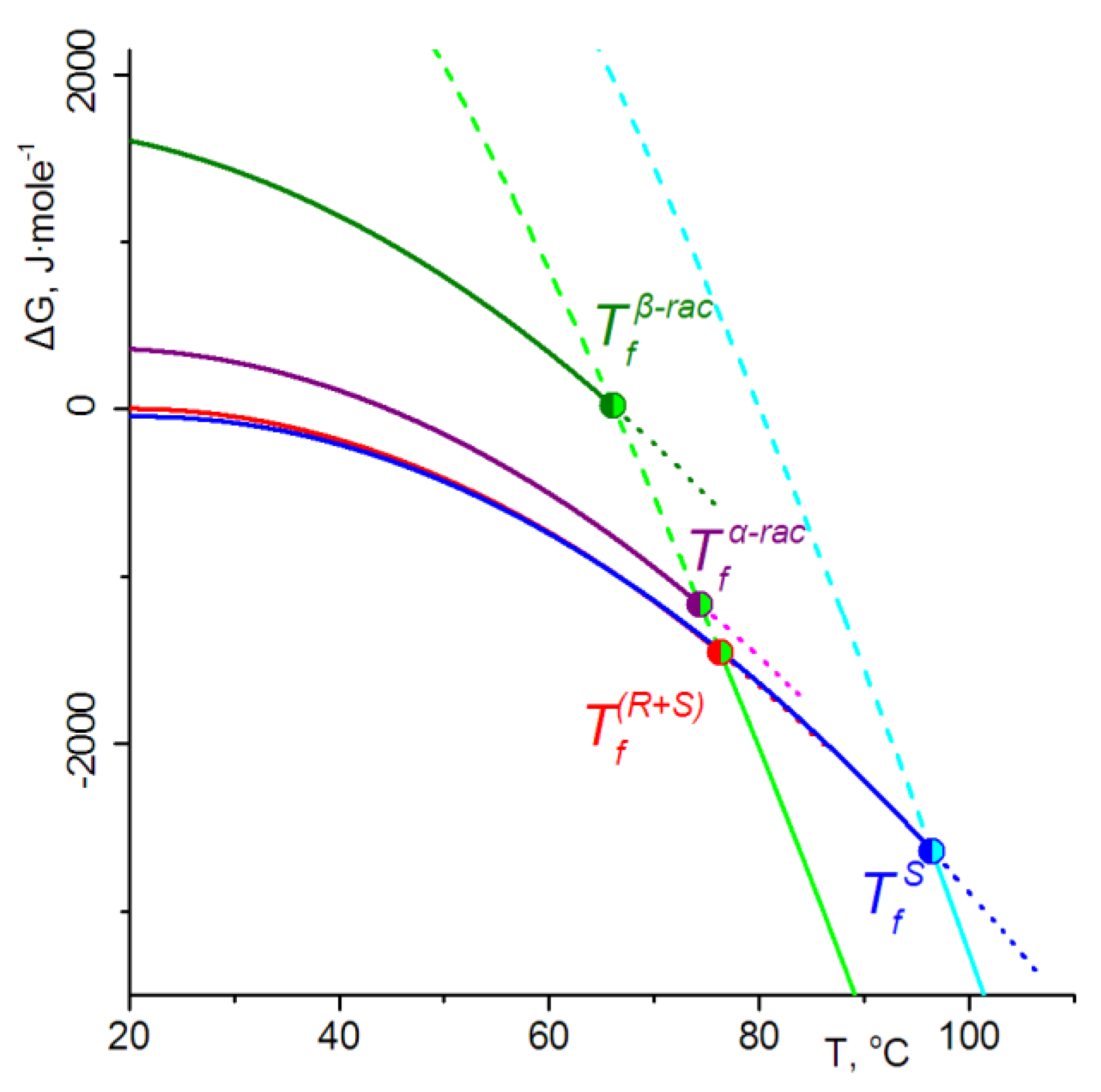
3.3. Phase Energetics
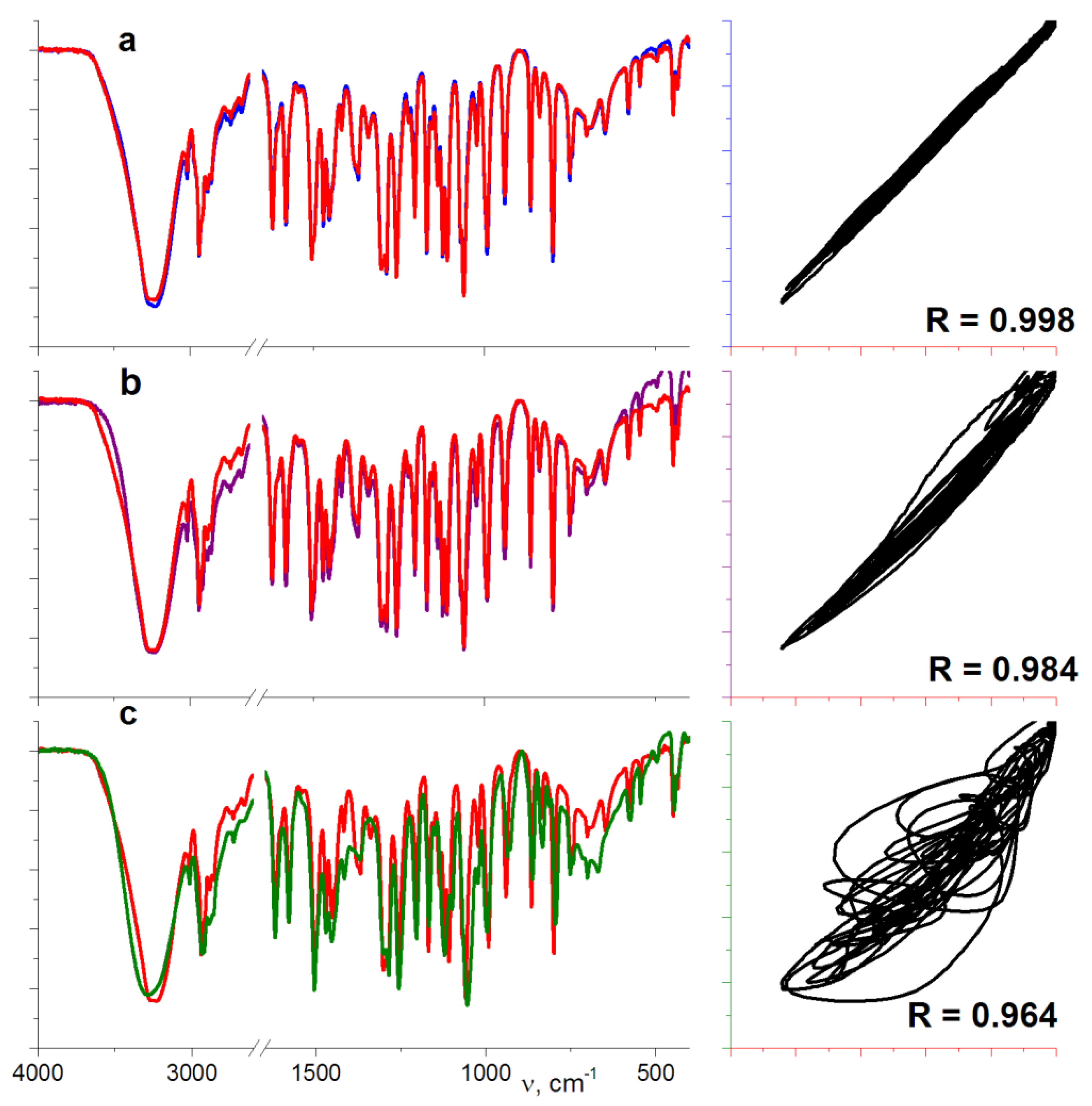
3.4. IR Investigations of All the Detected Phases
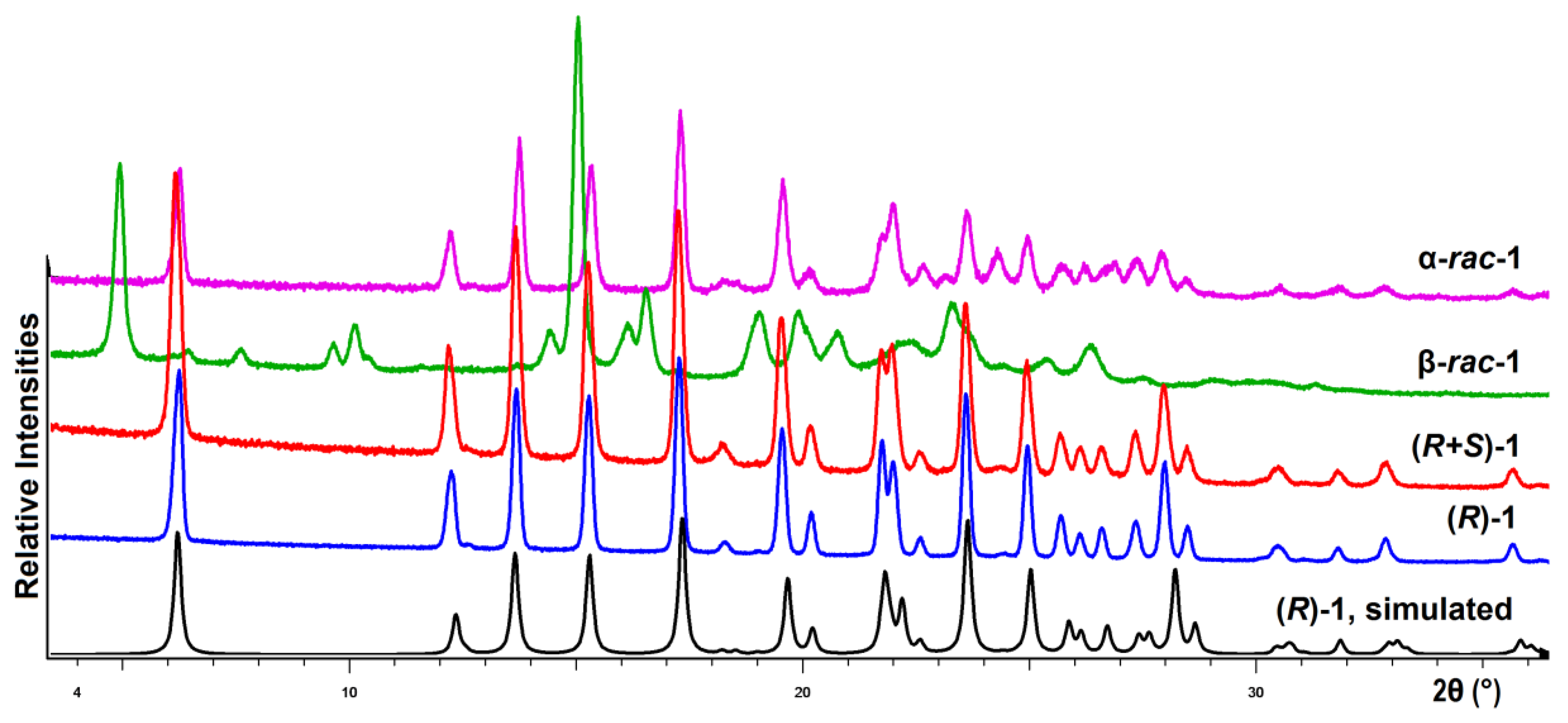
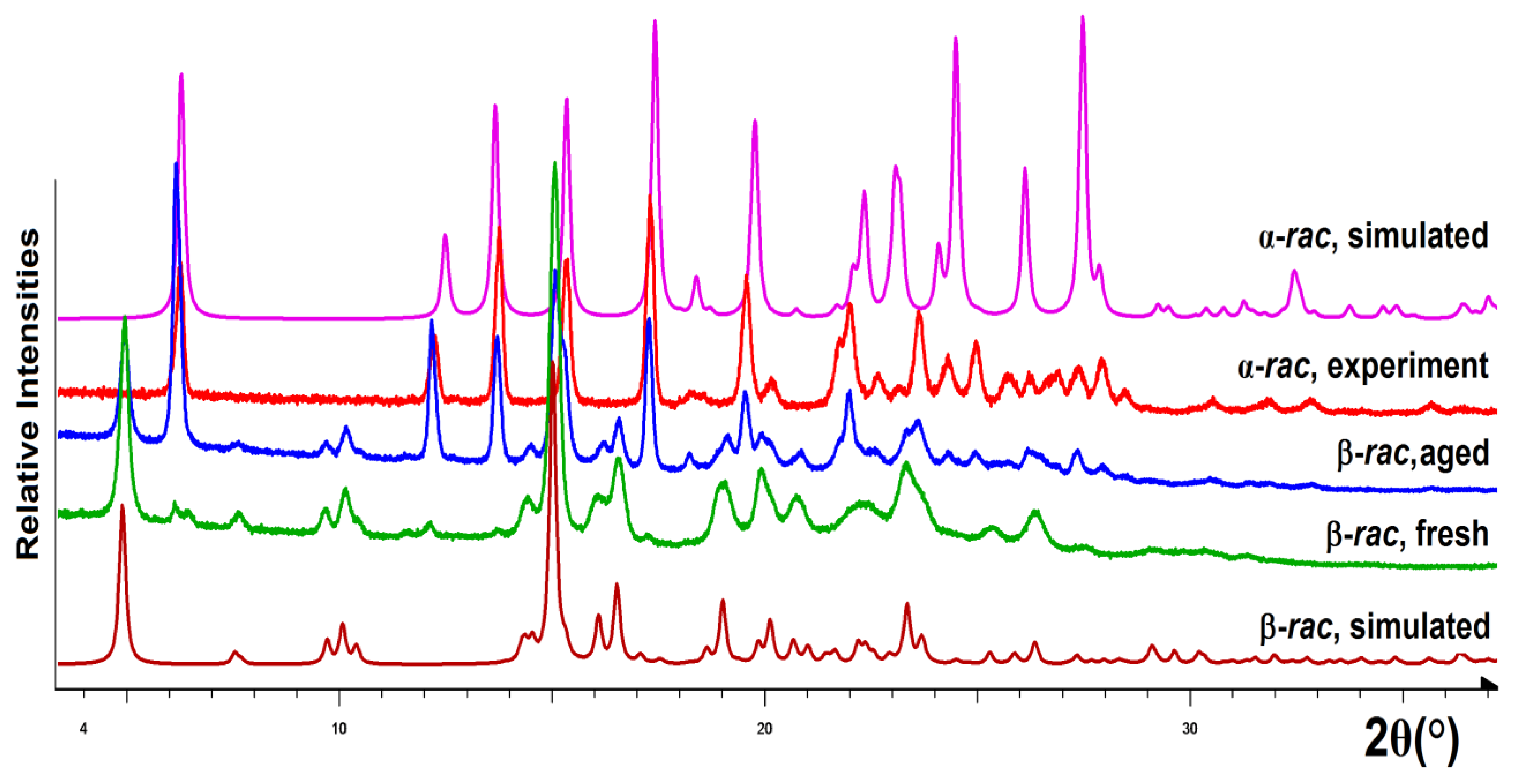
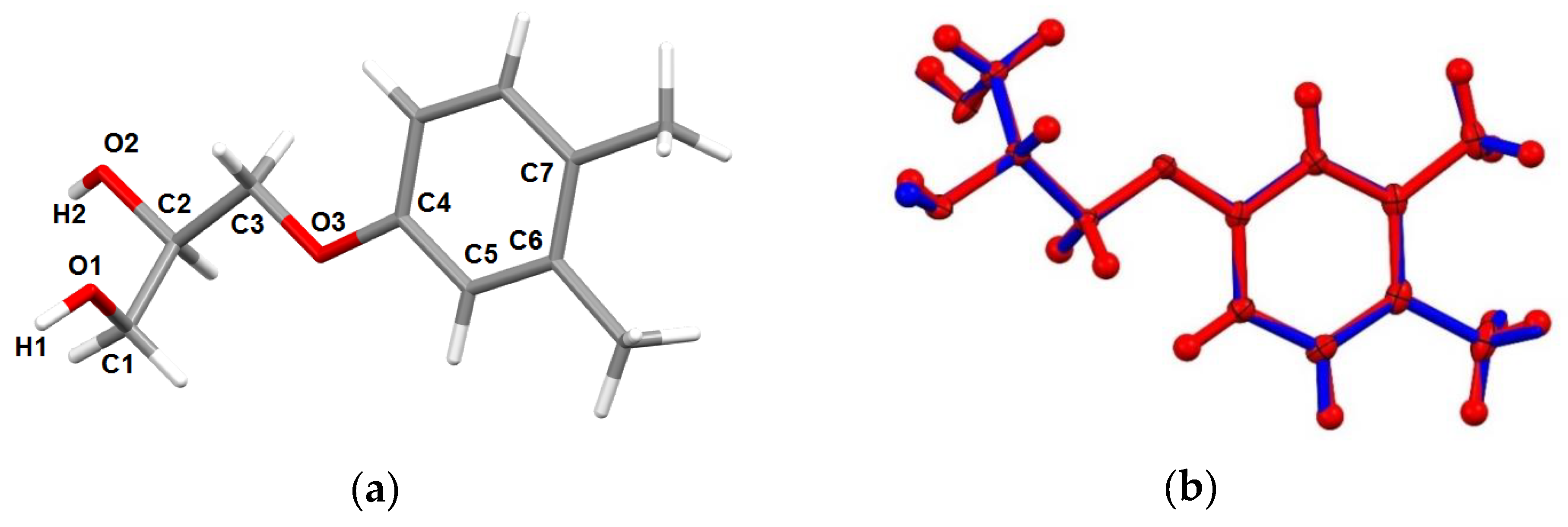
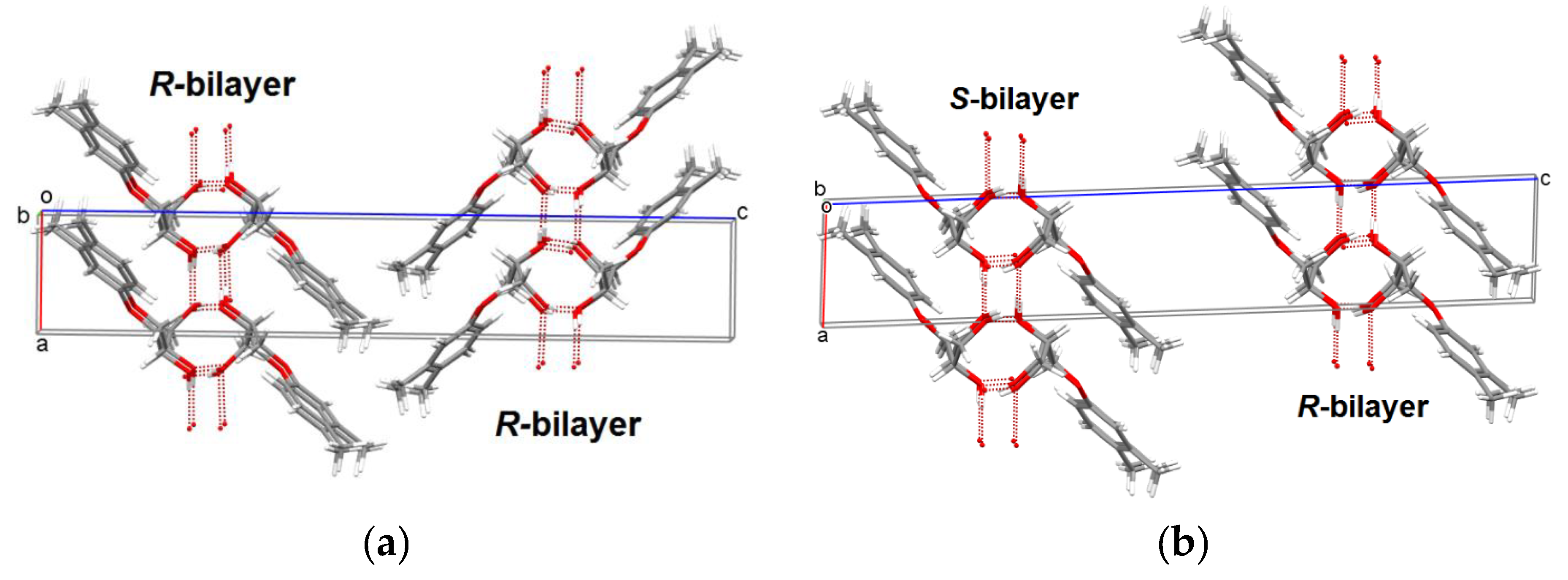
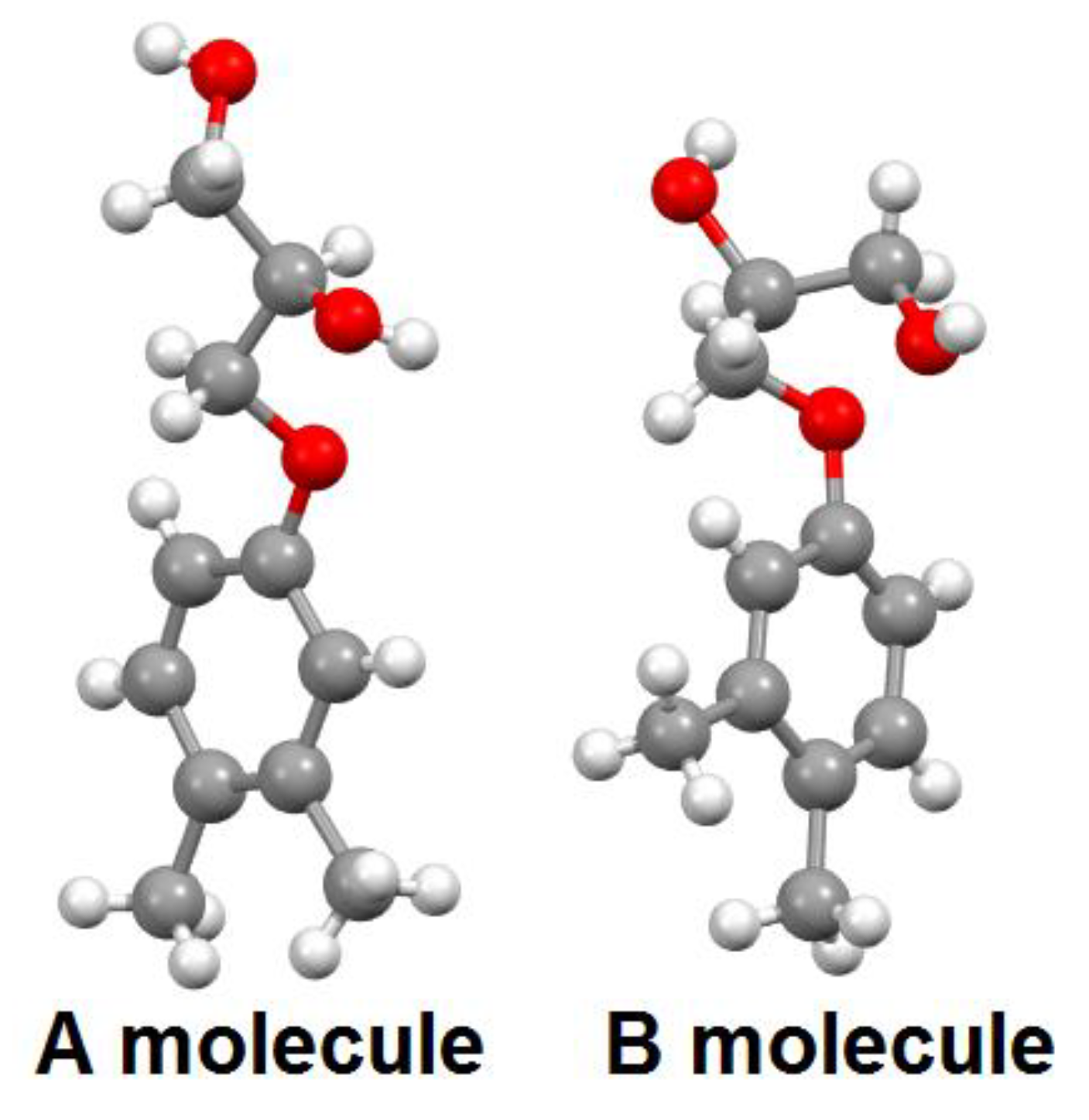
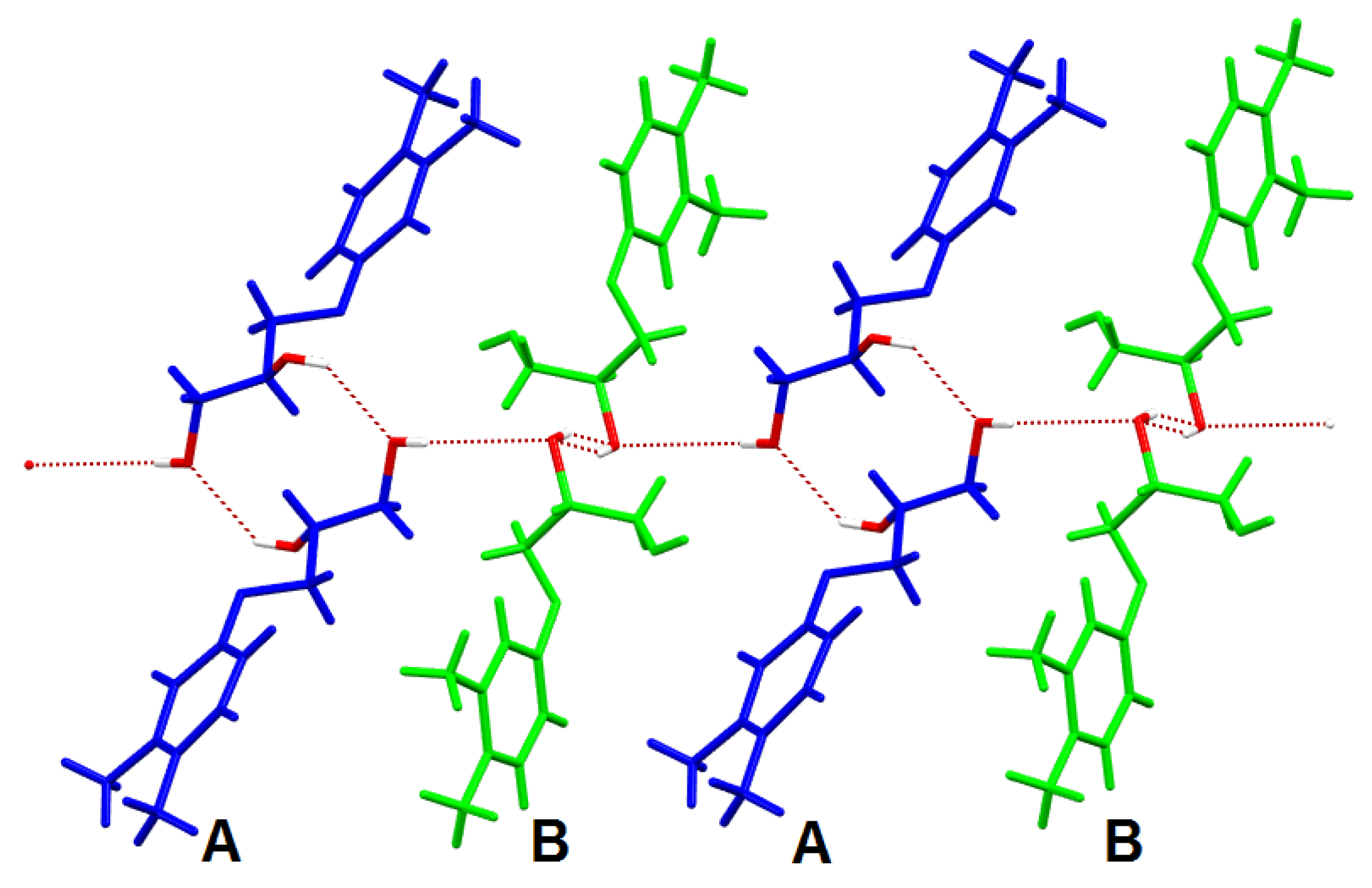
3.5. X-ray Diffraction Investigations
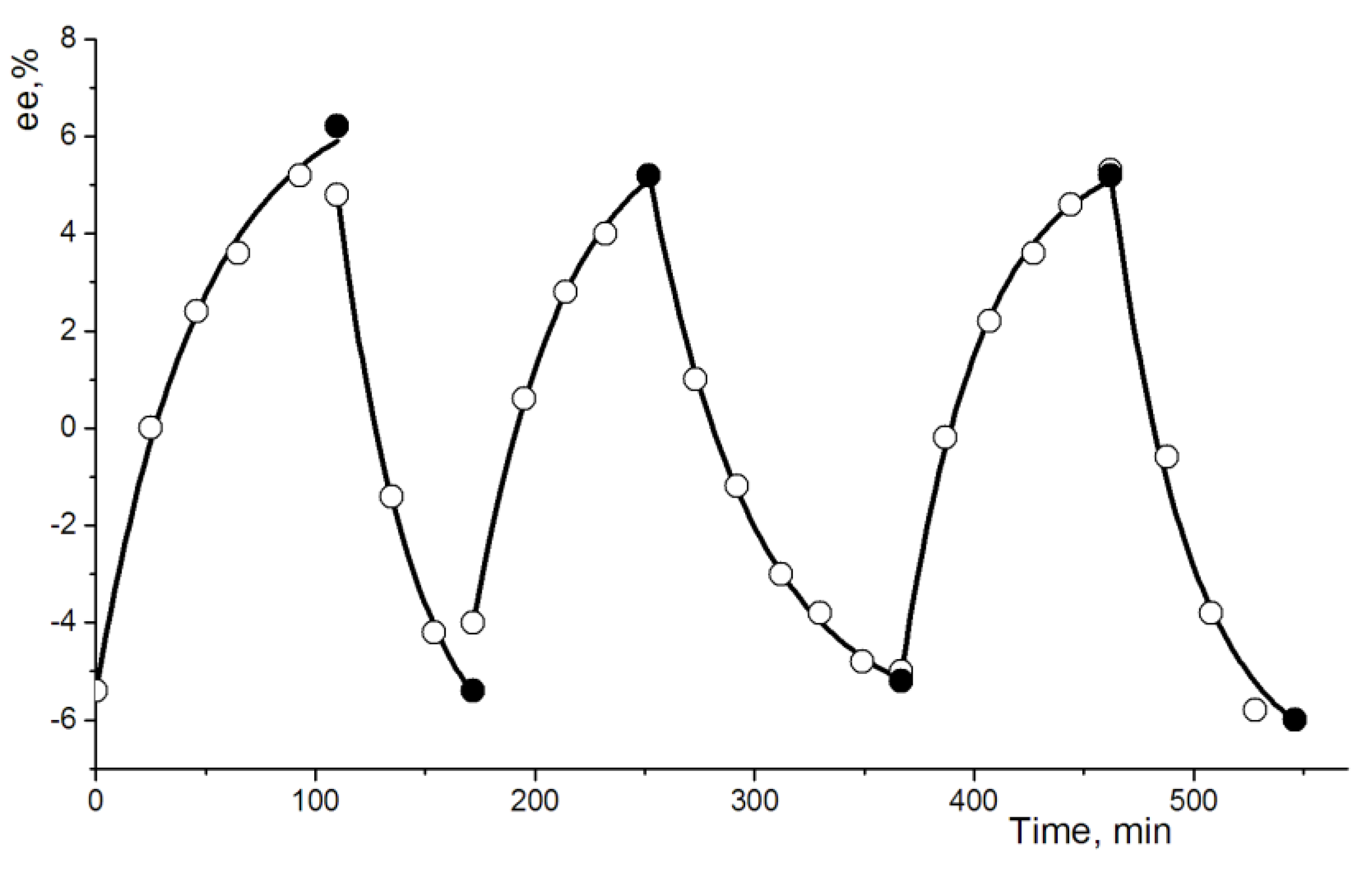
3.6. Direct Resolution of Rac-1 by Entrainment Procedure
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wagniere, G.H. On Chirality and the Universal Asymmetry: Reflections on Image and Mirror Image; Wiley-VCH: Hoboken, NJ, USA, 2007. [Google Scholar]
- Murakami, H. From racemates to single enantiomers—Chiral synthetic drugs over the last 20 years. Top. Curr. Chem. 2007, 269, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2018. An analysis of FDA drug approvals from the perspective of molecules. Molecules 2019, 24, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, B.; Girard, K.P.; Polster, C.S.; Croker, D.M. Progress to date in the design and operation of continuous crystallization processes for pharmaceutical applications. Org. Process Res. Dev. 2019, 23, 122–144. [Google Scholar] [CrossRef]
- Gao, Z.G.; Rohani, S.; Gong, J.B.; Wang, J.K. Recent developments in the crystallization process: Toward the pharmaceutical industry. Engineering 2017, 3, 343–353. [Google Scholar] [CrossRef]
- Lorenz, H.; Seidel-Morgenstern, A. Processes to separate enantiomers. Angew. Chem. Int. Ed. 2014, 53, 1218–1250. [Google Scholar] [CrossRef]
- Coquerel, G. Chiral discrimination in the solid state: Applications to resolution and deracemization. In Advances in Organic Crystal Chemistry; Tamura, R., Miyata, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 393–420. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, A. Crystallization-Based Separation of Enantiomers. In Stereoselective Synthesis of Drugs and Natural Products; Andrushko, V., Andrushko, N., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Faigl, F.; Fogassy, E.; Nogradi, M.; Palovics, E.; Schindler, J. Strategies in optical resolution: A practical guide. Tetrahedron Asymmetry 2008, 19, 519–536. [Google Scholar] [CrossRef]
- Palmans, A.R.A. Deracemisations under kinetic and thermodynamic control. Mol. Syst. Des. Eng. 2017, 2, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Bredikhin, A.A.; Bredikhina, Z.A. Stereoselective crystallization as a basis for single-enantiomer drug production. Chem. Eng. Technol. 2017, 40, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Levilain, G.; Eicke, M.J.; Seidel-Morgenstern, A. Efficient resolution of enantiomers by coupling preferential crystallization and dissolution. Part 1: Experimental proof of principle. Cryst. Growth Des. 2012, 12, 5396–5401. [Google Scholar] [CrossRef]
- Eicke, M.J.; Levilain, G.; Seidel-Morgenstern, A. Efficient resolution of enantiomers by coupling preferential crystallization and dissolution. Part 1: A parametric simulation study to identify suitable process conditions. Cryst. Growth Des. 2013, 13, 1638–1648. [Google Scholar] [CrossRef]
- Eicke, M.J.; Levilain, G.; Seidel-Morgenstern, A. Efficient resolution of enantiomers by coupling preferential crystallization and dissolution. Part 2: A parametric simulation study to identify suitable process conditions. Cryst. Growth Des. 2014, 14, 4872. [Google Scholar] [CrossRef]
- Cascella, F.; Seidel-Morgenstern, A.; Lorenz, H. Exploiting ternary solubility phase diagrams for resolution of enantiomers: An instructive example. Chem. Eng. Technol. 2020, 43, 329–336. [Google Scholar] [CrossRef]
- Oketani, R.; Marin, F.; Tinnemans, P.; Hoquante, M.; Laurent, A.; Brandel, C.; Cardinael, P.; Meekes, H.; Vlieg, E.; Geerts, Y.; et al. Deracemization in a complex quaternary system with a second-order asymmetric transformation by using phase diagram studies. Chem. Eur. J. 2019, 25, 13890–13898. [Google Scholar] [CrossRef] [Green Version]
- Binev, D.; Seidel-Morgenstern, A.; Lorenz, H. Continuous separation of isomers in fluidized bed crystallizers. Cryst. Growth Des. 2016, 16, 1409–1419. [Google Scholar] [CrossRef]
- Galan, K.; Eicke, M.J.; Elsner, M.P.; Lorenz, H.; Seidel-Morgenstern, A. Continuous preferential crystallization of chiral molecules in single and coupled mixed-suspension mixed-product-removal crystallizers. Cryst. Growth Des. 2015, 15, 1808–1818. [Google Scholar] [CrossRef]
- Cascella, F.; Temmel, E.; Seidel-Morgenstern, A.; Lorenz, H. Efficient resolution of racemic guaifenesin via batch-preferential crystallization processes. Org. Process Res. Dev. 2020, 24, 50–58. [Google Scholar] [CrossRef]
- Bredikhina, Z.A.; Novikova, V.G.; Zakharychev, D.V.; Bredikhin, A.A. Solid state properties and effective resolution procedure for guaifenesin, 3-(2-methoxyphenoxy)-1,2-propanediol. Tetrahedron Asymmetry 2006, 17, 3015–3020. [Google Scholar] [CrossRef]
- O’Neil, M.J. (Ed.) The Merck Index, 14th ed.; Merck and Co. Inc.: Whitehouse Station, NJ, USA, 2006. [Google Scholar]
- Saddique, F.A.; Zahoor, A.F.; Yousaf, M.; Irfan, M.; Ahmad, M.; Mansha, A.; Khan, Z.A.; Naqvi, S.A.R. Synthetic approaches towards the synthesis of beta-blockers (betaxolol, metoprolol, sotalol, and timolol). Turk. J. Chem. 2016, 40, 193–224. [Google Scholar] [CrossRef]
- Agustian, J.; Kamaruddin, A.H.; Bhatia, S. Single enantiomeric beta-blockers—The existing technologies. Process Biochem. 2010, 45, 587–1604. [Google Scholar] [CrossRef]
- Campo, C.; Llama, E.F.; Bermudez, J.L.; Sinisterra, J.V. Methodologies for the stereoselective synthesis of adrenergic beta-blockers: An overview. Biocatal. Biotransform. 2001, 19, 163–180. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Bredikhina, Z.A.; Zakharychev, D.V. Crystallization of chiral compounds: Thermodynamical, structural and practical aspects. Mendeleev Commun. 2012, 22, 171–180. [Google Scholar] [CrossRef]
- Bredikhina, Z.A.; Kurenkov, A.V.; Krivolapov, D.B.; Bredikhin, A.A. Stereoselective crystallization of 3-(2,6-dimethylphenoxy)propane-1,2-diol: Preparation of the single-enantiomer drug mexiletine. Tetrahedron Asymmetry 2015, 26, 577–583. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Bredikhina, Z.A.; Kurenkov, A.V.; Gubaidullin, A.T. Synthesis, crystal structure, and absolute configuration of the enantiomers of chiral drug xibenolol hydrochloride. Tetrahedron Asymmetry 2017, 28, 1359–1366. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Zakharychev, D.V.; Bredikhina, Z.A.; Kurenkov, A.V.; Krivolapov, D.B.; Gubaidullin, A.T. Spontaneous resolution of chiral 3-(2,3-dimethyl phenoxy)propane-1,2-diol under the circumstances of an unusual diversity of racemic crystalline modifications. Cryst. Growth Des. 2017, 17, 4196–4206. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Zakharychev, D.V.; Gubaidullin, A.T.; Bredikhina, Z.A. Solid phase behavior, polymorphism, and crystal structure features of chiral drug metaxalone. Cryst. Growth Des. 2018, 18, 6627–6639. [Google Scholar] [CrossRef]
- Bredikhina, Z.A.; Kurenkov, A.V.; Bredikhin, A.A. Nonracemic dimethylphenyl glycerol ethers in the synthesis of physiologically active aminopropanols. Russ. J. Org. Chem. 2019, 55, 837–844. [Google Scholar] [CrossRef]
- Hothersall, J.D.; Black, J.; Caddick, S.; Vinter, J.G.; Tinker, A.; Baker, J.R. The design, synthesis and pharmacological characterization of novel β2-adrenoceptor antagonists. Br. J. Pharmacol. 2011, 164, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Araujo, N.; Ferreira da Silva, A.; Qing, Y.; Lifino, M.; Russell, A.J.; Small, B.; Wade-Martins, R.; Wynne, G.M. Therapeutic Compounds. Int. Patent Appl. 2015. No. WO 2015/004485 A1. [Google Scholar]
- Sheldrick, G.M. SADABS, Program for Empirical X-ray Absorption Correction; Bruker-Nonius: Delft, The Netherlands, 2004. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Bruker AXS. DIFFRAC Plus Evaluation package EVA, Version 11. In User’s Manual; Bruker AXS: Karlsruhe, Germany, 2005; p. 258. [Google Scholar]
- Bruker AXS. TOPAS v3: General profile and structure analysis software for powder diffraction data. In Technical Reference; Bruker AXS: Karlsruhe, Germany, 2005; p. 117. [Google Scholar]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Meyerhoffer, W. Stereochemische Notizen. Ber. Dtsch. Chem. Ges. 1904, 37, 2604–2610. [Google Scholar] [CrossRef]
- Jaques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolutions; J. Wiley and Sons, Inc.: Hoboken, NJ, USA, 1981; p. 447. [Google Scholar]
- Li, Z.J.; Zell, M.T.; Munson, E.J.; Grant, D.J.W. Characterization of racemic species of chiral drugs using thermal analysis, thermodynamic calculation, and structural studies. J. Pharm. Sci. 1999, 88, 337–346. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Bredikhina, Z.A.; Akhatova, F.S.; Zakharychev, D.V.; Polyakova, E.V. From racemic compounds through metastable to stable racemic conglomerates: Crystallization features of chiral halogen and cyano monosubstituted phenyl glycerol ethers. Tetrahedron Asymmetry 2009, 20, 2130–2136. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Zakharychev, D.V.; Fayzullin, R.R.; Antonovich, O.A.; Pashagin, A.V.; Bredikhina, Z.A. Chiral para-alkyl phenyl ethers of glycerol: Synthesis and testing of chirality driven crystallization, liquid crystal, and gelating properties. Tetrahedron Asymmetry 2013, 24, 807–816. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Zakharychev, D.V.; Gubaidullin, A.T.; Fayzullin, R.R.; Samigullina, A.I.; Bredikhina, Z.A. Crystallization of chiral para-n-alkylphenyl glycerol ethers: Phase diversity and impressive predominance of homochiral guaifenesin-like supramolecular motif. Cryst. Growth Des. 2018, 18, 3980–3987. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding—Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | (R)-1 | α-rac-1 |
|---|---|---|
| Formula | C11H16O3 | C11H16O3 |
| Formula weight (g/mol) | 196.24 | 196.24 |
| Temperature, K | 296(2) | 296(2) |
| Crystal class | Orthorhombic | Monoclinic |
| Space group | P212121 | P21/n |
| Crystal size | 0.32 × 0.21 × 0.14 mm3 | 0.24 × 0.13 × 0.04 mm3 |
| Z, Z’ | 4, 1 | 4, 1 |
| Cell parameters | a = 4.9382(4) Å, b = 7.2807(6) Å, c = 28.728(2) Å | a = 4.9563(10) Å, b = 7.2972(18) Å, c = 28.491(7) Å β = 93.559(15)° |
| Volume, Å3 | 1032.88(14) | 1028.4(4) |
| F(000) | 424 | 424 |
| Calculated density, g/cm3 | 1.262 | 1.267 |
| μ, cm−1 | 0.91 | 0.91 |
| Theta range for data collection,, deg | 3.985–31.291 | 2.149–27.868 |
| Reflections measured | 14762 | 12777 |
| Independent reflections/R(int) | 3206/0.0374 | 2290/0.1433 |
| Number of parameters/restraints | 191/0 | 139/1 |
| Reflections [I > 2σ(I)] | 2669 | 1592 |
| Final R indices, R1/wR2 [I > 2σ(I)] | 0.0446/0.0965 | 0.1493/0.3564 |
| Final R indices, R1/wR2 (all reflections) | 0.0595/0.1026 | 0.1803/0.3791 |
| Goodness-of-fit on F2 | 1.042 | 1.372 |
| Largest diff. peak and hole, ρmax/ρmin (eÅ−3) | 0.236/−0.185 | 0.782/−0.535 |
| Run | Added Amount of rac-1, mg | Operation Amount of Enantiomers, mg | Resolution Time, min | (R)-1 and (S)-1 Obtained | ||||
|---|---|---|---|---|---|---|---|---|
| (R)-1 | (S)-1 | Yield, mg | ee1, % | YE2 | ||||
| mg | % | |||||||
| 1 | 1000 | 500 | 500 | 90 | (R) 23 | 72.7 | 12 | 2.4 |
| 2 | 18 | 495 | 505 | 138 | (S) 50 | 69.0 | 30 | 5.9 |
| 3 | 45 | 513 | 487 | 143 | (R) 46 | 82.2 | 33 | 6.4 |
| 4 | 41 | 488 | 512 | 303 | (S) 51 | 79.0 | 35 | 6.8 |
| 5 | 46 | 516 | 484 | 462 | (R) 55 | 85.6 | 42 | 8.1 |
| 6 | 50 | 487 | 513 | 157 | (S) 69 | 76.8 | 48 | 9.4 |
| 7 | 64 | 517 | 483 | 198 | (R) 94 | 69.4 | 60 | 11.6 |
| 8 | 89 | 481 | 519 | 236 | (S) 101 | 64.8 | 60 | 11.6 |
| Forms | Fusion Temperature (Tf), °C | Enthalpy of Fusion (ΔHf), J mole−1 |
|---|---|---|
| α-rac | 74.4 | 33.3 |
| β-rac | 66.1 | 27.8 |
| (R) | 96.5 | 38.2 |
| (R+S) | 76.3(76.2 1) | 34.9(35.2 1) |
| Run | Added Amount of rac-1, g | Operation Amount of Enantiomers, g | Resolution Time, min | (R)-1 and (S)-1 Obtained | ||||
|---|---|---|---|---|---|---|---|---|
| (R)-1 | (S)-1 | Yield, g | ee, % | YE2 | ||||
| g | % | |||||||
| 1 | 3.00 1 | 1.42 | 1.58 | 110 | (S) 0.50 | 73.8 | 0.29 | 18.5 |
| 2 | 0.42 | 1.57 | 1.43 | 65 | (R) 0.40 | 78.0 | 0.24 | 15.2 |
| 3 | 0.33 | 1.44 | 1.56 | 80 | (S) 0.40 | 78.0 | 0.24 | 15.3 |
| 4 | 0.33 | 1.58 | 1.42 | 115 | (R) 0.43 | 75.0 | 0.25 | 15.7 |
| 5 | 0.35 | 1.46 | 1.57 | 95 | (S) 0.43 | 74.8 | 0.25 | 15.6 |
| 6 | 0.35 | 1.59 | 1.41 | 85 | (R) 0.49 | 71.6 | 0.28 | 17.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bredikhin, A.A.; Zakharychev, D.V.; Bredikhina, Z.A.; Kurenkov, A.V.; Samigullina, A.I.; Gubaidullin, A.T. Stereoselective Crystallization of Chiral 3,4-Dimethylphenyl Glycerol Ether Complicated by Plurality of Crystalline Modifications. Crystals 2020, 10, 201. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030201
Bredikhin AA, Zakharychev DV, Bredikhina ZA, Kurenkov AV, Samigullina AI, Gubaidullin AT. Stereoselective Crystallization of Chiral 3,4-Dimethylphenyl Glycerol Ether Complicated by Plurality of Crystalline Modifications. Crystals. 2020; 10(3):201. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030201
Chicago/Turabian StyleBredikhin, Alexander A., Dmitry V. Zakharychev, Zemfira A. Bredikhina, Alexey V. Kurenkov, Aida I. Samigullina, and Aidar T. Gubaidullin. 2020. "Stereoselective Crystallization of Chiral 3,4-Dimethylphenyl Glycerol Ether Complicated by Plurality of Crystalline Modifications" Crystals 10, no. 3: 201. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst10030201