
Molecular Simulation of External Electric Fields on the Crystal State: A Perspective


School of Chemical and Bioprocess Engineering, University College Dublin, Belfield, Dublin 4, Ireland
Crystals 2021, 11(11), 1405; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111405
Submission received: 24 September 2021
/
Revised: 8 November 2021
/
Accepted: 12 November 2021
/
Published: 18 November 2021
(This article belongs to the Special Issue Feature Papers on "Hybrid and Composite Crystalline Materials" 2021-2022)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Unpacking the mechanistic insights into how externally applied electric fields affect the physicochemical properties of crystals represents a challenge of great importance for a plethora of natural phenomena, in addition to a broad array of industrial operations and technologies. As such, the key goals in such field effect studies centre around how thermodynamic and kinetic relaxation processes in crystals are affected, including charge carrier conduction and energy transfer processes, and this is a very recent area of fundamental scrutiny. Indeed, in recent years, there has been a steadily mounting number of reports of field-manipulated crystal-state phenomena. Taking as the background a range of natural phenomena, phenomenological theory, state-of-the-art experiments and technological observations, the present review examines the role of nonequilibrium molecular simulation in its scrutiny of intra-crystal phenomena from an atomistic viewpoint, in addition to providing a framework for a predictive molecular design philosophy by which to refine field crystal understanding.
1. Introduction
In recent years, the effect of externally applied fields of different kinds (e.g., acoustic, electric and electromagnetic) on the behaviour of crystals has become a more studied area, both in terms of the fundamentals and technological applications. One reason has been due to how external fields are known to alter crystals’ underlying thermodynamic and dynamical properties.
Despite different kinds of externally applied fields being applied to regulate the vibrational behaviour of the crystalline state, the underlying dynamics of this are, in general, less well-understood. This is particularly so for electric fields, which have seen a gradually mounting number of reports of electro-manipulated crystalline behaviour and intra-crystal phenomena. Nonequilibrium molecular simulation, performed in explicit externally applied fields, has, in recent years, contributed a great deal towards our insight into atomistic out-of-equilibrium phenomena, especially in from the viewpoint of qualitative mechanisms and in how polymorphic energy landscapes and chemical reaction barriers, in tandem with dynamical and intra-crystal fluctuation phenomena themselves, are shifted [1]. The scope for in-field, nonequilibrium molecular simulations to enhance mechanistic insights into underpinning intra-crystal phenomena is most exciting. From a more utilitarian viewpoint, a nonequilibrium molecular simulation acts as a pragmatic and predictive “molecular design” prototyping method, allowing for understanding a crystal’s response to electric fields and, of course, the redesign and optimisation of crystalline materials’ field feedback characteristics and “resonance” (e.g., for polymorphic sequencing and selection). This exciting, enabling outlook shall be explored in the present review and ramifications digested vis-à-vis a final vision of how this area of out-of-equilibrium, external field endeavours can progress in the years and decades ahead in the context of crystalline materials science and engineering.
Before focussing the remainder of this review on the topic of how electric fields influence crystal-state behaviours leveraging nonequilibrium, in-field molecular simulations, it is very helpful to reflect, albeit briefly, on how other kinds of externally applied fields help to manipulate the crystal state. Following the seminal work by Kushwaha et al. on phononic crystals [2], itself building on the pioneering work of Veselago [3], an important review of acoustically manipulated crystal behaviours in the area of phononic crystals and metamaterials was published by Liu et al. [4], which explored, inter alia, how ultrasound approaches can resonate with phonon modes and serve to shift dynamical properties. Indeed, other important reviews have considered acoustic cavitation vis-à-vis the underlying mechanisms of “sono-crystal” phenomena induced by pressure waves [5].
2. Crystal Response Theory in External Electric Fields
Understanding how crystallisation phenomena in externally applied electric fields (termed electro-crystallisation) is pivotal for inducing crystallisation and manipulating the crystal polymorphic outcome and nonequilibrium molecular simulation’s role has been discussed very recently in this context in a comprehensive review [6]. The scope of the present review, rather, focuses on how electric fields alter intra-crystal phenomena in crystalline materials without any phase change, i.e., with the crystal structure order being preserved; if interested in field-influenced crystallisation, the reader is referred to Reference [6]. As we shall discuss, (nonequilibrium) molecular simulation can provide a great deal of fundamental insights into field response intra-crystal behaviours. Prior to discussing this, per se, we put this into the general context of the crystal lattice response theory in external electric fields.
2.1. Thermodynamic Properties
In the presence of external electric fields, as regards the perturbation theory of crystal thermodynamic properties (such as Gibbs’ energies and enthalpies), Makogon [7] and Kaschiev [8] devised expressions for shifts in the thermodynamic equilibrium conditions using clathrate hydrates and ice. The field strength would need to pertain to the general vicinity of 0.01 V/nm to have any significant effect, confirmed by the nonequilibrium molecular dynamics (NEMD) work of English and MacElroy [9]. Amadei et al. formulated a quasi-Gaussian entropy (QGE) framework, expressing alterations in free energy vis-à-vis moment-generating functions in terms of the properties’ fluctuations [10]. Importantly, this provides thermodynamic equations of the states dependent explicitly on both the field strength and temperature. In terms of further thermodynamic-modelling progress, Aragones et al. adopted a Monte Carlo simulation and Gibbs–Duhem approach in building a TIP4P/2005 phase diagram in 0.3-V/nm external fields [11]. By general definition, ; therefore, we can infer from field-adjusted Gibbs’ energy that we can derive in-field entropies [7,8,9,10,11]. In any event, these advances, both as regards (in-field) thermodynamic phase shift theory and the associated (nonequilibrium) molecular simulations, lead to a more thorough understanding of how thermodynamic landscapes themselves are altered in terms of “electro-response theory”.
2.2. Theory of Interaction of Electric Fields with Crystals
In this section, the underlying theory of the interaction of matter with an external electromagnetic (e/m) field shall be discussed and then applied to the field effects on crystal lattices. Firstly, equations describing the dependence between the electric field , displacement , current density j and magnetic induction/field (B and H, respectively) shall be defined:
for which alongside , and denotes the medium’s dielectric constant, is its relative permeability and represents the conductivity. and represent the respective permittivity and permeability of a vacuum. Considering an area featuring a zero charge, Maxwell’s relations are
From Equations (1) and (2), the equations for e/m waves may be found. These are
with an analogous relationship for H.
Equation (3) describes plane e/m waves of angular frequency , wherein both components are mutually orthogonal and are given by
and the wave vector
and u is the phase velocity of the e/m wave.
A parallel relationship pertains to the magnetic analogue subject to
The wave number k may be related to the properties of the absorbing crystal:
The complex refractive index of the absorbing medium, , is defined as
where c is the velocity of the propagation in vacuo, and . Using Equation (8) gives
Experimentally, the weakening of strength I, is given by the Beer–Lambert law:
where z is the path length of the radiation, and is the absorption coefficient.
The imaginary refractivity may be gauged vis-à-vis an imaginary dielectric constant or an imaginary . If one writes as either or , then one may define and :
The polarisation P may be expressed vis-à-vis dielectric constant and conductivity by
Therefore, the complex dielectric constant becomes
For gases and cubic crystals (e.g., clathrate hydrates), the dielectric constant and conductivity are isotropic. In more general situations, they adopt tensor characteristics.
One may define a quantity:
as the power transfer per unit area along the wave’s plane. For a nonconducting medium (one for which ), the intensity I of the e/m field is given by
The classical absorption theories are unable to predict the resonant frequencies of a substance with an applied field, so one must apply quantum mechanics for a satisfactory treatment.
2.3. Quantum Mechanical Treatment of Absorption
The Schrödinger equation describing the wavefunction of a particle of mass m moving linearly on the x-axis under the action of a potential is
where . If one takes the usual Hamiltonian function as
and replaces the x-direction momentum by the differential operator and E by the operator , then Equation (16) becomes
abbreviated as . This may be generalised to three dimensions by .
By separating the wave function into a product of two functions dependent on x and t alone, i.e., , one may find a general solution for Equation (16):
where the coefficients have imaginary number dependence on time. Normalising the wave function implies that
The coefficients give the likelihood of energy at t. changes with time, given that the likelihood of the systems being in various energy configurations alters. If one supposes that, at time , the system is in such a state l that
The quantity represents the likelihood of transferring from configuration l to m, so that, if the energy from an external e/m field participated in this transfer process, its intensity would clearly be related to the transition probability.
In calculating the transition probability for a given case, one requires an expression for the part of the Hamiltonian that reflects the interaction of the e/m field and the system. The Hamiltonian is composed of
and the interaction term may be treated classically [12] or in a fully quantum manner [13], with a quantum treatment for the other two components. The fully quantum theory accounts for spontaneous transitions in the absence of a field, but the semiclassical theory is able to provide all of the main results quite accurately. Following the semiclassical approach of Houghton and Smith [12], the Hamiltonian for an electron in the presence of a field becomes
where e is electronic charge, p is momentum and the vector potential A is such that , where B is the magnetic component of the e/m field, and the scalar potential is such that .
If one neglects the magnetic component’s minor effect, then the interaction Hamiltonian becomes
for an x-polarised electric field (one for which , ). Therefore, the electric dipole transitions accounted for by the theory arise from an interaction term of the form , where M is the total electric dipole moment of the system.
The Hamiltonian interaction of Equation (24) may be used to calculate the probability of absorption (or, conversely, emission) of e/m radiation in an external field, resulting in a change of state.
In an e/m field, the Schrödinger equation for a particle’s motion becomes
where the perturbing Hamiltonian is small compared with the unperturbed Hamiltonian . The set of wave functions of the unperturbed system are given by
whilst the altered system wave functions may be expressed as :
Following Reference [12], the substitution of Equation (27) into Equation (25) and multiplication, integrated in space and followed by the substitution of the Hamiltonian interaction of Equation (26), results in the transition probability per unit time, , being defined as
where the energy density for an electric field polarised in the x-direction is given by
and the levels l and m exhibit degeneracy featuring their respective statistical weighting and . is the transition dipole for :
and the x-component of the matrix element of the dipole moment is given by
with analogous expressions of the y and z components.
For crystals, the energy levels are usually continuous, giving rise to band spectra. Crystalline solid wave functions adopt the lattice’s periodicity, so it is more expedient to adopt the momentum operator .
Following Houghton and Smith [12], if one writes from Equation (24) with and , then is proportional to for the transition and
These levels are labelled by the lattice wave . The x-component of is related to the electron wavelength in terms of . A solid featuring dimension L big in comparison to that of the spacing between atoms, the amount of levels n per band, when treated as a continuum, is .
Following Reference [12], the absorption coefficient for the material at the frequency is given by
where the energy E has been written for for the transition , and n in Equation (33) is the refractive index. Equation (33) may be generalised in terms of sidestepping the use of by its three-dimensional analogue:
In some applications to the solid state, the matrix element may be considered as changing sluggishly; in which case, absorptivity is via the density of states . This depends greatly on the nature of the lattice vibrations.
2.4. Lattice Vibrations
In this section, the underlying theory of lattice vibrations of crystals shall be discussed, and then, the effect of external e/m fields on exciting these modes shall be examined. The vibrational modes of crystals are attributable to collective motions of the constituent atoms and occur usually in the far-infrared (IR) (or submillimetre) frequency region. IR spectroscopy or incoherent inelastic neutron scattering (IINS) experiments shed much light on a lattice’s modes and underlying atomic interactions governing such collective motions. Specifically, IR spectroscopy gives extensive information on the coupling of these modes with an external e/m field, i.e., the technique reveals the resonant frequencies.
The motion of the periodic atoms and the associated boundary conditions of the periodic system impose only certain “allowed” frequencies with an associated energy . These quanta of elastic vibrational energy are “phonons”. The allowed solutions for the wave equation have an associated relationship between the angular frequency and the wave vector ; the relation is not usually linear but has “dispersion”, i.e., the phonon group velocity is dependent on the frequency (unlike , which is applied in the continuum theory of elastic waves, i.e., for long-wavelength phonons, for which ).
If there is an alteration of the state with a crystal’s electric moment shifting, coupling can occur between such motions and the e/m wave. This process occurs in ionic systems or molecular crystals (e.g., clathrate hydrates). Such absorption would be described by an equation similar to Equation (34), containing the density of the states .
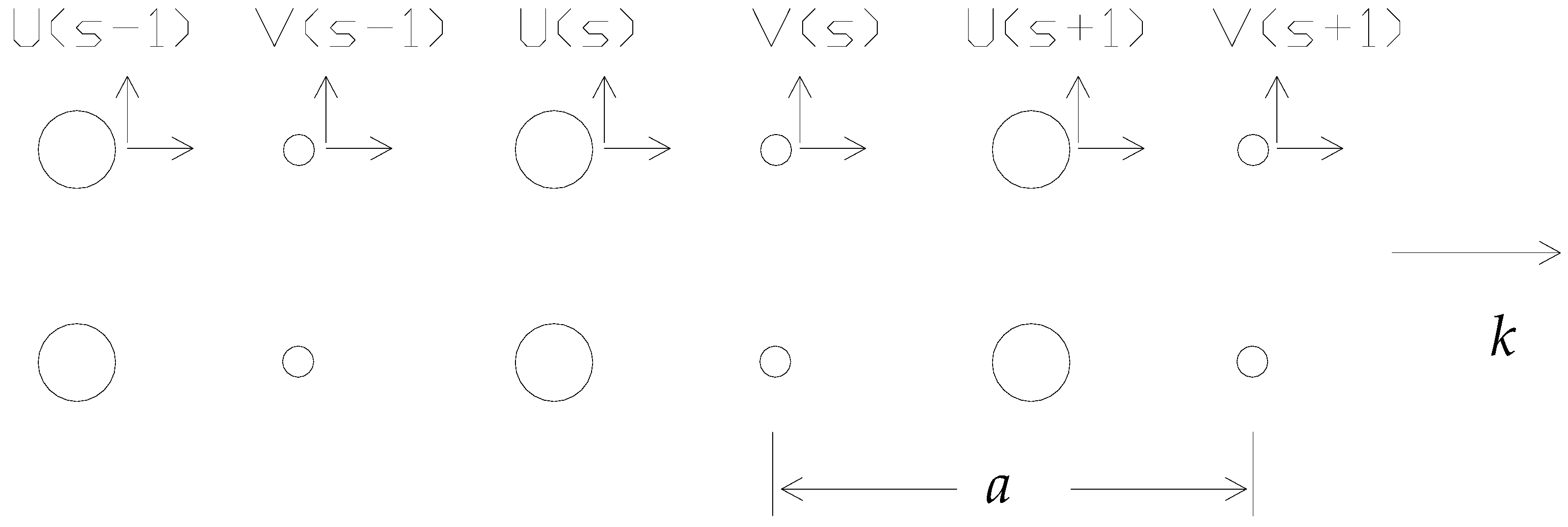
As a prototypical example of a lattice vibration, consider a diatomic crystal structure with masses and connected by a Hooke’s law force constant C between adjacent planes, with an alternating pattern of planes for each type of atom (1 and 2) (Figure 1).
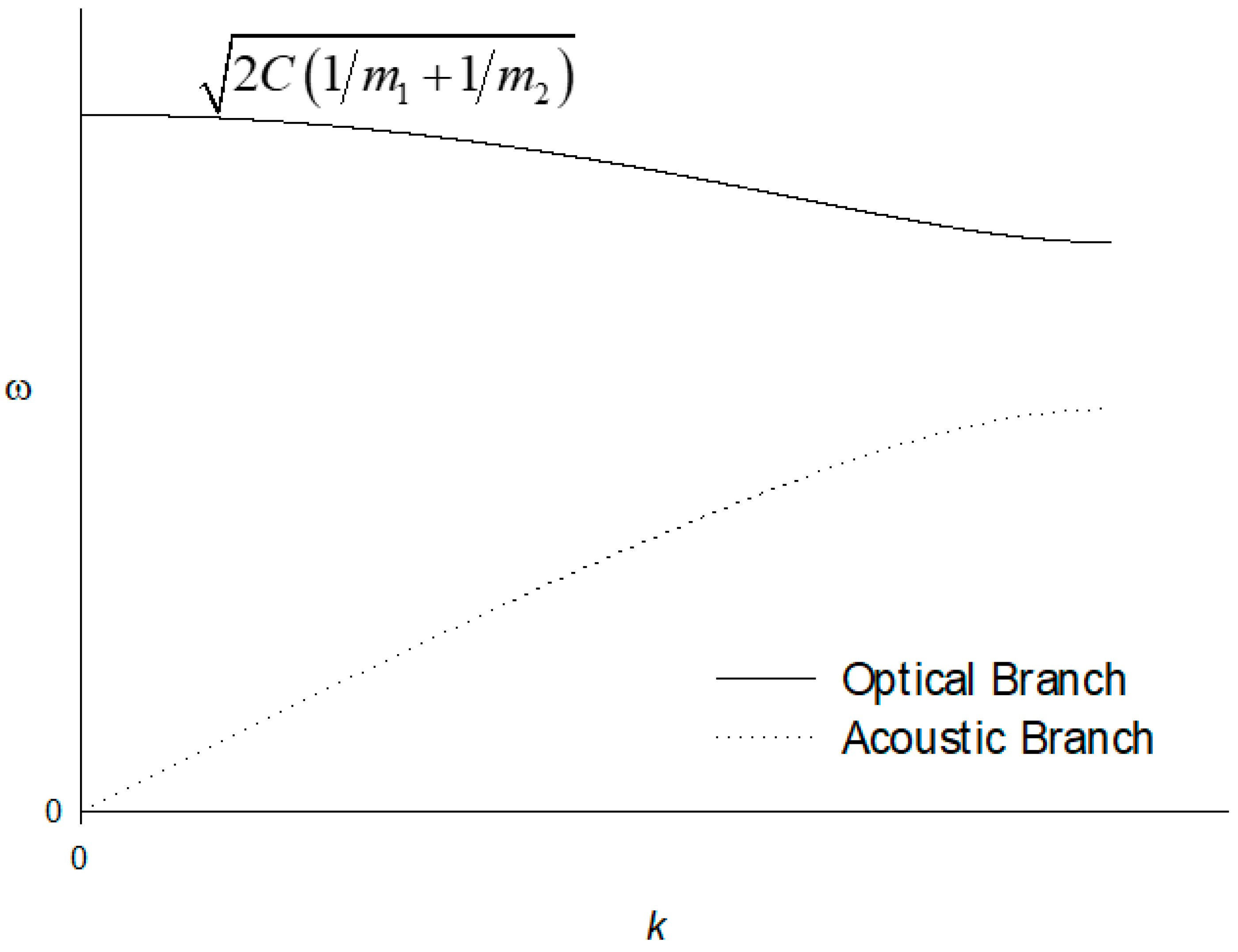
The repeat distance is a in the direction of the wave vector k, and the displacements of atoms are denoted by , , , … for planes s − 1, s and s + 1, whilst those of atoms are given by , and . The problem is then one-dimensional. For each wave vector, there are three modes, one of longitudinal polarisation and two of transverse polarisation. For each polarisation mode, the dispersion relation develops two branches, known as acoustic and optical branches. There are longitudinal LA and transverse acoustic TA modes and longitudinal LO and transverse optical TO modes. For a total of p atoms per unit cell, there are 3p branches to the dispersion relation: three acoustic branches and optical branches.
Newton’s equations of motion assume each plane interacts only with its closest neighbours:
If one looks for solutions in the form of a travelling wave, with different amplitudes u and v on alternate planes, then one obtains
and, upon the substitution of Equation (36) into (35), one has
The homogenous linear equations can be solved only if the determinant of the coefficients of unknowns u and v can disappear:
Equation (38) may be solved exactly for , although considering the limiting situations, and at the edge of the first Brillouin zone are more straightforward [14]. For small ka, one has , and the two roots are
The extent of the first Brillouin zone is . At , the roots are
The full dependence of on k, i.e., the dispersion relation, for the case is shown in Figure 2.
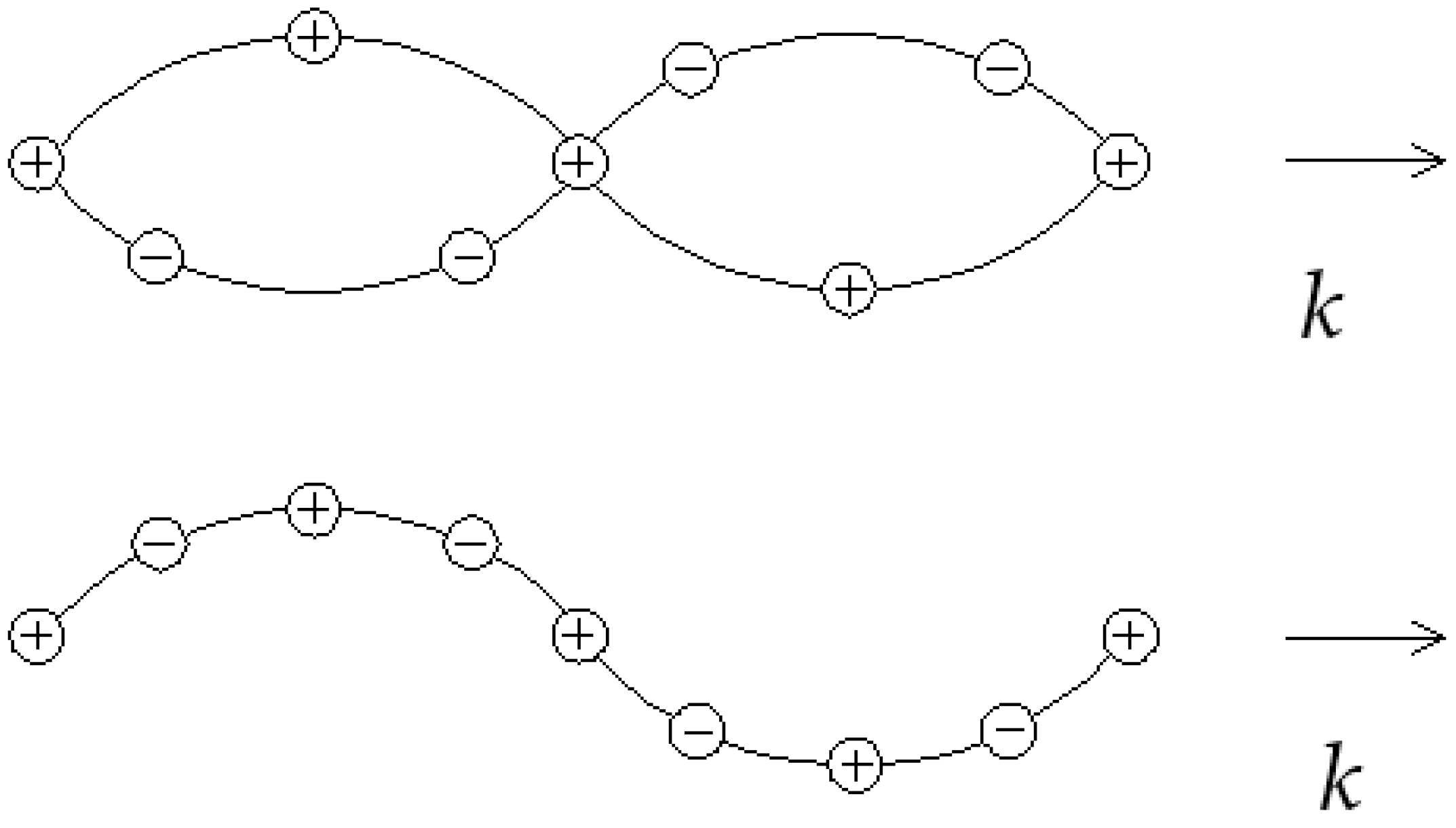
The particle displacements in the transverse acoustic and transverse optical branches are shown in Figure 3. For the optical branch at , one finds the substitution of Equation (39) into Equation (37):
If the two atoms carry opposite charges, as in Figure 3, then the motion of this type of wave may be excited by the electric field of an e/m wave (hence the term “optical” for its interaction with light waves). The limit of Equation (40) gives the solution : the atoms, along with their centre of mass, move together, as in long-wavelength acoustic vibrations (hence the term “acoustic”). It can be seen in Figure 3 that wavelike solutions do not exist for certain frequencies between and ; if one seeks solutions in the frequency gap for real , then the wave vector k shall be complex, meaning that the wave is damped in space.
In the case of a three-dimensional crystal, the equations of motion have plane–wave solutions of type , restricted no longer along a given chain of atoms, and may be resolved into purely transverse or longitudinal vibrations in certain directions of high symmetry. In this three-dimensional case, the density of the states or the number of phonon states per unit frequency range are given by [14]
where is an element of area on a constant frequency surface in k-space.
2.5. Electric Field Absorption
Absorption does not coincide with vibrational spectra (i.e., the density of modes); such field-induced motion produces an oscillating dipole moment, as described by a similar equation to Equation (34).
There are two main mechanisms of absorption, depending on the nature of the crystal:
- Restrahl absorption in ionic crystals only via the generation of single phonons;
- Multi-phonon absorption, wherein two or more phonons interact to give rise to an electric moment with which radiation may couple.
Restrahl resonance occurs in ionic crystals when the frequencies and wave vectors of both the incident radiation and the phonons are approximately equal. More specifically, transverse optical (TO) phonons may interact with the transverse e/m waves. At resonance, the phonon–photon coupling changes the characteristics of phonon propagation, and a forbidden frequency band is established for reasons that have nothing to do with the periodicity of the lattice. The quantum of the coupled phonon–photon transverse wave field is known as a “polariton”. The typical resonant frequencies for Restrahl absorption occur well below those of multi-phonon absorption at the Brillouin zone boundaries.
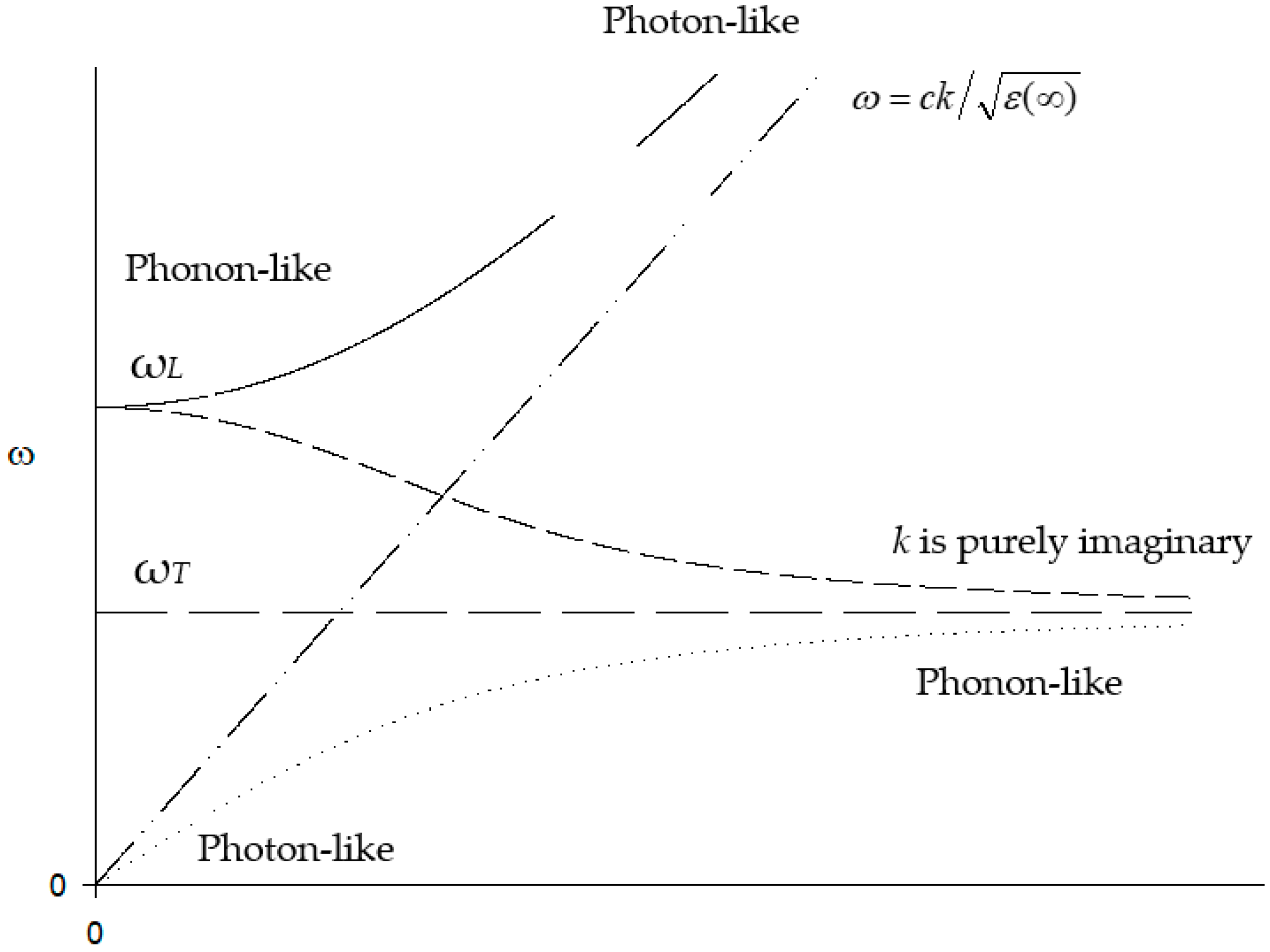
Following Reference [14], if one takes the TO phonon as having a frequency in the absence of coupling with photons and take the polarisation P as , where there are N ion pairs of an effective charge q and reduced mass m per unit volumes with a displacement u of the positive ions relative to the negative ions, then the polariton dispersion relation is given by the solution of
The dispersion relation is plotted in Figure 4.
At , there are two roots, for the photon and
for the polariton. As may be seen in Figure 4, there is a forbidden frequency band between and , for which there is a complex wave vector solution for real , leading to a damping of the wave.
The LO phonons vibrate at a higher frequency than the transverse waves, given by
where and are the static and high-frequency dielectric constants. However, the e/m radiation couples only to the lower frequency TO branch in polar crystals.
Multi-phonon absorption is significantly weaker than one-phonon Restrahl absorption and occurs at higher frequencies. It is thought to involve one vibrational mode inducing charges on the atoms and a second (or third, etc.) mode causing a vibration simultaneously of the induced charges, thus producing an electric moment that may couple to the applied electric field.
3. Molecular Simulation in External Electric Fields
Having described basic crystal–field interaction theory from a molecular quantum vantage and how the underlying thermodynamic and dynamical quantities underpinning this may be shifted and perturbed by external electric fields, we now turn to a mechanistic description of how we may apply external electric fields in a nonequilibrium molecular simulation (such as nonequilibrium molecular dynamics, NEMD) prior to then reviewing how this advanced tool has been used to shed light on intra-crystal phenomena in recent decades. As the literature to date has used a primarily classical method of coupling external fields to molecular systems (such as crystals), we restrict our present attention to this, although we shall reflect in closing as to the prospect of the pervious quantum-mechanical treatment of polariton excitations being coupled to nonequilibrium molecular simulations of the crystal state.
In NEMD, the system’s Hamiltonian, H, is perturbed by adding the term HE to represent the external field, e.g., for dipolar particles with a field E acting along the z-axis, this is [1,6]:
where μtot is the total, collective dipole vector. E(t) may be either static (time-independent) or time-dependent (e.g., an oscillating electric field, with an electromagnetic (e/m) field being a good example). Of course, the details of the field perturbation, per se, depend on the details of the system’s computational handling and on how the energy and forces are computed (e.g., whether as a system of classical point charges or via electronic structure approaches, etc.).
In the (spatial) derivative formulation of field perturbations of the Hamiltonian system of Equation (47), we have Equation (48) below, expressed for the example of an e/m field (i.e., with a magnetic component also). If an e/m or electric field force, fi,elec = qiE, is applied to each partial charge site i with charge qi (static or fluctuating), Newton’s Second Law becomes:
and fi arises from V by standard vector differentiation [15]. Taking the e/m field in Equation (48) as an example, both components occur along the laboratory x and y directions, respectively, so the polarisation plane is x–y, with propagation along the z-axis from Maxwell’s equations. Further, the time-varying e/m-field electric component vector is given by
in terms of the maximum of the electric and magnetic component intensities Fmax and Bmax. Now, Emax/Bmax = c/n, and the root mean square (r.m.s.) electric field intensity for an e/m field is given by Erms = Emax/, where n is the system’s refractive index, and c is the speed of light.
For time-varying, cyclic fields (e.g., e/m fields), an important point in NEMD is to simulate enough cycles in a given NEMD simulation for a sufficient statistical sampling of the field effects, and especially so in lower-frequency alternating fields, as the oscillating field period is long compared with the system’s underlying (translational) frequency modes [1]. Another important point for such a NEMD is that the intensities required to observe tangible external field effects are often much higher, by definition, than the underlying local or intrinsic electric fields in the system itself (typically of the order of 1–3 V/Å [16,17]); this necessary disparity is driven by the need to span the gulf between experimental and simulation timescales. In any event, there is a general linear response regime (for dipolar and other, e.g., dynamical, properties) in the region of external-field intensities up to ~0.5 V/nm [1], for which the external field torque or forces are no more than a few percentages of the intrinsic ones (e.g., in Equation (48)).
Thermostatting is of huge importance in NEMD in crystallisation (whether propagated via empirical potentials or ab initio MD). For instance, in “real-world” systems (typically approximating imperfectly “sluggish” NPT or NVT conditions), there will be a (slow) dissipation of the field-imparted thermal energy generated by the friction of molecules or atoms rotating or translating [1]; only under strictly adiabatic conditions extending to the molecular scale would the non-use of any thermostatting in NEMD be appropriate (e.g., for the study of field-induced heating).
The underlying foundations of ab initio molecular dynamics (AIMD) typically involve classical propagation during the time of a Hamiltonian system, which is now itself computed via electronic structure methods, with (Hellmann–Feynman) forces. Umari and Pasquarello formulated the underlying theory for (ground-state) Density-Functional Theory in external fields under periodic boundary conditions (PBC) by a “Berry-phase” approach to polarisation theory [18,19]. A key advantage of in-field DFT under PBC is that condensed-phase, periodic systems can be simulated in EEFs with fully self-consistent electron-density rearrangement, which is essential for the accurate handling of electronically delocalised systems. However, nonequilibrium AIMD, as we shall see, has been applied much less to study field-manipulated crystal states, per se, although its initial usage to model field-driven intra-crystal phenomena is certainly encouraging (vide infra). The present review does not consider the computation of intrinsic electric fields in DFT [17] but, rather, the imposition of external electric fields; as References [1,6] note astutely, the magnitude of externally applied electric fields in a nonequilibrium molecular simulation, which is needed typically to lead to noticeable thermodynamic and dynamical property shifts, are about 1 to 2% of the fields that are present intrinsically. Of course, it is important in DFT and AIMD, whether using equilibrium or nonequilibrium propagation of the system (in the respective absence and presence of external fields), to choose functionals judiciously [20].
4. Crystal Systems in External Electric Fields
In this section, the limited investigations into crystal systems and growth in external electric fields shall be discussed briefly, i.e., “electro-crystal” phenomena. As pointed out previously, in the case of e/m fields, the electric component of external fields is dominant, and therefore, the magnetic field component was not examined in some of the following studies. Although we shall examine some “fluid-like” properties of proton hopping in the present review, we will not consider how electric fields induce (fluid-)phase changes in electric fields [21], much less crystallisation itself [1,6]; the crystallographic Wyckhoff positions remain fully intact in the work reviewed in the present case.
4.1. Electric-Field-Driven Heating
Blanco and Auerbach [22] performed nonequilibrium MD simulations to study microwave heating on various zeolites and zeolite guest systems. Industrially important zeolites, which constitute high-charge (NaY) and low-charge (dealuminated Y = DAY and silicate) materials, were studied. The zeolite systems were modelled using a bespoke force field [23], and the Ewald technique was utilised for electrostatic interactions with fixed partial charges [15]. A time-varying electric field was introduced as a forcing function in Newton’s equations of motion, as described previously, and the field frequency was set at 23.9 GHz (or = 0.15 rad/ps) at the blue end of the microwave spectrum. A time-dependent system temperature was defined (by proper normalisation of the system kinetic energy at each MD time step) to investigate the microwave heating effects on each system. To disentangle such distributions in the equilibrium from microwave-driven cases, a temperature for each atom type was determined. It was determined [22] that the stochastic velocity replacing of Andersen [24] was better than Nosé–Hoover thermostatting [25]. It was found that the thermostat parameters had to be adjusted at each new field intensity. Furthermore, Blanco and Auerbach found that different steady-state temperatures could be maintained for the host and guest in simulations of methanol in both of the siliceous zeolites [22]. This may also be rationalised in terms of inefficient energy transfer from the guest to the host framework.
Reference [22] was interesting and significant, in that it used a nonequilibrium molecular simulation to establish that the energy distributions in microwave-driven zeolite guest systems are qualitatively different from conventional heating. One limitation of the study may be the use of fixed partial charges in the potential models, particularly for ionic species, which would be expected to exhibit substantial polarisability in the presence of intense external electric fields. However, Blanco and Auerbach also carried out a NEMD simulation of further zeolite guest systems to more explicitly study the loading dependence of athermal effects [26]. In a further exciting work, Gharibeh et al. studied microwave-induced temperature distributions across different atomic species in zeolites [27], providing more detailed and quantitative mechanistic views of how microwave field-driven athermal distributions arise from the standpoint of energy dissipation dynamics, and, as we shall reflect upon later, developments in NE-AIMD and polarisable force fields have much to offer in terms of a future outlook for NEMD applied to crystals in general. Jobic et al. also confirmed, by experiment, these NEMD predictions of microwave-induced temperature heterogeneity, an important athermal effect [28].
In further NEMD probing of microwave-induced athermal behaviour in crystals, English et al. performed NEMD simulations of rutile–titania in external electric fields with the frequency in the microwave-to-far-infrared range, carrying out both thermostatted and nonthermostatted NEMD, with electromagnetic fields applied along both a and c crystallographic directions [29]. It was observed that the ions responded rapidly to the applied fields—not only with heating for nonthermosatted NEMD but, also, the structural response. The peaks and troughs of the Ti–Ti radial distribution functions were sharpened vis-à-vis the zero field case at the moments of maximal applied field intensity during the e/m-field cycles [29]. Interestingly, in the case of nonthermostatted NEMD simulation producing heating, as in References [22,26,27,28], 500 GHz was found to be the most effective frequency in exciting vibrational modes, inducing the fastest rate of temperature rise. Above 2000–2500 K, microwave-induced crystal structure melting was encountered [29]. For thermostatted NEMD, the 50-GHz case led to the most substantial enhancement in ionic translational mobility, given that this external field coupled more effectively with the underlying translational modes in the crystal density of states, as opposed to the vibrational modes (which tended to lead instead to the greatest level of heating [29]).
4.2. Field-Induced Vibrational and Structural Perturbations in Crystals
Aside from electric field heating, per se, arising from NEMD inducing the vibrational (and translational) coupling of applied electric fields, there is the important matter of how electric fields perturb crystals’ wider properties, often originating from vibrational coupling and field-induced “resonance”. To this end, Reference [30] carried out NEMD simulations of methane hydrates over a range of e/m fields, examining the frequencies of the system mass density fluctuations with three major contributions: water molecules’ liberations giving rise to the main one at 720 cm−1, regardless of any applied field. One of the more minor system density fluctuations arises at 10–12 cm−1, owing to the propagation of local density fluctuations independent of e/m fields. The final system density fluctuation arises from the e/m fields, and it only becomes the case for intensities of 0.12 V/Å or higher—close to the field intensity threshold for electro-dissociation [1]. This modal frequency is double that of the applied e/m field, owing to a field–crystal “resonance” relationship [1], and it was shown that the main qualitative features of the translational and liberational densities of states (DOSs) were unaffected by applying the fields. There was also similar vibrational coupling found in the case of configurational energy values, as well as system mass density. There was a level of structural “smearing” identified in the cycle-averaged radial distribution functions, as was also seen in Reference [29].
Inspired by surprising experiments, Skačej and Zannoni scrutinised elastomer liquid crystals and their significant structural deformations and swelling by applying perpendicularly applied electric fields using nonequilibrium Monte Carlo simulations, studying an off-lattice mode [31]. The resultant crystal deformation mode applied to the nematic phase, as well as a mode switching between orientationally disordered isotropic and field-aligned nematic phases. Critical behaviour similar to that of a liquid crystal in an external field was observed, with typical switching thresholds that were higher than for the (semi)soft actuation mode involving director rotation [31].
4.3. Field-Imparted Grotthus-like Proton Hopping in Superionic Ice Polymorphs
The impact of external electric fields on various polymorphs of ice has been shown recently by NE-AIMD; here, the intramolecular break-up of water molecules was facilitated by the applied external electric fields, which led to Grotthus-type migration of such “electro-liberated” protons in now-superionic ice polymorphs. Cassone et al. applied NE-AIMD in static electric fields to ices Ih and XI (the proton-ordered version of Ih), finding that respective field intensities of around 0.25 and 0.22 V/Å were required as threshold levels for which water molecules break up, and a field-sustained proton flow was generated through the remaining intact crystal lattice, with the corresponding intramolecular dissociation and proton flow value in liquid water being about 0.36 V/Å [32]. In the crystal state, with oxygen atoms remaining in the crystallographically distinct lattice sites, the external static electric fields were able to induce and sustain protonic currents at lower field strengths than the liquid state by virtue of crystal symmetry [32]. This “fluid-like” finding of field-induced superionic Grotthus-style protonic conduction was also witnessed by Futera et al. in their NE-AIMD simulations for a case of static fields applied to high-pressure ice VII polymorphs, which are thought to be present in large-planet mantles (e.g., Uranus) and exoplanets [33].
5. Conclusions
The present review discussed how external electric fields alter crystal-state phenomena, such as thermodynamics, selective heating, proton hopping, structural aspects and kinetic/diffusional barriers, from the viewpoint of nonequilibrium molecular simulation, considering the mechanistic coupling of field-mediated forces of ions and torques on molecules. The perturbation of hydrogen bonding between water molecules in ice and hydrates, how this affects aqueous solubility [21,34] and the electrostatic interactions [35] are important cases in point.
In terms of future outlooks, although the present review mentioned (nonequilibrium) DFT and AIMD as being important for the study of (polarisable) crystal structures (whether as ionic or molecular crystals), this is a still-underexplored area in field-manipulated phenomena. In particular, we note with interest the recent developments in electronically excited-state and other ab initio methods for time-dependent simulations for photochemical and photophysical phenomena [36,37,38,39], and a key future development will lie in how to couple the external field formulations outlined in the present work to these excited-state electronic structure approaches. To make further progress, developments in polarisable force fields and neural network potentials may have much to offer in allowing for more accurate responses of systems to the applied electric fields, e.g., in terms of the rearrangement of the shapes of “electron clouds” and the wider electronic degrees of freedom. In addition, the adoption of a truly quantum treatment of electric and electromagnetic field absorption, per se, along the lines of polaritons discussed earlier in this review, as opposed to the more classical Hamiltonian perturbation approaches favoured to date in NEMD and NE-AIMD, would also be beneficial and worth exploring in terms of future outlooks on field-perturbed crystal-state phenomena. This will allow for, amongst other things, full quantum rigour in modelling the next generation of modern (ferro-electric and otherwise) field effect nanotransistors and neuromorphic devices, given that quantum effects, per se, are becoming inherently important at the nanometre-length scale in ever-smaller field-responsive crystal-state devices. As such, the present review lays down an important vision and outlook for this formidable and challenging undertaking.
Funding
N.J.E. thanks Science Foundation Ireland for the funding (SFI 17/NSFC/5229).
Conflicts of Interest
The authors declare no conflict of interest.
References
- English, N.J.; Waldron, C.J. Perspectives on external electric fields in molecular simulation: Progress, prospects and challenges. Phys. Chem. Chem. Phys. 2015, 17, 12407. [Google Scholar] [CrossRef]
- Kushwaha, M.S.; Halevi, P.; Dobrzynski, L.; Djafari-Rouhani, B. Acoustic band structure of periodic elastic composites. Phys. Rev. Lett. 1993, 71, 2022–2025. [Google Scholar] [CrossRef]
- Veselago, V.G. The electrodynamics of substances with simultaneously negative values of e and μ. Sov. Phys. Usp. 1968, 10, 509–514. [Google Scholar] [CrossRef]
- Liu, J.; Guo, H.; Wang, T. A Review of Acoustic Metamaterials and Phononic Crystals. Crystals 2020, 10, 305. [Google Scholar] [CrossRef]
- Na Kim, H.; Suslick, K.S. The Effects of Ultrasound on Crystals: Sonocrystallization and Sonofragmentation. Crystals 2018, 8, 280. [Google Scholar] [CrossRef] [Green Version]
- English, N. Molecular Simulation of Crystallisation in External Electric Fields: A Review. Crystals 2021, 11, 316. [Google Scholar] [CrossRef]
- Makogon, Y.F. Hydrates of Hydrocarbons; PennWell Books: Tulsa, OK, USA, 1997. [Google Scholar]
- Kashchiev, D. On the influence of the electric field on nucleation kinetics. Philos. Mag. 1972, 25, 459. [Google Scholar] [CrossRef]
- English, N.J.; MacElroy, J.M.D. Theoretical studies of the kinetics of methane hydrate crystallization in external electromagnetic fields. J. Chem. Phys. 2004, 120, 10247. [Google Scholar] [CrossRef]
- Amadei, A.; Apol, M.E.F.; Brancato, G.; Di Nola, A. Theoretical equations of state for temperature and electromagnetic field dependence of fluid systems, based on the quasi-Gaussian entropy theory. J. Chem. Phys. 2002, 116, 4437. [Google Scholar] [CrossRef]
- Aragones, J.L.; MacDowell, L.G.; Siepmann, J.I.; Vega, C. Phase diagram of water under an applied electric field. Phys. Rev. Lett. 2011, 107, 155702. [Google Scholar] [CrossRef] [Green Version]
- di Bartolo, B.; Powell, R.C. Phonons and Resonances in Solids; Wiley: New York, NY, USA, 1976. [Google Scholar]
- Houghton, J.T.; Smith, S.D. Infrared Physics; Clarendon Press: Oxford, UK, 1966. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics, 7th ed.; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids, 2nd ed.; Clarendon Press: Oxford, UK, 2017. [Google Scholar]
- Cao, H.; English, N.J.; MacElroy, J.M.D. Diffusive hydrogen inter-cage migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates. J. Chem. Phys. 2013, 138, 094507. [Google Scholar] [CrossRef]
- McDonnell, K.; Wadnerkar, N.; English, N.J.; Rahman, M.; Dowling, D. Photo-active and optical properties of bismuth ferrite (BiFeO3): An experimental and theoretical study. Chem. Phys. Lett. 2013, 572, 78–84. [Google Scholar] [CrossRef]
- Umari, P.; Pasquarello, A. Ab initio molecular dynamics in a finite homogeneous electric field. Phys. Rev. Lett. 2002, 89, 157602. [Google Scholar] [CrossRef] [PubMed]
- Umari, P.; Pasquarello, A. Density functional theory with finite electric field. Int. J. Quant. Chem. 2004, 101, 666. [Google Scholar] [CrossRef]
- Dev, P.; Agrawal, S.; English, N.J. Functional Assessment for Predicting Charge-Transfer Excitations of Dyes in Complexed State: A Study of Triphenylamine–Donor Dyes on Titania for Dye-Sensitized Solar Cells. J. Phys. Chem. A 2013, 117, 2114–2124. [Google Scholar] [CrossRef] [Green Version]
- Ghaani, M.R.; Kusalik, P.G.; English, N.J. Massive generation of metastable bulk nanobubbles in water by external electric fields. Sci. Adv. 2020, 6, eaaz0094. [Google Scholar] [CrossRef] [Green Version]
- Blanco, C.; Auerbach, S.M. Microwave-Driven Zeolite−Guest Systems Show Athermal Effects from Nonequilibrium Molecular Dynamics. J. Am. Chem. Soc. 2002, 124, 6250–6251. [Google Scholar] [CrossRef]
- Jaramillo, E.; Auerbach, S.M. New force field for Na cations in faujasite-type zeolites. J. Phys. Chem. B 1999, 103, 9589–9594. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Blanco, C.; Auerbach, S.M. Nonequilibrium molecular dynamics of microwave-driven zeolite− guest systems: Loading de-pendence of athermal effects. J. Phys. Chem. B 2003, 107, 2490–2499. [Google Scholar] [CrossRef]
- Gharibeh, M.; Tompsett, G.; Lu, F.; Auerbach, S.M.; Yngvesson, K.S.; Conner, W.C. Temperature Distributions within Zeolite Precursor Solutions in the Presence of Microwaves. J. Phys. Chem. B 2009, 113, 12506–12520. [Google Scholar] [CrossRef]
- Jobic, H.; Santander, J.E.; Conner, W.C.; Wittaker, G.; Giriat, G.; Harrison, A.; Ollivier, J.; Auerbach, S.M. Experimental Evidence of Selective Heating of Molecules Adsorbed in Nanopores under Microwave Radiation. Phys. Rev. Lett. 2011, 106, 157401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, N.J.; Sorescu, D.C.; Johnson, J.K. Effects of an external electromagnetic field on rutile Tio2: A molecular dynamics study. J. Phys. Chem. Solids 2006, 67, 1399–1409. [Google Scholar] [CrossRef]
- Waldron, C.J.; English, N.J. Global-density fluctuations in methane clathrate hydrates in externally applied electromagnetic fields. J. Chem. Phys. 2017, 147, 024506. [Google Scholar] [CrossRef] [PubMed]
- Skačej, G.; Zannoni, C. Molecular simulations elucidate electric field actuation in swollen liquid crystal elastomers. Proc. Nat. Acad. Sci. USA 2012, 109, 10193–10198. [Google Scholar] [CrossRef] [Green Version]
- Cassone, G.; Giaquinta, P.V.; Saija, F.; Saitta, A.M. Proton Conduction in Water Ices under an Electric Field. J. Phys. Chem. B 2014, 118, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Futera, Z.; Tse, J.S.; English, N.J. Possibility of realizing superionic ice VII in external electric fields of planetary bodies. Sci. Adv. 2020, 6, eaaz2915. [Google Scholar] [CrossRef]
- English, N.J.; Carroll, D.G. Prediction of Henry’s Law Constants by a Quantitative Structure Property Relationship and Neural Networks. J. Chem. Inf. Comput. Sci. 2001, 41, 1150–1161. [Google Scholar] [CrossRef]
- English, N.J.; MacElroy, J.M. Atomistic simulations of liquid water using Lekner electrostatics. Mol. Phys. 2002, 100, 3753–3769. [Google Scholar] [CrossRef]
- Kronik, L.; Neaton, J.B. Excited-State Properties of Molecular Solids from First Principles. Annu. Rev. Phys. Chem. 2016, 67, 587–616. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; English, N.J. Electronic structure of cation-codoped TiO2 for visible-light photocatalyst applications from hybrid density functional theory calculations. App. Phys. Lett. 2011, 98, 142103. [Google Scholar] [CrossRef]
- Agrawal, S.; Dev, P.; English, N.J.; Thampi, K.R.; MacElroy, J.M.D. A TD-DFT study of the effects of structural variations on the photochemistry of polyene dyes. Chem. Sci. 2012, 3, 416–424. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Band gap engineering of double-cation-impurity-doped anatase-titania for visible-light photo-catalysts: A hybrid density functional theory approach. Phys. Chem. Chem. Phys. 2011, 13, 13698–13703. [Google Scholar] [CrossRef]
Figure 1.
A linear diatomic lattice.

Figure 2.
Optical and acoustic branches of the dispersion relation for a diatomic linear lattice. Here, m1 > m2, and the branch for m2 is above m1.
Figure 2.
Optical and acoustic branches of the dispersion relation for a diatomic linear lattice. Here, m1 > m2, and the branch for m2 is above m1.

Figure 3.
TO (upper) and TA (lower) phonons in a linear diatomic lattice.

Figure 4.
Coupling of the photons and TO phonons. denotes phonons without coupling to the external e/m field, whilst that labelled corresponds to e/m waves in the crystal that are uncoupled to the phonons . The continuous and dotted lines are the dispersion relations for the resonance between the phonons and e/m radiation. One effect of the coupling is to produce a frequency gap between and ; within this gap, the wave vector k has a purely imaginary magnitude given by the dashed line in the figure. It may be seen that the characters of the branches vary with k and that there are regions of mixed electrical–mechanical aspects at the nominal crossover.
Figure 4.
Coupling of the photons and TO phonons. denotes phonons without coupling to the external e/m field, whilst that labelled corresponds to e/m waves in the crystal that are uncoupled to the phonons . The continuous and dotted lines are the dispersion relations for the resonance between the phonons and e/m radiation. One effect of the coupling is to produce a frequency gap between and ; within this gap, the wave vector k has a purely imaginary magnitude given by the dashed line in the figure. It may be seen that the characters of the branches vary with k and that there are regions of mixed electrical–mechanical aspects at the nominal crossover.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
English, N.J. Molecular Simulation of External Electric Fields on the Crystal State: A Perspective. Crystals 2021, 11, 1405. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111405
AMA Style
English NJ. Molecular Simulation of External Electric Fields on the Crystal State: A Perspective. Crystals. 2021; 11(11):1405. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111405
Chicago/Turabian StyleEnglish, Niall J. 2021. "Molecular Simulation of External Electric Fields on the Crystal State: A Perspective" Crystals 11, no. 11: 1405. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111405
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.