Exploration of Bis(nickelation) of 1,1′-Bis(o-carborane)



Institute of Chemical Sciences, School of Engineering & Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK
*
Authors to whom correspondence should be addressed.
†
Present address: Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore.
Crystals 2021, 11(1), 16; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11010016
Submission received: 10 December 2020
/
Revised: 22 December 2020
/
Accepted: 22 December 2020
/
Published: 27 December 2020
(This article belongs to the Special Issue Carborane: Dedicated to the Work of Professor Alan Welch)
Abstract
:The metalation of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10], with the smaller {Ni(dmpe)} fragment sourced from [Ni(dmpe)Cl2], is explored. The bis(metalated) products are obtained as a diastereoisomeric mixture. These isomers were separated, fully characterised spectroscopically and crystallographically and identified as rac-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (1) and meso-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (2). Previously, these 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 architectures (where both cages are not isomerised), were inaccessible, and thus new structures can be achieved during bis(nickelation) with {Ni(dmpe)}. Further, the metalation of the tetra-thallium salt with the bulky {Ni(dppe)} fragment sourced from [Ni(dppe)Cl2] was also studied. These bis(nickelated) products were also fully characterised and are afforded as the stereospecific species rac-[1-(1′-3′-(dppe)-3′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (3) and [1-(2′-4′-(dppe)-4′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (4α). In the latter metalation, compound 3 shows intramolecular dihydrogen bonding, contributing to the stereospecificity, whereas isomerisation from 3,1,2 to 4,1,2- in the 4α is related to steric relief.
1. Introduction
Since the discovery of bis(carboranes) in 1964, the chemistry of 1,1′-bis(o-carborane) (Scheme 1, a) has evolved rapidly, particularly once a high-yielding synthetic route was devised in 2003. Bis(carborane) offers a versatile building block in designing three-dimensional molecules, an array of homogeneous catalyst precursors, luminescent materials and organic derivatives via Ccage-H or Bcage-H functionalisation [1,2,3,4,5,6].
Since 2010, the Welch group have established many variations of the metalation chemistry of 1,1′-bis(o-carborane), one approach being cage expansion chemistry via reduction-metalation of this species [1], whilst another metalation strategy explored broadly within the group is deboronation/metalation of 1,1′-bis(o-carborane).
The single deboronation/metalation of 1,1′-bis(o-carborane) has been reported for cobalt, nickel and ruthenium metal fragments and afforded a wide range of mono-metallic-bis(carborane) isomers [7,8]. In further developments, double deboronation/metalation was achieved, such that both cages became metallacarboranes. The bimetallic metallacarboranes derived from the metalation of doubly deboronated 1,1′-bis(o-carborane) with both rhodium and ruthenium fragments are 3,1,2-MC2B9-2′,1′,8′-MC2B9 [M = {Rh}, {Ru}] species, in which one of the cages has isomerised (Scheme 1, b and c). There are limited examples of the parent 3,1,2-MC2B9-3′,1′,2′-MC2B9 form [9,10]. Variation in the isomer type with cobalt has been achieved by varying the metalation source, forming either 3,1,2-CoC2B9-3′,1′,2′-CoC2B9 (non-isomerised) or 3,1,2-CoC2B9-2′,1′,8′-CoC2B9 (isomerised) (Scheme 1, c) products [9]. Notably, an example of stepwise deboronation/metalation-deboronation/heterometalation is also reported [11]. Here we document an expansion of the bimetallic metallacarboranes library via the double deboronation/nickelation of 1,1′-bis(o-carborane).
2. Materials and Methods
2.1. General Considerations
Experiments were carried out under dry, oxygen-free N2, using standard Schlenk techniques, although subsequent manipulations were performed at ambient condition. Tetrahydrofuran (THF) was dried and distilled under sodium/benzophenone, whilst petrol was distilled from sodium wire before use. DCM was purified in an MBRAUN SPS-800(Dieselstr. 31, D-85748 Garching,). Degassing of solvents was performed (3 × freeze-pump-thaw cycles) before reaction. Preparative TLC used Kieselgel F254 glass plates (20 × 20 cm). 1H (400.1 MHz), 31P (162.0 MHz) or 11B (128.4 MHz) NMR spectra and 1H-31P Heteronuclear Multiple Bond Correlation (HMBC) experiment (in Supplementary Materials) were run on a Bruker DPX-400 spectrometer (Bruker BioSpin AG, Fallenden, Switzerland). The precursors 1,1′-bis(o-carborane) [12], its deboronated derivative [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10] (Tl4-salt) (WARNING: Thallium is extremely toxic, appropriate precautions are required when handling thallium compounds) [9] and [NiCl2(dmpe)] (dmpe = 1,2-bis(dimethylphosphino)ethane) [13] were prepared by modified literature methods. [NiCl2(dppe)] (dppe = 1,2-bis(diphenylphosphino)ethane) and the remaining reagents were purchased commercially.
2.1.1. Synthesis and Characterisation of rac-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (1) and meso-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (2)
The Tl4-salt (0.60 g, 0.56 mmol) was taken into THF (15 mL). The yellow suspension was degassed by freeze-pump-thaw (three cycles). [NiCl2(dmpe)] (0.31 g, 1.1 mmol) was transferred at −196 °C. The reaction suspension was allowed to warm and was stirred at room temperature. After overnight stirring, the mixture turned dark green. All volatiles were removed in vacuo. The residue was dissolved in DCM and passed through a small pad of silica. The filtrate was reduced in volume under low pressure and purified by preparative TLC using DCM and petrol (80:20) to afford two dark green bands, which were collected as solids. The upper green band with Rf = 0.84 afforded rac-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (1) (80 mg, 21%) and a lower green band with Rf = 0.63 gave meso-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (2) (76 mg, 20%).
Compound 1: 1H NMR (CD2Cl2): δ 2.43 (d, 3JPH = 14.0 Hz, 2H, CHcage), 2.14–1.90 (m, 8H, P{CH2}2P), 1.83 (d, 2JPH = 10.0 Hz, 6H, CH3), 1.67 (d, 2JPH = 10.0 Hz, 6H, CH3), 1.55 (d, 2JPH = 10.0 Hz, 12H, CH3). 1H{31P} NMR (CD2Cl2): δ 2.43 (s, 2H, CHcage), 2.12–1.89 (m, 8H, P{CH2}2P), 1.83 (s, 6H, CH3), 1.67 (s, 6H, CH3), 1.56 (s, 12H, CH3). 31P{1H} NMR (CD2Cl2): δ 43.1 (d, 2JPP = 29.2 Hz, 2P), 33.0 (d, 2JPP = 29.2 Hz, 2P). 11B{1H} NMR (CD2Cl2): δ −2.7 (2B), −4.1 (2B), −7.3 (2B), −11.8 (2B), −13.6 (2B), −15.8 (4B), −21.1 (4B).
Compound 2: 1H NMR (CD2Cl2): δ 2.48 (d, 3JPH = 10.0 Hz, 2H, CHcage), 2.16–1.79 (m, 8H, P{CH2}2P), 1.73 (d, 2JPH = 10.0 Hz, 6H, CH3), 1.70 (d, 2JPH = 8.0 Hz, 6H, CH3), 1.57 (d, 2JPH = 10.0 Hz, 6H, CH3), 1.55 (d, 2JPH = 8.0 Hz, 6H, CH3). 1H{31P} NMR (CD2Cl2): δ 2.49 (s, 2H, CHcage), 2.14–1.78 (m, 8H, P{CH2}2P), 1.74 (s, 6H, CH3), 1.70 (s, 6H, CH3), 1.57 (s, 6H, CH3), 1.55 (s, 6H, CH3). 31P{1H} NMR (CD2Cl2): δ 43.7 (d, 2JPP = 28.4 Hz, 2P), 33.4 (d, 2JPP = 28.4 Hz, 2P). 11B{1H} NMR (CD2Cl2): δ−1.5 (2B), −4.0 (2B), −10.4 (2B), −11.5 (2B), −14.5 (3B), −15.9 (5B), −20.4 (2B).
2.1.2. Synthesis and Characterisation of rac-[1-(1′-3′-(dppe)-3′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (3) and [1-(2′-4′-(dppe)-4′,1′,2′-closo–NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (4α)
A yellow suspension of Tl4-salt (0.60 g, 0.56 mmol) in THF (20 mL) was frozen at −196 °C. [NiCl2(dppe)] (0.59 g, 1.1 mmol) added to the frozen mixture. After overnight stirring, the reaction mixture gave a green suspension. All volatiles were evaporated off under reduced pressure. The mixture was taken into DCM and filtered through silica. The filtrate was reduced in a minimum amount under low pressure. The mixture was purified by preparative TLC with DCM and petrol (60:40). This yielded, along with a trace amount of purple band with Rf = 0.39, two major mobile components. The lower army-green band with Rf = 0.28 collected as a solid, and after crystallisations, yielded rac-[1-(1′-3′-(dppe)-3′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (3) (130 mg, 20%). The upper army green-band with Rf = 0.35 afforded [1-(2′-4′-(dppe)-4′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (4α) (122 mg, 19%).
Compound 3: 1H NMR (CD2Cl2): δ 7.80–7.25 (m, 40H, C6H5), 3.29–1.96 (m, 8H, P{CH2}2P), 1.83 (d, 3JPH = 11.6 Hz, 2H, CHcage). 1H{31P} NMR (CD2Cl2): δ 7.79–7.25 (m, 40H, C6H5), 3.29–1.97 (m, 8H, P{CH2}2P), 1.84 (s, 2H, CHcage). 31P{1H} NMR (CD2Cl2): δ 51.6 (d, 2JPP = 19.9 Hz, 2P), 39.2 (d, 2JPP = 19.9 Hz, 2P). 11B{1H} NMR (CD2Cl2): δ 4.9 (2B), −2.5 (4B), −9.0 (2B), −14.7 (5B), −18.0 (5B).
Compound 4α: 1H NMR (CD2Cl2): δ 8.01–7.02 (m, 40H, C6H5), 3.67–2.26 (m, 8H, P{CH2}2P), 1.94 (s, 1H, CHcage), 1.66 (d, 3JPH = 11.2 Hz, 1H, CHcage). 1H{31P} NMR (CD2Cl2): δ 8.02–7.01 (m, 40H, C6H5), 3.68–2.25 (m, 8H, P{CH2}2P), 1.95 (s, 1H, CHcage), 1.67 (s, 1H, CHcage). 31P{1H} NMR (CD2Cl2): δ 62.9 (s, 2P), 49.4 (d, 2JPP = 16.6 Hz, 1P), 47.5 (d, 2JPP = 16.6 Hz, 1P). 11B{1H} NMR (CD2Cl2): δ 5.9 (1B), 1.7 (1B), −0.2 (1B), −4.6 (3B), −9.8 to −20.4 multiple overlapping resonances with maxima at −9.8, −13.5, −15.6, −16.8, −20.4 (total integral of last five resonances 12B).
2.2. Crystallographic Studies
X-ray diffraction quality crystals of 1–4α were obtained by solvent diffusion at 5 °C using DCM and petrol 40–60 as antisolvent. Intensity data for compounds 1–3 were collected on a Bruker X8 APEXII diffractometer, whereas for compound 4α on a Rigaku FRE+ equipped with VHF Varimax confocal mirrors and an AFC10 goniometer and HG Saturn 724+ detector using Mo-Kα X-radiation at the UK National Crystallography Service. All crystals were mounted in inert oil on a cryoloop and cooled to 100 K by an Oxford Cryosystems Cryostream. Indexing, data collection and absorption correction were performed using the APEXII suite of programmes [14]. Structures were solved with the SHELXS programme [15] and refined by full-matrix least-squares (SHELXL), using OLEX2 [16]. Table 1 summarises the crystallographic parameters. The location of the CH vertices in all cases was established by the Vertex to Centroid (VCD) method developed by the Welch group [17]. The VCD method is based on the fact that, in a carborane cage, the C vertices are closer to the cage centroid than the B vertices. The CH vertex’s location was corroborated by the Boron-to-Hydrogen distance method (“B”–H bond lengths for actual C atoms refined to ca. <0.8Å) in all cases except 4α, where the positions of the H atoms could not be freely refined against the weak and twinned diffraction data. In this case, B-H distances were restrained to 1.10(2) Å.
1 was treated as a two-component crystal and refined with hklf 5 data. In 4α, disordered phenyl carbon atoms were constrained to have equal displacement parameters. The crystal of 4α was not single and refined as a two-component twin with the twin law 1 0 0 0 –1 0 0 0 –1 against HKLF 4 data, which were themselves treated with the solvent mask procedure implemented in OLEX2. HKLF 5 data were not available from the National crystallography service for this structure. A refinement model of the solvent showed approximately three molecules of highly disordered CH2Cl2 per bis carborane.
3. Results and Discussion
3.1. Characterisation of Compounds 1 and 2
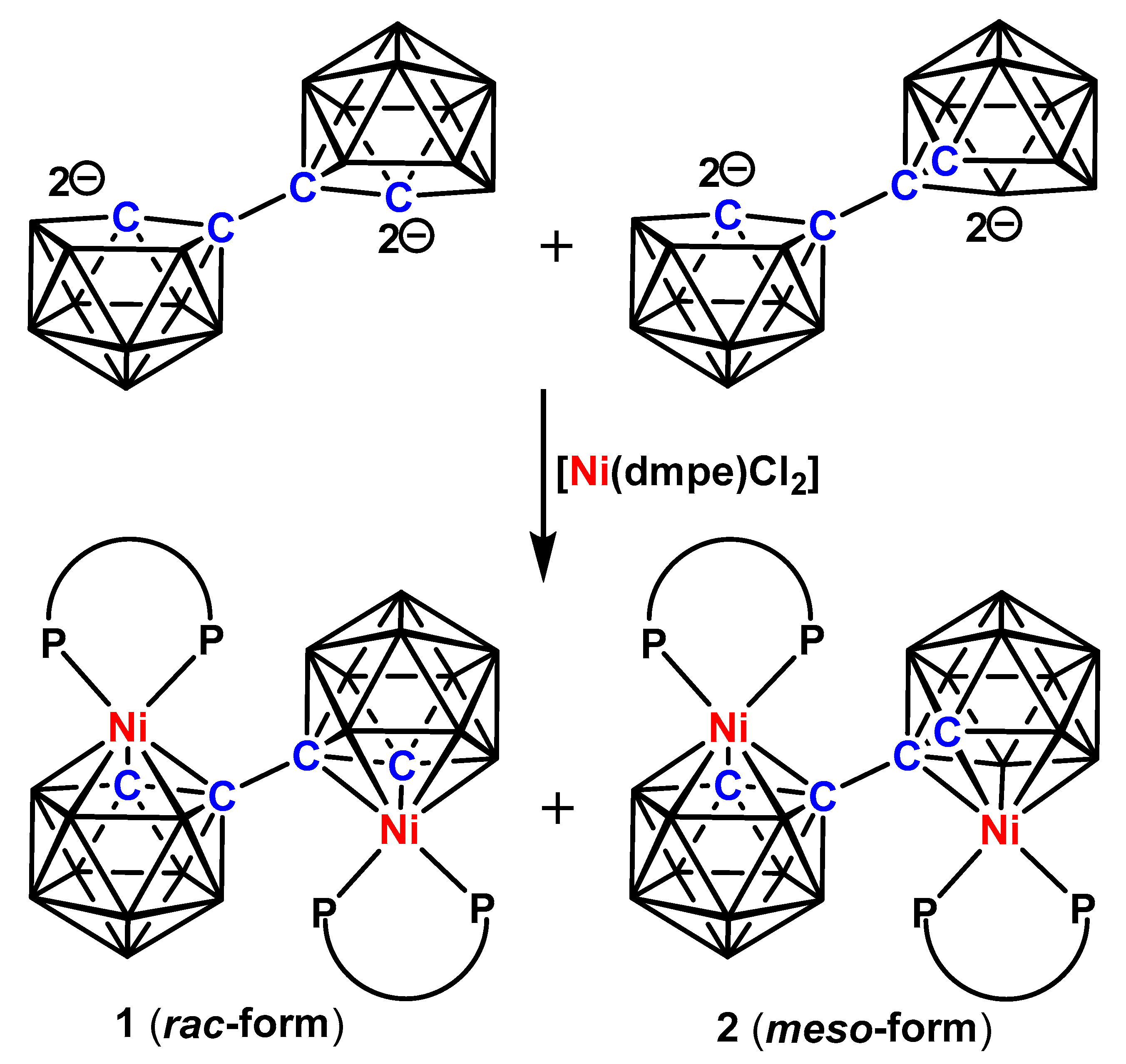
Previously, the double deboronated [7-(7′-7′,8′-nido-C2B9H10)-7,8-nido-C2B9H10]4− tetraanion derived from [1-(1′-1′,2′-closo-C2B10H11)-1,2-closo-C2B10H11], commonly called 1,1′-bis(o-carborane), was isolated as [HNMe3]+ or [BTMA]+ [BTMA = benzyltrimethylammonium] salts. Later, [Tl]+ salts were chosen for metalation because of their generally better yields [9]. We have reacted a suspension of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10] in THF with [Ni(dmpe)Cl2] at room temperature. Workup and purification of the dark-green suspension involving preparative thin-layer chromatography (TLC) resulted in two dark green bands, compounds 1 and 2, in moderate yield (Scheme 2). These were fully characterised by a variety of spectroscopic and crystallographic analyses.
An interesting feature of the double deboronation-metalation of 1,1′-bis(o-carborane) is that it gives diastereomeric metallcarborane products. Indeed, compound 1 turns out to be racemic, whilst compound 2 is in the meso form. These observations are also consistent with the NMR spectroscopic analysis. The 1H NMR spectrum of compound 1 reveals resonances arising from the methylene bridge of the two dmpe ligands, whilst the methyl groups of the dmpe ligands produce three doublet resonances. The first two are of integral-6 at δ 1.83, 1.67 ppm with coupling 2JPH = 10.0 Hz, and the last one is of integral-12 at δ 1.55, 2JPH = 10.0 Hz. On the contrary, the 1H NMR spectrum of compound 2 reveals, in addition to the signals for methylene bridge of the dmpe ligands, four doublets assigned to the methyl protons of the dmpe fragments at δ 1.73 (2JPH = 10.0 Hz), 1.70 (2JPH = 8.0 Hz), 1.57 (2JPH = 10.0 Hz) and 1.55 (2JPH = 8.0 Hz) ppm, each of integral-6. These resonances collapse to the corresponding singlets in the 1H{31P} spectrum. In the 1H NMR spectra there is also a single CHcage resonance of integral two for compound 1 appearing as a doublet δ 2.43 (3JPH = 14.0 Hz) ppm, confirmed as arising from coupling to phosphorus, since it collapses to a singlet on broad-band 31P decoupling, whilst the CHcage signal of integral two for compound 2 appears at a higher frequency, δ 2.48 (3JPH = 10.0 Hz) ppm, and collapses to a singlet on broad-band 31P decoupling. Therefore, these two isomers show slightly different characteristics in their proton NMR spectra. Moving to the 31P{1H} NMR spectrum of compound 1, this shows two mutual doublets with the integral ratio of 1:1 at δ 43.1 and 33.0 ppm and a coupling constant 2JPP = 29.2 Hz. This indicates that in each cage the two phosphorus atoms are magnetically inequivalent. Moreover, the two pairs of phosphorus atoms in different cages are also magnetically equivalent. This clearly shows that the metallacarborane cages are asymmetric. Similarly, the 31P{1H} NMR spectrum of compound 2 reveals two mutually coupled doublets of integral two at δ 43.7 and 33.4 ppm with coupling 2JPP = 28.4 Hz, not very different from that of compound 1. Notably, from the 1H-31P HMBC experiment, the splitting of CHcage arises from the trans phosphorus at δ 33.0 ppm for compound 1 and the trans phosphorus at δ 33.4 ppm for compound 2. Therefore, the resonances δ 43.1 ppm and δ 43.7 ppm correspond to the phosphorus atoms cis to the CHcage for compound 1 and compound 2, respectively. The 11B{1H} NMR spectrum of compound 1 consists of seven resonances with relative integrals 2:2:2:2:2:4:4 from high frequency to low frequency, whereas compound 2 shows a distinctly different pattern comprising seven resonances in the integral ratio 2:2:2:2:3:5:2 from high frequency to low frequency. However, this spectroscopic information is not conclusive in determining the exact structures of the racemic and meso isomers.
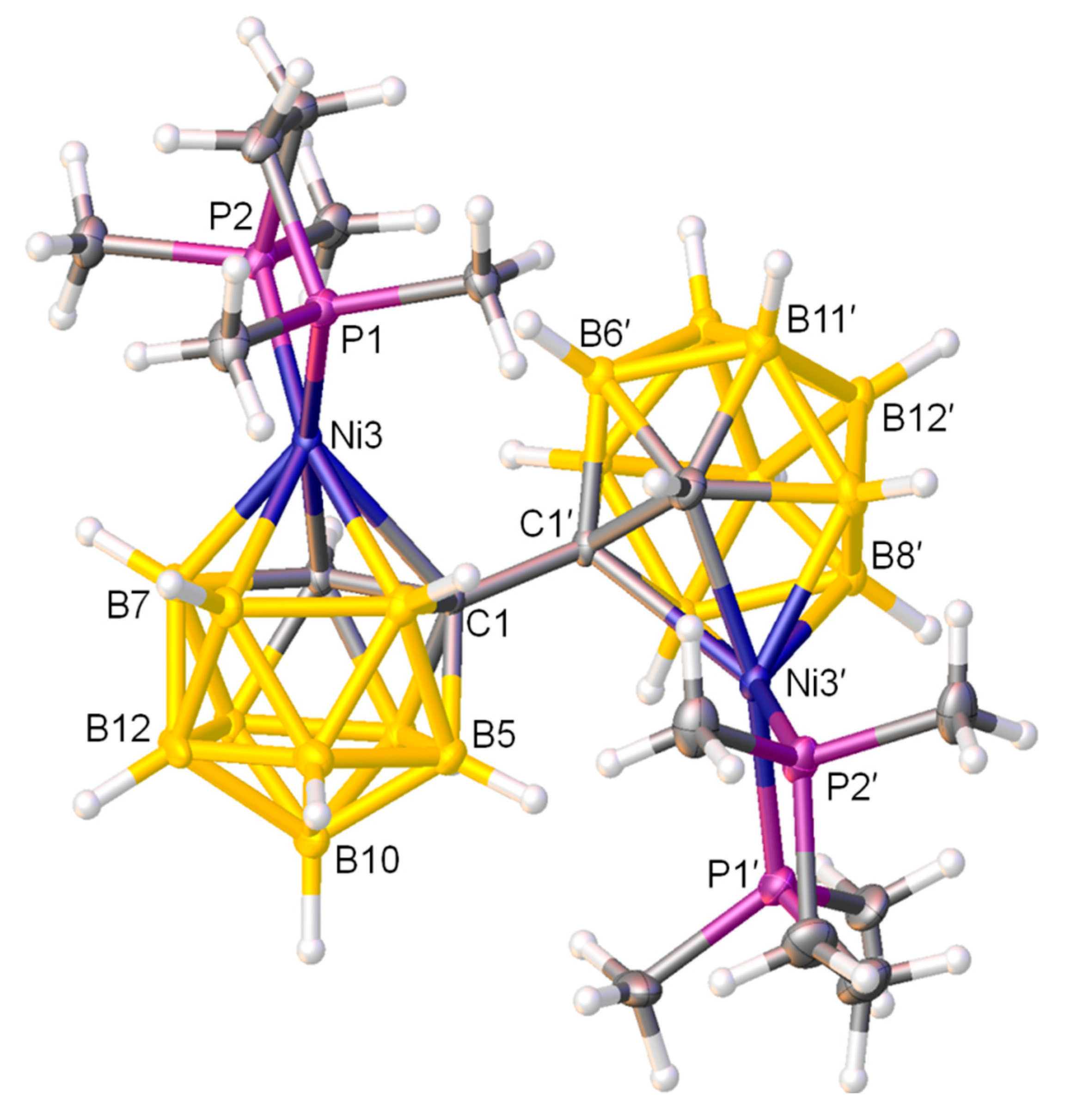
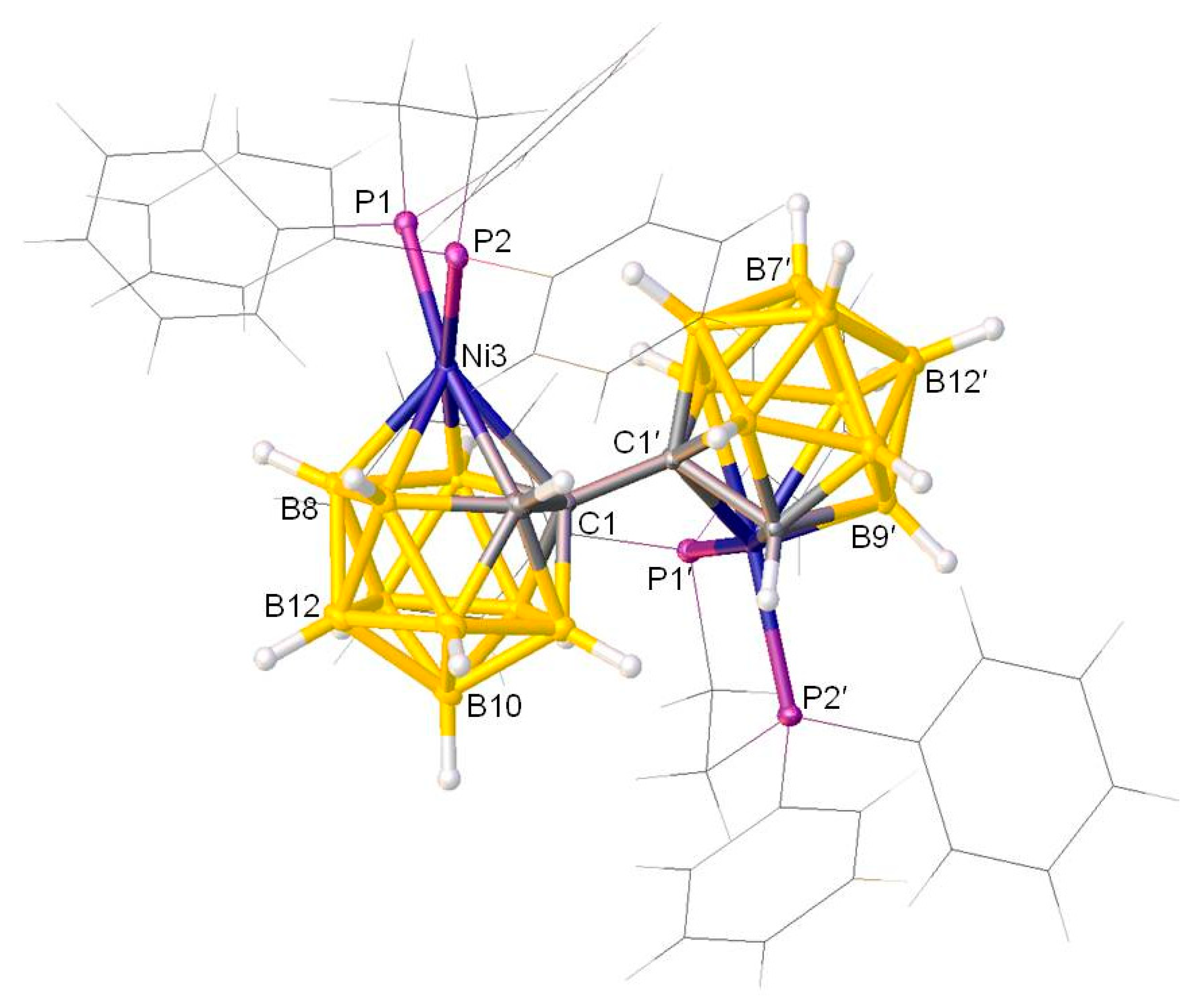
A crystallographic study was carried out for both compounds 1 and 2. It is envisaged that the double deboronation-metalation of 1,1′-bis(o-carborane) with {Ni(dmpe)} fragments generates racemic and meso mixtures with 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 architectures. Indeed, both cages of compounds 1 and 2 are singly-metalated and in the 3,1,2-NiC2B9 form. As regards the identification of the diastereoisomer, a crystallographic C2 axis passes perpendicular to the C1-C1′ bond, leading to the same chirality for both cages and meaning that compound 1 is a racemic isomer, whereas a non-crystallographic inversion centre i can be imagined at the mid-point of the C1-C1′ bond in 2, meaning different chirality for each cage and showing that compound 2 is a meso isomer. Figure 1 shows a perspective view of a single molecule of racemic-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (1), whereas a perspective view of a single molecule of meso-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (2) is presented in Figure 2. These diastereoisomers are the first such examples in which both the metallacarborane moieties remain unisomerised, i.e.,1 and 2 are 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 in racemic and meso forms, previously inaccessible. Both compounds 1 and 2 show clear evidence of internal crowding, since both cages have 3,1,2-NiC2B9 architectures. The presence of the {(dmpe)NiC2B9} substituents on C1 or C1′ inhibit the free rotation of the {Ni(dmpe)} fragment on both the primed and non-primed cages. It is confirmed that in each cage the phosphorus atoms are inequivalent, giving rise to two doublets observed in the 31P NMR spectra (previously discussed). Since both cages are of the 3,1,2-NiC2B9 form, the two metallacarborane units at C1 and C1′ push each other away, so that the {Ni(dmpe)} fragment is bent away from its ideal orientation within the cages. In principle, the ideal orientation of the {NiP1P2} fragment in a NiC2B9 icosahedron is perpendicular to the vertical mirror plane through the C2B9 unit, conveniently defined by the interplane dihedral angle (θ) of 90°. [8] For compound 1, θ is found to be 55.36(8)° for both the primed cage and non-primed cage, whilst for compound 2, θ is 57.59(15)° and 49.58(18)° for the primed cage and the non-primed cage, respectively. These were calculated using the planes through Ni3P1P2 and Ni3′P1′P2′ and through B6B8B10 and B6′B8′B10′for the non-primed and primed cages, respectively. Thus, the dihedral angles are clearly twisted away from the idealised 90°. We also note that in compound 1 the {NiPP} plane is bent away from the perpendicular to the bottom pentagonal B5B6B11B12B9 plane by.6(7)°, whereas in compound 2 the corresponding angles are 6.1(12)° for the non-primed cage and 21.5(15)° for the primed cage. The internal steric crowding between two (dmpe)NiC2B9 units is also evidenced by the elongated Ni3-C1 distances compared to Ni3–C2 [compound 1 primed cage: 2.2977(19) versus 2.0657(18) Å]. This is also evidenced in compound 2, Ni3-C1: 2.326(4) [Ni2-C2: 2.066(4) Å] and Ni3′-C1′: 2.299(4) [Ni2′-C2′: 2.103(3) Å].
3.2. Characterisation of Compounds 3 and 4α
The treatment of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10] in THF with [Ni(dppe)Cl2] at room temperature followed by work up and purification involving preparative TLC afforded two army-green bands, compounds 3 and 4α (Scheme 3). The compounds were characterised spectroscopically as well as by single crystal XRD.
Earlier we noted a diastereoisomeric mixture resulting from the deboronation-metalation of 1,1′-bis(o-carborane) with the {Ni(dmpe)} fragment. However, with the {Ni(dppe)} fragment, additionally, isomerisation occurred in the case of a mono-metalated [3,1,2-(dppe)-NiC2B9-1,2-C2B10] species which transformed to [4,1,2-(dppe)-NiC2B9-1,2-C2B10] due to the stereo-electronic nature of the dppe. [8] Therefore, with double metalation using the{Ni(dppe)} fragment, the products could be diastereoisomers, stereospecific products and other isomers thereof. Although the 1H NMR spectra of 3 and 4α are more complex than those of compounds 1 and 2, there are resonances from phenyl protons and methylene bridge protons of the dppe. In the proton spectrum of 3, there is also a CHcage resonance of integral two which appears at δ 1.84 ppm as a doublet J = 11.6 Hz, whilst in the proton spectrum of 4α, there are two CHcage resonances, each of integral-1, one at δ 1.94 ppm appearing as a singlet and the other at δ 1.67 ppm appearing as a doublet J = 11.2 Hz. The doublets collapse to singlets in the 1H{31P} spectra. Notably the CHcage resonance of 3 appears at a lower frequency than that of 1 (δ 2.43 ppm) or 2 (δ 2.48 ppm). The two doublet CHcage resonances clearly indicate that both cages of compound 3 are nickellated and could be of 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 architecture with reference to the proton spectra of compounds 1 and 2. The doublet and singlet CHcage resonances indicate that nickelation occurred for both cages of compound 4α where one cage could be unisomerised, i.e., the {3,1,2-NiC2B9} form and another cage could be isomerised to either a {4,1,2-NiC2B9} or a {2,1,8-NiC2B9} form. These inferences are further supported by the 31P NMR spectra of compounds 3 and 4α. The 31P{1H} NMR spectrum of compound 3 consists of two mutual doublets with integral ratio 1:1 at δ 51.6 and 39.2 ppm with coupling J = 19.1 Hz; on the contrary, the 31P{1H} NMR spectrum of 4α consists of a singlet of integral-2 at δ 62.9 ppm and two mutual doublets, each of integral-1 at δ 49.4 and 47.5 ppm, with a coupling J = 16.6 Hz. This signifies the asymmetric nature of each metallacarborane cage but the symmetric nature of the whole molecule. The 11B{1H} spectrum of compound 3 reveals five resonances with the relative integrals 2:4:2:5:5 from high frequency to low frequency, a different pattern to that for compounds 1 or 2, whilst the 11B{1H} NMR spectrum of 4α consists of multiple overlapping resonances with a total integral of 18B and is different in pattern to that of compounds 1, 2 and 3, thus preventing the identification of the exact isomer present and thereby requiring single crystal XRD analysis.
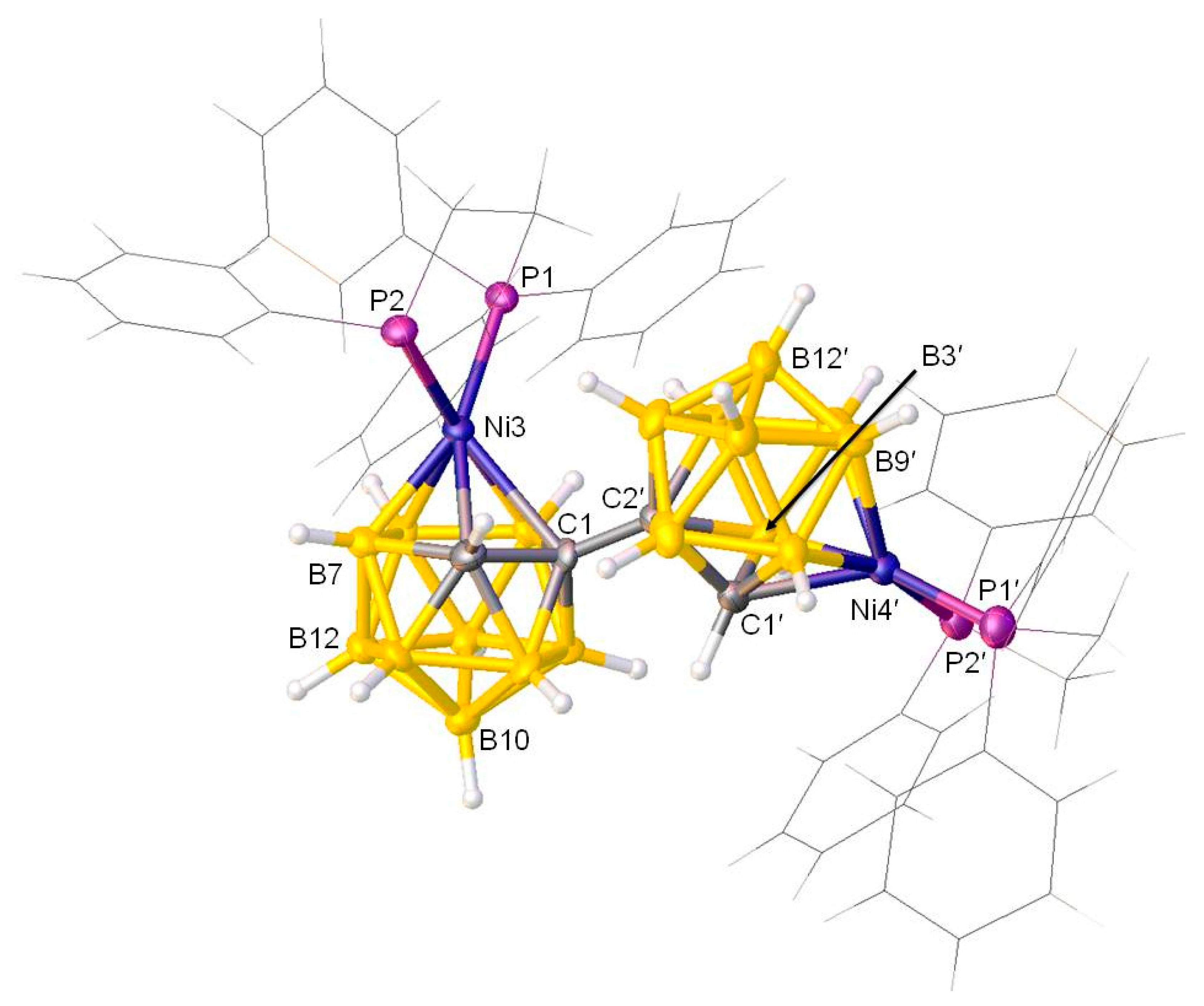
The precise natures of 3 and 4α were confirmed by crystallographic analysis. The racemic form of compound 3 is confirmed by a crystallographic C2 axis passing through the mid-point of the C1-C1′ bond, and thus requiring both cages to be of the same chirality. Figure 3 shows a perspective view of a single molecule of rac-[1-(1′-3′-(dppe)-3′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (3). However, it is clear that for compound 4α one of the cages has isomerised. A perspective view of a single molecule of [1-(2′-4′-(dppe)-4′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (4α) is shown in Figure 4. For compound 3, both cages are nickellated by {Ni(dppe)} fragments and are of 3,1,2-NiC2B9 architecture. As noted, the midpoint of the C1-C1′ bond lies on a crystallographic 2-fold axis. This leads to two bulky {(dppe)NiC2B9} units, connected at C1 and C1′, pushing each other apart. There is a clear indication of internal crowding which is obvious from the orientation of the {NiP1P2} fragment in the NiC2B9 icosahedra. In 3 (for the non-primed cage), the θ (previously discussed) between the plane containing Ni3P1P2 and the plane through B6B8B10 is 60.72(9)°, which deviates significantly from 90°. Further evidence of steric congestion is demonstrated by the fact that the plane of the {NiP1P2} fragment deviates from perpendicularity to the plane through B5B6B11B12B9 vertices by 6.6(8)°. Additionally, the longer Ni3–C1 distance compared to Ni3–C2 [2.336(2) versus 2.124(2) Å] again supports significant steric crowding in the molecule. The steric congestion in 3 would be slightly relieved if the 3,1,2-NiC2B9 cage would isomerise to either the 4,1,2-NiC2B9 form or the 2,1,8-NiC2B9 form. Indeed, this has been experimentally observed in 4α, which is of 3,1,2-NiC2B9-4′,1′,2′-NiC2B9 form, where one of the cages has isomerised. The molecule of 4α is partly disordered (two orientations for a phenyl ring), but the atomic connectivity is nonetheless clear. The internal crowding slightly reduces as, for compound 4α (for 3,1,2-NiC2B9 cage), the θ between the Ni3P1P2 and B6B8B10 planes is 64.8(4)°, closer to the idealised 90°. However, in 4α, the non-primed (3,1,2-NiC2B9 cage) still has significant internal crowding, as indicated by the longer distances Ni3-C1: 2.327(9) Å versus Ni2-C2: 2.129(9) Å. The relaxation of steric congestion in the (4′,1′,2′-NiC2B9 cage) of 4α is reflected in the slightly shorter distance Ni4′-C1′ is 2.167(8) Å. Furthermore the dihedral angle between the Ni4′P1′P2′ and C1′B11′B12′ planes for the primed cage is 86.1(4)°. Thus, the metalation of doubly deboronated species from 1,1′-bis(o-carborane) with {Ni(dppe)} results in bis-nickelated products, and although in principle both racemic and meso products were anticipated, only stereospecific racemic-3 and 4α were observed. The meso form was not found. The rationale for the formation of the rac form is discussed below.
3.3. Dihydrogen Interaction in 3 and Isomerisation in 4α
The reaction of the [7-(7′-7′,8′-nido-C2B9H10)-7,8-nido-C2B9H10]4− tetraanion with {Ni(dmpe)} fragments affords diastereomeric mixture products, i.e., rac (1) and meso (2) forms of 3,1,2-NiC2B9-3′,1′,2′-NiC2B9, whereas the same reaction with the more bulky {Ni(dppe)} fragment results in two isolable stereospecific products, i.e., rac 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 (3) and 3,1,2-NiC2B9-4′,1′,2′-NiC2B9, in compound 4α. The isolated rac-3 displays intramolecular dihydrogen bonding, as was found previously during the rationalisation of the stereospecific {CoCp} fragment metalation reaction with the [7-(7′-7′,8′-nido-C2B9H10)-7,8-nido-C2B9H10]4− tetranion to form the only racemic form of the product 3,1,2-CoC2B9-3′,1′,2′-CoC2B9 [9]. The formation of intramolecular dihydrogen bonding associated with CH atoms of one cage and BH atoms of the other cage results from the relatively protonic and hydridic nature of the CH and BH atoms, respectively. 11B NMR analysis of individual vertices for 3,1,2-MC2B9 compounds established that the most hydridic BH atoms are H5 and H6 [18]. In the case of rac-3, the orientation of two {(dppe)NiC2B9} cages enables two sets of intramolecular dihydrogen bonds, CH2⋯BH6i 2.07(3) Å (where i = 1 − x, y, 0.5 − z) and its symmetry equivalent, CH2i⋯BH6. Notably, the hypothetical analogous meso form could only allow for one set of such intramolecular dihydrogen bonding, whatever the rotameric arrangement, thereby rendering the meso isomer less favourable.
Crowding between the bulky {(dppe)(3,1,2-NiC2B9)} substituent on C1′ and the dppe ligand on Ni3 of an unisomerised 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 species is likely to be the major cause of the isomerisation observed in compound 4α. Isomerisation moves the {(dppe)(3,1,2-NiC2B9)} substituent down to the lower pentagonal belt further away from the {Ni(dppe)2} on the other cage. Indeed, we have already observed steric crowding in the unisomerised compound 3. In regards to the {3′,1′,2′-NiC2B9} to {4′,1′,2′-NiC2B9} isomerisation, this can likely be related to electronic factors, specifically the electron withdrawing nature of the dppe ligand. A similar 3,1,2- to 4,1,2- isomerisation of mono-metalated nickelacarboranes with dppe and PPh2Me ligands has been previously established [8].
4. Conclusions
Four new bis(nickelated) species are documented from the metalation of doubly deboronated 1,1′-bis(o-carborane), and their identity was confirmed by both spectroscopic and crystallographic means. The metalation of the {Ni(dmpe)} fragment with the Tl4-salt gives diastereoisomeric products with the unusual crowded architecture of 3,1,2-NiC2B9-3′,1′,2′-NiC2B9 as racemic and meso forms. In contrast, metalation with the {Ni(dppe)} fragment results in the 3,1,2-NiC2B9-4′,1′,2′-NiC2B9 species, a stereospecific racemic product. The racemic product in the latter case shows intramolecular dihydrogen bonding, hence explaining the stereospecific reaction, whereas the stereo-electronic nature of the bis(phosphine) ligand influences the formation of the isomerised 3,1,2-NiC2B9-4′,1′,2′-NiC2B9 species.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4352/11/1/16/s1, NMR spectra of all new compounds are available online along with the crystallographic data, free of charge. Crystallographic information for all compounds here has been deposited in the Cambridge Crystallographic Data Centre as supplementary publications nos. CCDC 2048463, 2048465, 2,048,466 and 2,048,464 (compounds 1, 2, 3 and 4α).
Author Contributions
Syntheses and original draft preparation, and editing, D.M.; final structure analysis and manuscript editing, G.M.R. All authors have read and agreed to the published version of the manuscript.
Funding
D.M. is grateful to the Heriot-Watt University for financial assistance; a James-Watt Ph.D. studentship was awarded to D.M. from 2013 to 2016. We thank EPSRC for funding the Bruker X8Apex2 diffractometer.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work is taken from the Ph.D. thesis (2016) of D.M., supervised by Alan J. Welch at Heriot-Watt University. We thank EPSRC for funding the Bruker X8Apex2 diffractometer and the U.K. National Crystallography Service for data collection of compound 4α.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sivaev, I.B.; Bregadze, V.I. 1,1′-Bis(ortho-carborane)-based transition metal complexes. Co-ord. Chem. Rev. 2019, 392, 146–176. [Google Scholar] [CrossRef]
- Yruegas, S.; Axtell, J.C.; Kirlikovali, K.O.; Spokoyny, A.M.; Martin, C.D. Synthesis of 9-borafluorene analogues featuring a three-dimensional 1,1′-bis (o-carborane) backbone. Chem. Commun. 2019, 55, 2892–2895. [Google Scholar] [CrossRef] [PubMed]
- Jeans, R.J.; Chan, A.P.Y.; Riley, L.E.; Taylor, J.; Rosair, G.M.; Welch, A.J.; Sivaev, I.B. Arene-Ruthenium complexes of 1,1′-bis (ortho-carborane): Synthesis, characterization, and catalysis. Inorg. Chem. 2019, 58, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.P.Y.; Parkinson, J.A.; Rosair, G.M.; Welch, A.J. Bis (phosphine) hydridorhodacarborane derivatives of 1,1′-bis (ortho-carborane) and their catalysis of alkene isomerization and the hydrosilylation of acetophenone. Inorg. Chem. 2020, 59, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Kirlikovali, K.O.; Axtell, J.C.; Anderson, K.P.; Djurovich, P.I.; Rheingold, A.L.; Spokoyny, A.M. Fine-tuning electronic properties of luminescent Pt (II) complexes via vertex-differentiated coordination of sterically invariant carborane-based ligands. Organometallics 2018, 37, 3122–3131. [Google Scholar] [CrossRef]
- Wu, J.; Cao, K.; Zhang, C.-Y.; Xu, T.-T.; Ding, L.-F.; Li, B.; Yang, J. Catalytic oxidative dehydrogenative coupling of cage B–H/B–H bonds for synthesis of bis (o-carborane)s. Org. Lett. 2019, 21, 5986–5989. [Google Scholar] [CrossRef] [PubMed]
- Thiripuranathar, G.; Man, W.Y.; Palmero, C.; Chan, A.P.Y.; Leube, B.T.; Ellis, D.; McKay, D.; MacGregor, S.A.; Jourdan, L.; Rosair, G.M.; et al. Icosahedral metallacarborane/carborane species derived from 1,1′-bis(o-carborane). Dalton Trans. 2015, 44, 5628–5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, D.; Man, W.Y.; Rosair, G.M.; Welch, A.J. Steric versus electronic factors in metallacarborane isomerisation: Nickelacarboranes with 3,1,2-, 4,1,2- and 2,1,8-NiC 2 B 9 architectures and pendant carborane groups, derived from 1,1′-bis(o-carborane). Dalton Trans. 2016, 45, 15013–15025. [Google Scholar] [CrossRef] [PubMed]
- Thiripuranathar, G.; Mandal, D.; Argentari, M.; Chan, A.P.Y.; Man, W.Y.; Rosair, G.M.; Welch, A.J. Double deboronation and homometalation of 1,1’-bis (ortho-carborane). Dalton Trans. 2017, 46, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Behnken, P.E.; Marder, T.B.; Baker, R.T.; Knobler, C.B.; Thompson, M.R.; Hawthorne, M.F. Synthesis, structural characterization, and stereospecificity in the formation of bimetallic rhodacarborane clusters containing rhodium-hydrogen-boron bridge interactions. J. Am. Chem. Soc. 1985, 107, 932–940. [Google Scholar] [CrossRef]
- Chan, A.P.Y.; Rosair, G.M.; Welch, A.J. Heterometalation of 1,1′-bis (ortho-carborane). Inorg. Chem. 2018, 57, 8002–8011. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Xie, Z. A facile and practical synthetic route to 1,1′-bis (o-carborane). Organometallics 2008, 27, 5167–5168. [Google Scholar] [CrossRef]
- Booth, G.; Chatt, J. 590. Some complexes of ditertiary phosphines with nickel (II) and nickel (III). J. Chem. Soc. 2004, 3238–3241. [Google Scholar] [CrossRef]
- Bruker AXS. APEX2-Software Suite for Crystallographic Programs; Bruker AXS, Inc.: Madison, WI, USA, 2009. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta. Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- McAnaw, A.; Scott, G.; Elrick, L.; Rosair, G.M.; Welch, A.J. The VCD method-A simple and reliable way to distinguish cage C and B atoms in (hetero) carborane structures determined crystallographically. Dalton Trans. 2013, 42, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Bown, M.; Plešek, J.; Baše, K.; Štibr, B.; Fontaine, X.L.R.; Greenwood, N.N.; Kennedy, J.D. Assigned cluster 11B and 1H NMR properties of [3-(η6-C6Me6)-closo-3,1,2-RuC2B9H11]. Magn. Reson. Chem. 1989, 27, 947–949. [Google Scholar] [CrossRef]
Scheme 1.
Line diagrams of a–c. Species a is 1,1′-bis(o-carborane), whereas b (α-form) and c (β-form) were derived via double deboronation/metalation of a. Unlabelled vertices are B.
Scheme 1.
Line diagrams of a–c. Species a is 1,1′-bis(o-carborane), whereas b (α-form) and c (β-form) were derived via double deboronation/metalation of a. Unlabelled vertices are B.

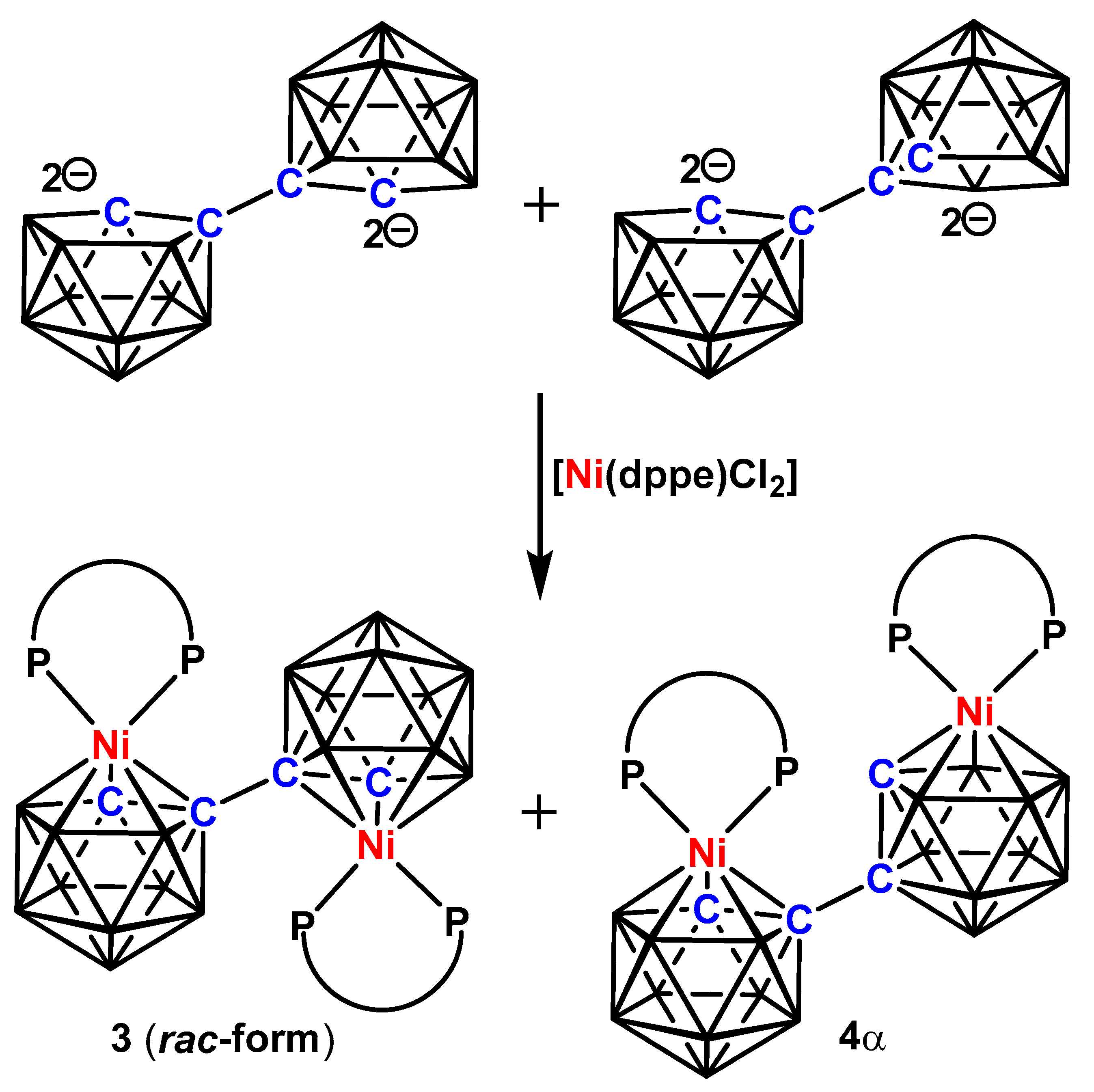
Scheme 2.
Synthesis of 1 and 2 from the nickelation of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10]. NiPP = {Ni(dmpe)}.
Scheme 2.
Synthesis of 1 and 2 from the nickelation of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10]. NiPP = {Ni(dmpe)}.

Figure 1.
Molecular structure of rac-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (1). Atoms with dashed suffixes are generated by the symmetry operation 1.5-x, y, 0.5-z.
Figure 1.
Molecular structure of rac-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (1). Atoms with dashed suffixes are generated by the symmetry operation 1.5-x, y, 0.5-z.

Figure 2.
Molecular structure of meso-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (2).
Figure 2.
Molecular structure of meso-[1-(1′-3′-(dmpe)-3′,1′,2′-closo-NiC2B9H10)-3-(dmpe)-3,1,2-closo-NiC2B9H10] (2).

Scheme 3.
Synthesis of 3 and 4α from the nickelation of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10]. NiPP = {Ni(dppe)}.
Scheme 3.
Synthesis of 3 and 4α from the nickelation of [Tl]2[1-(1′-3′,1′,2′-closo-TlC2B9H10)-3,1,2-closo-TlC2B9H10]. NiPP = {Ni(dppe)}.

Figure 3.
A perspective view of rac-[1-(1′-3′-(dppe)-3′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (3) (all phenyls and the –CH2–CH2– bridge of dppe are in wireframe for clarity). Atoms with dashed suffixes are generated by the symmetry operation 1 − x, y, 0.5 − z.
Figure 3.
A perspective view of rac-[1-(1′-3′-(dppe)-3′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (3) (all phenyls and the –CH2–CH2– bridge of dppe are in wireframe for clarity). Atoms with dashed suffixes are generated by the symmetry operation 1 − x, y, 0.5 − z.

Figure 4.
Molecular structure of [1-(2′-4′-(dppe)-4′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (4α) (all phenyls and the –CH2-CH2- bridge of dppe are in wireframe for clarity).
Figure 4.
Molecular structure of [1-(2′-4′-(dppe)-4′,1′,2′-closo-NiC2B9H10)-3-(dppe)-3,1,2-closo-NiC2B9H10] (4α) (all phenyls and the –CH2-CH2- bridge of dppe are in wireframe for clarity).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic data for compounds 1–4α.
| 1 | 2 | 3 | 4α | |
|---|---|---|---|---|
| Formula | C16H52B18Ni2P4 | C19H58B18Cl6Ni2P4 | C59H74B18Cl6Ni2P4 | C59H74B18Ni2P4Cl6 |
| M | 680.45 | 935.23 | 1431.76 | 1431.76 |
| Crystal system | monoclinic | triclinic | monoclinic | orthorhombic |
| Space group | P2/n | P-1 | C2/c | P212121 |
| a/Å | 13.6195(9) | 11.4867(6) | 25.0952(8) | 10.2816(4) |
| b/Å | 8.7239(5) | 12.5327(6) | 14.9165(5) | 20.3356(9) |
| c/Å | 14.3241(9) | 15.0938(8) | 19.7053(6) | 32.3413(14) |
| α/° | 90 | 96.986(3) | 90 | 90 |
| β/° | 100.316(3) | 90.037(3) | 113.620(2) | 90 |
| γ/° | 90 | 91.277(2) | 90 | 90 |
| U/Å3 | 1674.41(18) | 2156.21(19) | 6758.4(4) | 6762.0(5) |
| Z | 2 | 2 | 4 | 4 |
| Dcalcg/cm3 | 1.350 | 1.440 | 1.407 | 1.406 |
| µ(Mo-Kα)/mm −1 | 1.328 | 1.412 | 0.929 | 0.928 |
| F(000)/e | 708 | 960 | 2944 | 2944 |
| 2Θ range/° | 5.572 to 52.748 | 4.462 to 52.206 | 5.584 to 58.27 | 4.2 to 55.12 |
| Data measured | 46,391 | 29,045 | 59,135 | 45,269 |
| Unique data, n | 3479 | 8280 | 8989 | 15,482 |
| Variables | 216 | 520 | 445 | 792 |
| S (all data) | 1.053 | 1.031 | 1.015 | 0.946 |
| Rint | 0.0436 | 0.0517 | 0.0510 | 0.1211 |
| R, wR2 [all data] | 0.0258, 0.0498 | 0.0752, 0.0996 | 0.0598, 0.0974 | 0.1200, 0.1892 |
| Emax, Emin/e Å−3 | 0.29/−0.26 | 0.87/−0.79 | 0.65/−1.01 | 0.52/−0.72 |
| Flack parameter | - | - | - | 0.12(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mandal, D.; Rosair, G.M. Exploration of Bis(nickelation) of 1,1′-Bis(o-carborane). Crystals 2021, 11, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11010016
AMA Style
Mandal D, Rosair GM. Exploration of Bis(nickelation) of 1,1′-Bis(o-carborane). Crystals. 2021; 11(1):16. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11010016
Chicago/Turabian StyleMandal, Dipendu, and Georgina M. Rosair. 2021. "Exploration of Bis(nickelation) of 1,1′-Bis(o-carborane)" Crystals 11, no. 1: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11010016
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.