1. Introduction
The application of deep-ultraviolet (DUV) laser light sources such as photorefractive keratectomy and 195 nm photolithography have stimulated interest and efforts to develop non-linear optical (NLO) crystals for DUV generation. Unfortunately, the highly competitive environment also causes many of the researchers to be unforthcoming about their efforts. Based on the limited data available, the field can be roughly divided into two halves by what Schunemann describes as a “200 nm wall in terms of deep-UV generation” [
1].
In order to break the 200 nm wall, an anionic group theory was developed in 1989 by Chen [
2], which greatly promoted the discovery of more than ten borate UV and DUV crystals including LiB
3O
5 (LBO),
β-BaB
2O
4 (BBO), CsB
3O
5 (CBO), K
2Al
2B
2O
7 (KABO), and KBe
2BO
3F
2 (KBBF) [
3]. Apart from KBBF, a DUV laser cannot be generated in other borate crystals by direct second-harmonic generation (SHG). Therefore, as Hassaun said: “
Deep-UV applications await improved nonlinear optics” [
4].
At present, only a few non-toxic DUV NLO materials have been discovered including the diamond-like structure of LiZn(OH)CO
3 [
5] as well as alkali metal borate systems CsLiB
6O
10 (CLBO) and K
2[B
3O
3(OH)
5] [
6]. However, high quality stoichiometric single crystals of these novel non-toxic DUV NLO materials are still extremely rare due to the high volatility of Li
2O, B
2O
3, and the flux (e.g., CaB
4O
7, CaO, and PbO) resulting in phase transitions or great deviations from the stoichiometry [
7]. Recently, alkaline earth metal fluorooxoborate compounds such as Ca
2B
10O
14F
6 (CBOF) have been studied and the chemically stable and NLO-favorable layered NCS beryllium-free borates might meet most of the structural requirements of DUV NLO crystals [
8]. Recently, single crystals of CBOF with micrometer sizes were grown by the flux method, which might have good linear and NLO properties and might be valuable for further investigations [
9].
To the best of our knowledge, no comprehensive theoretical understanding of CBOF has been reported in detail until now. Herein, we report the electronic structure, formation energy, and defect-induced optical absorption properties based on the
first principle theory. We performed a prediction for the crystal growth habits based on Bravais–Friedel Donnay–Harker (BFDH) [
10] and Hartman–Perdok theories [
11]. We found that the interstitial F (Fi) and antisite defect O
F are the dominant defects in the CBOF material due to the low formation energies and the defects of F and O vacancies (V
F, V
O) as well as interstitial O (O
i), which may be responsible for inducing the optical absorption centers in CBOF.
2. Method
The ab initio electronic structure calculations were performed using the CASTEP code [
12] and the total energy pseudopotential approach [
13]. We set the energy cutoff of the plane-wave basis functions to be 490 eV, resulting in the convergence of the total energy for greater than 1.2 meV per atom. For the exchange-correlation energy [
14], ultrasoft pseudopotentials [
15] and the Perdew–Burke–Ernzerhof (PBE) gradient-corrected functional [
16] were used. The forces between the atoms were calculated by the Hellmann–Feynman method [
17] and the total energy of the whole system was minimized using the density-mixing conjugate gradient approach [
18]. An isolated point defect was calculated in the periodic boundary conditions by using a repeated supercell created from a conventional cell of CBOF, which consisted of 64 ± 1 atoms.
The formation energy (E
f) of the point defects and the equilibrium concentrations can be calculated by the total energy. Regarding the vacancy and interstitial defects, the formation can be written as [
19]:
EN±1 is the total energy for a supercell with a point defect,
EN is the total energy of the supercell without defects, and
μ is the chemical potential of the atom, which were calculated by a cubic cell with lattice constants of 20 Å. The equilibrium concentration (
n/
N) of the point defects at room temperature were also calculated as [
20]:
Here, E is the formation energy for the point defect, T is the temperature, and k is the Boltzmann constant.
CBOF crystallizes in a non-centrosymmetric orthorhombic structure, space group C
mc2
1, with
a = 9.911 Å,
b = 8.402 Å,
c = 7.966 Å, and
α =
β =
γ = 90° [
21]. The crystal morphologies of the CBOF crystals were quantitatively investigated via the
first principle calculations using attachment energy model BFDH theories [
22] and using the molecular interactions to calculate the growth rates for the different crystal faces (PBC method).
For the CBOF crystal faces, we calculated their attachment energy based on the ab initio method for the slices {
hkl}, which were determined by the Donnay–Harker prediction [
23]. Regarding the calculations for attachment energy, we chose the {020} face as the example. The first step was to construct a slab with ten growing slices. Afterwards, we moved the top {020} slice with the interplanar distance
dhkl = 4.201 Å to 150 Å from the surface of the slab to decrease the interaction between the slice and the slab. The energy for the slab and the separated slice were calculated and the attachment energies were then deduced. In this paper, the attachment energy (
Eatt) was deduced by
Eatt =
Elatt −
Eslice where
Elatt is the energy of the combined system,
Eslice is the energy for the growth slice, and the growth rate is proportional to the
Eatt.
Periodic bond chains (PBCs) are defined as the periodic chains with strong bonds along a certain crystalline direction, which is the basis of the BFDH theory. The miller faces can be divided to be the F (flat) faces containing no less than two non-parallel PBCs in the thickness of
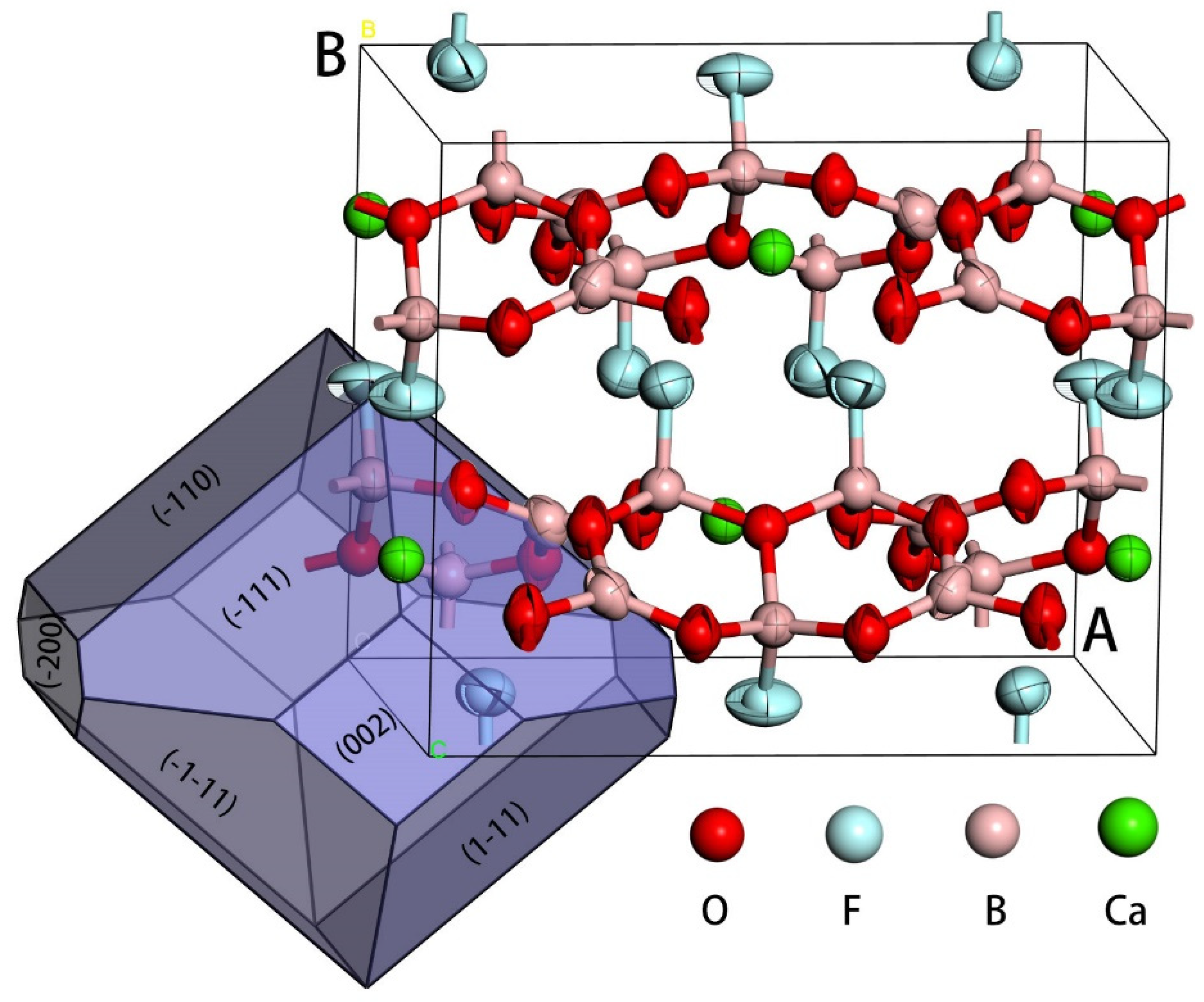
dhkl. Face S is defined as having only one PBC whereas K faces contain no PBC. The growth rate was ranked as faces K > S > F; therefore, F faces were the main forms on the real crystal growth process. Based on the structure of CBOF, as shown in
Figure 1, three main PBCs were found as <001>, <100>, and <101>.
3. Results and Discussion
Table 1 lists the various {
hkl} faces of CBOF, the
dhkl, the
Eatt attachment energies, the PBCs that were parallel to the corresponding {
hkl} faces, and the actual experimental observed faces. From this data, we found that two F faces were defined and both of them had more than two non-parallel PBCs. The {110} faces were parallel to three main PBCs, <001>, <100>, and <101>; the {111} faces were parallel to <100> and <101> and were named the F faces. The {200}, {020}, and {002} faces were parallel to only <001>, <100>, and <101>, respectively, which were referred to as the S faces. F faces are normally the best developed, i.e., the growth rate is relatively slow compared with the K and S faces. We concluded, therefore, that the S faces would be the first to disappear during the growth of the crystal.
Figure 1 shows one of the predicted shapes. Based on the attachment energy calculations, we considered that the order of morphology relevance for the miller faces was {110} > {111} > {200}. It has been shown that {110} and {111} faces are usually well-developed whereas {200} faces appear only under a very special condition [
9].
For the electronic structure ab initio calculations, we searched the equilibrium configuration by the relaxation of both the lattices and atomic positions; the space group C
mc2
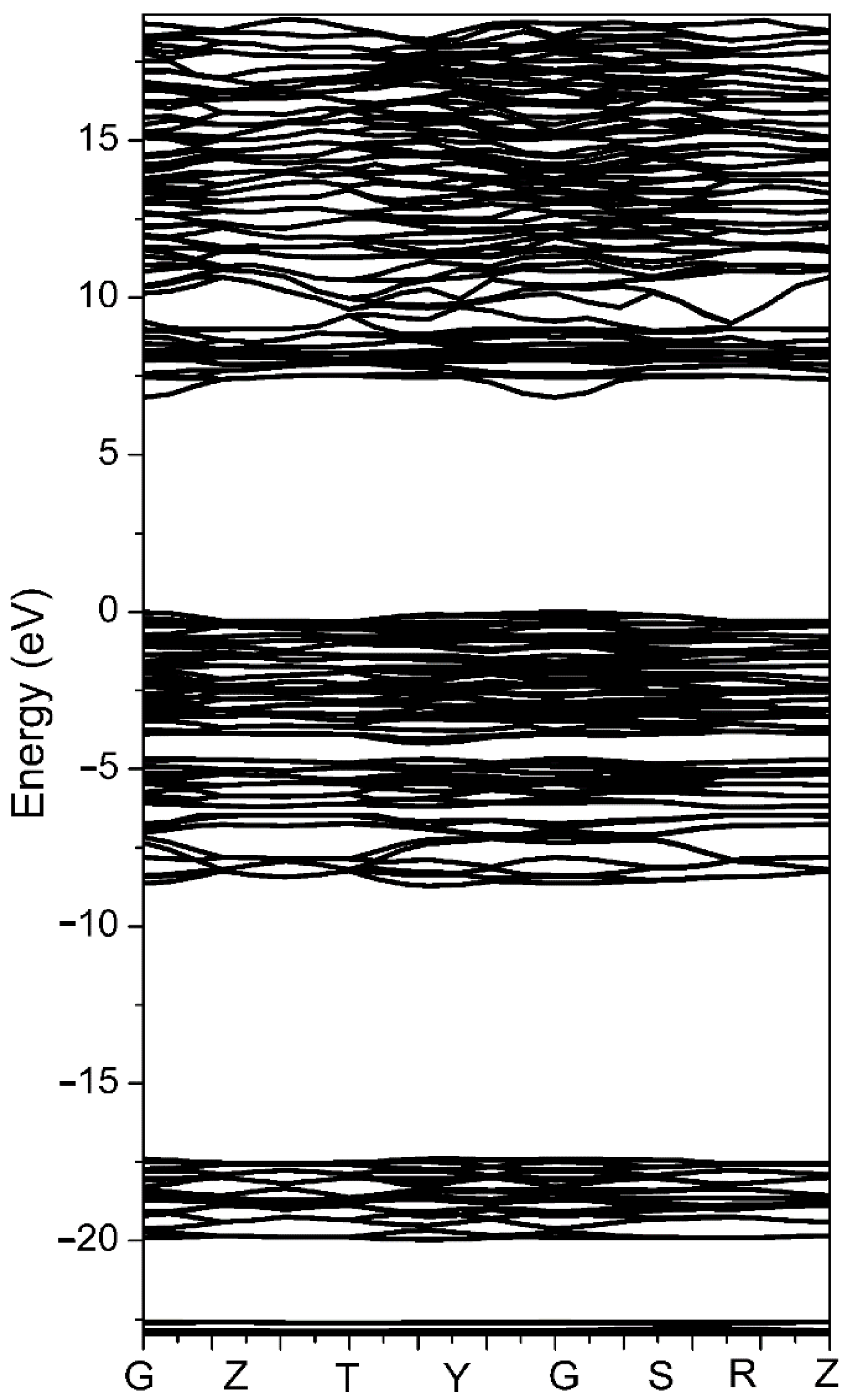
1 was fixed. The density of states (DOS) and the responding electronic structures of CBOF were calculated. The band structure is shown in
Figure 2, from which we observed that the energy gap of CBOF was approximately 6.90 eV, which was 1.2 eV lower than previously reported due to our correction of the GGA underestimate of the band gap.
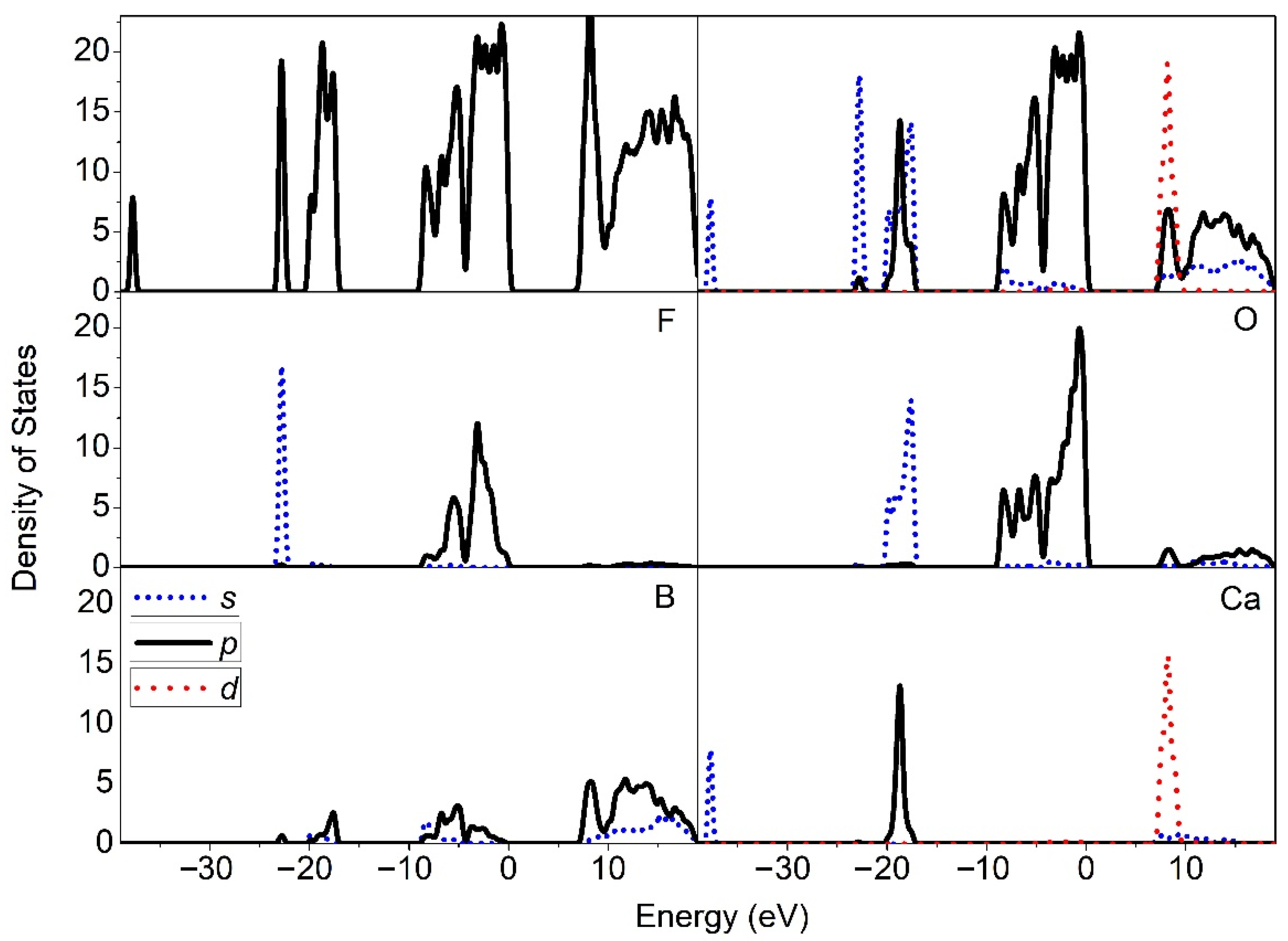
The total density of states (DOS) is shown in
Figure 3. We plotted the partial DOS of the atoms O, F, B, and Ca, respectively, in CBOF. From this, we observed that the valence band states were mainly occupied by the
p states of the O and F atoms. The lowest conduction states (c.a. 6.9 eV) above the Fermi energy E
f were dominated by the
p states of the B and
d-derived states of the Ca atoms. At the energy states approximately 20 eV below E
f, there were primarily the
p of Ca and the
s of the O character. At −24 eV in the deep valence state, the
s states with a bonding character of the F atoms were occupied.
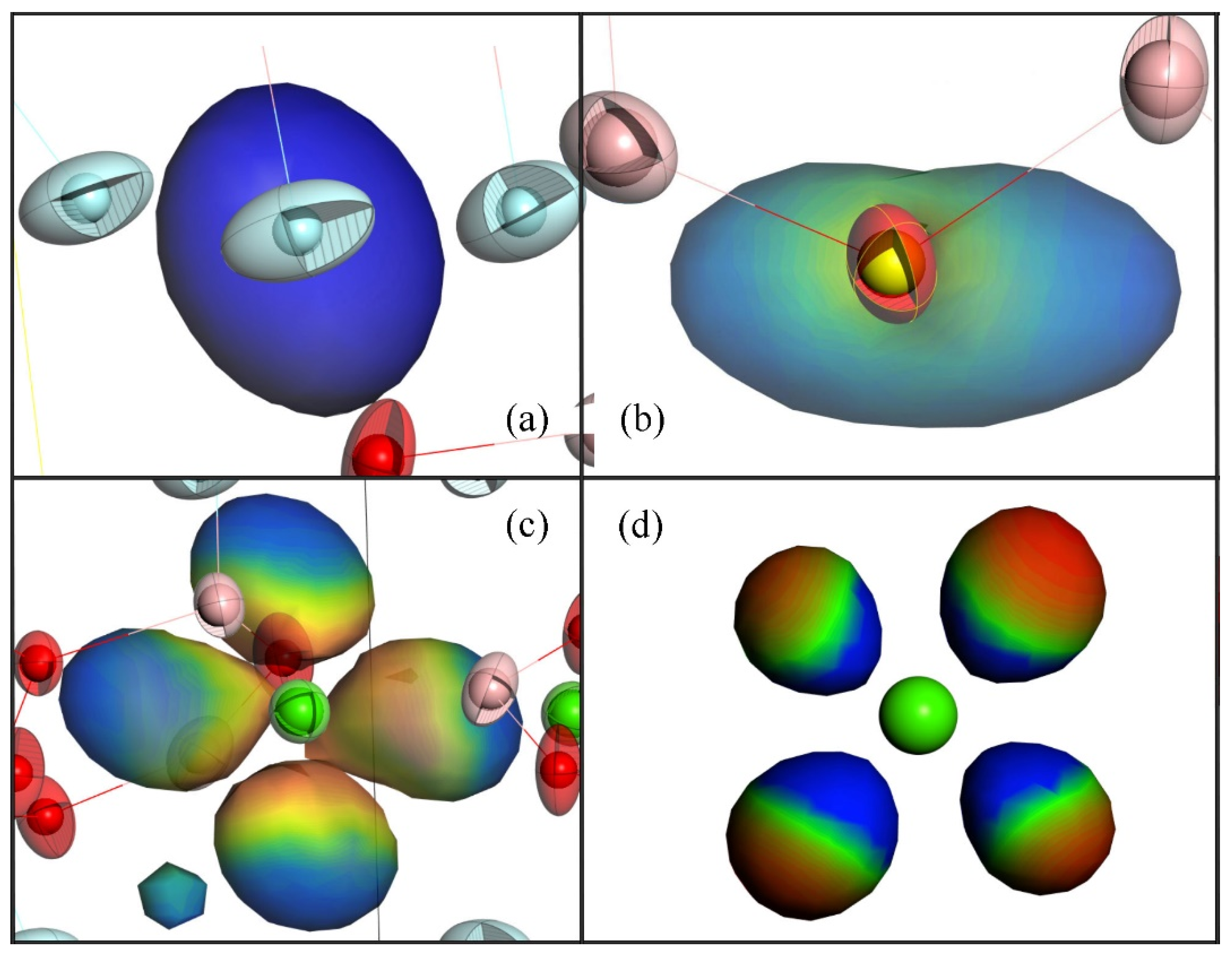
Figure 4 shows the wave function distributions of the orbitals for the energy levels at the positions −24.165, −20.763, 0, and 6.984 eV in the DOS for CBOF. It can be seen that the
s character of F occupied the energy level of −24.165 eV in the valence band state (
Figure 4a). From
Figure 4b, it can be observed that the occupied levels at the Fermi states were mainly composed of the bonding
py-derived states of the O and F atoms. The unoccupied states at 6.984 eV above the Fermi level were composed of B
p and Ca
d-derived states (
Figure 4c). At the position of 20.763 eV below the Fermi level, the σ molecular orbitals with a bonding character induced by the
px-derived states of Ca and the
s states of O atoms were primarily occupied (
Figure 4d).
From the crystal structure, we observed that O and F were tightly bound between the O–B and F–B covalent bonds in CBOF. The calculated overlap populations averaged at 0.78 and 0.45, respectively; therefore, both required more energy to be absorbed to escape from their lattice positions in CBOF. The formation energies calculated for O
v and F
v were 12.04 and 8.69 eV, respectively, and their equilibrium concentrations at a temperature of 298 K were calculated to be approximately 3.27 and 5.24 × 10
−24 mol%. For a more detailed investigation, we calculated the bond populations and the atomic charges via a Mulliken analysis [
24]. The calculated overlap and charge population for the atoms are listed in
Table 2. As there were several different BO
3 and BO
2F groups with a covalent character, the distances of B–O and B–F were not identical. We observed four different B–O
(1,2,3,4) bond lengths with 1.364, 1.367, 1.371, and 1.435 Å. The corresponding overlap populations were calculated to be 0.75, 0.78, 0.73, and 0.64, respectively. The bond lengths for two different B–F bonds were 1.436 and 1.450 Å and their overlap populations were 0.45 and 0.44, respectively. The bonding between the BO
3 and BO
2F groups with the Ca atoms was very weak. We calculated the bond length and overlap population for Ca–F to be 2.265 Å and 0.09, respectively, indicating that the ionic bonding characters were present along the crystalline
y direction due to the small charge density distribution (see
Figure 1 and
Table 2). The above-mentioned covalent and ionic bonding characters were in good agreement with the analysis results based on the Mulliken population and charge density approach.
For the defect-induced absorption properties, we calculated the electronic structures of fifteen point defects including interstitial, vacancy, antisite, Frenkel, and Schottky of the Ca, O, F, and B atoms in CBFO. Their formation energies are listed in
Table 3. The formation energies for the interstitial F (Fi) and antisite defect O
F were 0.33 and 0.83 eV and, as a result, the equilibrium concentrations at 0 K were nearly 8.29 and 6.14 × 10
−16 mol%, respectively. Considering the energetics of the system, we suggested that the point defects native in the CBOF crystal could be dominated by F
i and O
F. Based on the defective calculations, we found that the optical absorption centers could be induced by the point defect O and F vacancies (V
F, V
O) as well as interstitial O (O
i) due to the induced occupied energy levels in the band gap of the perfect CBOF. The band gaps of the system containing the point defects V
F, V
O, and O
i were reduced to 2.9, 4.1, and 1.8 eV, respectively, compared with the gap value of 6.9 eV calculated by DFT for the perfect CBOF. The results indicated that the damage threshold might be lowered by these point defects in CBOF. The laser damage threshold of CBOF might also be decreased by the defect Ca vacancy due to its increasing ionic conductivity.
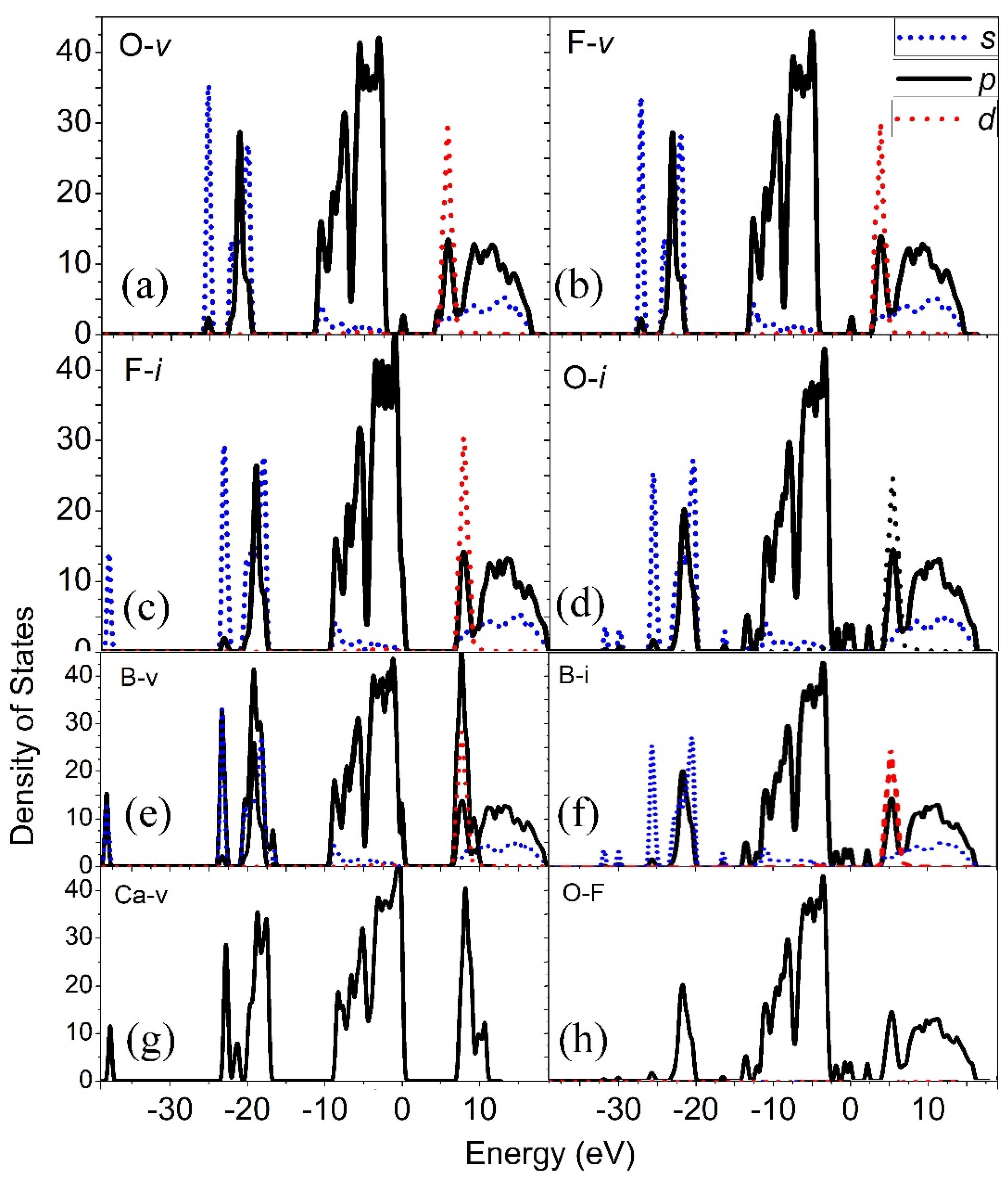
Figure 5 shows the representative of the density of states (DOS) and the local density of states (LDOS) of the fifteen point defects in CBOF. From the LDOS for the defect vacancies O (O
v) (
Figure 5a), we observed that the extra level induced by the defects in the band gap was introduced into the bottom area of the conduction band and the band gap was lowered to be 4.1 eV from the defect-free band gap 6.9 eV, indicating that a defect-induced optical absorption center might be induced by the vacancy of the O defect; therefore, it might be responsible for the deterioration of the laser damage character of the CBOF single crystals. We found that the F vacancy (V
F) also induced the defective levels in the band gap; therefore, the gap was lowered to be 2.9 eV (
Figure 5b). The induced defect states were composed of the
p orbitals of F atoms with ionic bonding states. However, no optical absorption centers were introduced in the band gap by the defect interstitial F atom (F
i) (
Figure 5c); therefore, we suggested that it had no effect on the optical absorption properties of the CBOF crystal although it had the lowest formation energy of 0.33 eV and a relatively high concentration at room temperature.
From
Figure 5d, we noted that the defect of O
i also induced optical absorption centers in the gap and lowered the band gap to 1.8 eV. Regarding the defect of interstitial O and its wave function distributions located on the positions of 0 and 1.750 eV, respectively, in the LDOS, we observed that the occupied energy levels at E
f were mainly composed of the p
y orbitals with a non-bonding character. The occupied states located at −1.750 eV were primarily composed of the σ molecular orbital with a bonding character formed by the O
px states. We suggested that the O defects might be partially removed by a thermal annealing in the O
2 atmosphere and, therefore, increase the damage threshold of the CBOF crystals.
For the B vacancies (V
B), as seen in
Figure 5e, we observed that there were no defect states in the band gap. Its formation energy was also high; up to 18.74 eV due to tight bonding to the surrounding O and F atoms. Therefore, the B vacancy had no contribution to the laser-induced damage threshold for the CBOF crystal. An interstitial B (B
i) with the formation energy of 1.73 eV induced the defect states at the location of 1.3 eV above the Fermi level (
Figure 5f), which are mainly composed of the
p-derived states of B atoms with a non-bonding character.
From
Figure 5g, we observed that there was no defect state generated in the band gap for the Ca vacancy (V
Ca) system. The formation energy of V
Ca (14.31 eV) was much higher than that of the interstitial Ca defect (Ca
i with 5.19 eV). The overlap populations for Ca–O and Ca–F were calculated to be only 0.09 and 0.04, respectively, leading to a weak bonding with the neighboring O and F atoms. A door might have been opened by the 2D channels in CBOF and, therefore, increased the possibility for Ca atoms to diffuse out of the lattice. We suggested that the defect of the vacancy Ca might also exist in a real CBOF system but with a low concentration due to its high formation energy. We calculated the full relaxation of the atoms and lattices and found that the lattices of CBOF were enlarged by a vacancy of the Ca defect to be approximately Δ
a = +0.9%, Δ
c = +0.6%, and Δ
b = +1.3%.
The cavity surrounded by the F and O atoms was enlarged by the vacancy Ca defect and the distances were averagely elongated to be approximately +1.6% from the Vca defect to the neighboring F and O atoms, resulting in a condition favorable to the fast movement of the Ca and, therefore, promoting the cavity in CBOF being filled in by extraneous impurities. Furthermore, the ionic conductivity of CBOF could be increased by increasing the Ca migration rate, resulting in a decrease of the laser damage properties for this material.
In a crystal, antisite defects occasionally exist where one atom is substituted by another type of atom but with the same lattice position. For antisite point defect O substituting F (O
F) in CBOF, the formation energy was calculated to be approximately 0.83 eV. We suggested that O
F might exist in CBOF with a relative high concentration. Based on the analysis of the LDOS of O
F and the impurity levels induced in the band gap for O
F, we derived that the impurity states in the band gap at c.a. 0.9 eV were primarily introduced from the
p-derived states of the antisite O with an unbounding character (
Figure 5h). We suggested that an O
F antisite point defect might play an important role in the deterioration of optical properties if it exists in CBOF.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}