
Theoretical Investigations of a BN Polymorph with sp2 + sp3 Hybridizations

1
Department of Mechanical and Electrical Engineering, Hetao College, Bayannur 015000, China
2
CAAC Key Laboratory of Civil Aircraft Airworthiness Technology, Civil Aviation University of China, Tianjin 300300, China
3
Research and Application Center of Automation, Hetao College, Bayannur 015000, China
*
Author to whom correspondence should be addressed.
Crystals 2021, 11(12), 1574; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121574
Submission received: 9 November 2021
/
Revised: 13 December 2021
/
Accepted: 15 December 2021
/
Published: 16 December 2021
Abstract
:The crystal structure, mechanical anisotropy, elastic properties and electronic characteristics, as well as the stability, of P4/m BN are predicted by means of density functional theory. In this work, BN in the P4/m phase demonstrates mechanical and dynamical stability. Compared with the values of bulk B, E and G in the P4/m phase, the B of BN in the P4/m phase is greater than that of dz4 BN, while the G and E of P4/m BN are greater than those of Pnc2 BN and dz4 BN. The ratio of the bulk-to-shear modulus for P4/m BN is less than 1.75 and dz4 BN, dz2 BN and lzlz2 BN, indicating that P4/m BN is more brittle than dz4 BN, dz2 BN and lzlz2 BN. P4/m BN exhibits stronger mechanical anisotropy in G and E than Pbca BN, P42/mnm BN and Pm-3m BN but much weaker mechanical anisotropy than P4/mbm BN, B7N7, B11N11 and B15N15. In addition, P4/m BN is a quasi-direct bandgap semiconductor, and the difference between the direct and the indirect bandgap is 0.008 eV. In order to obtain further characteristics of P4/m BN for future synthetic verification, the X-ray diffraction (XRD) patterns for P4/m BN are also calculated. Given its properties, P4/m BN is a good candidate for photoelectric devices.
1. Introduction
As an isoelectronic structure in carbon materials, boron nitride has many polymorphs, such as sp3 hybridizations [1,2,3,4], sp2 hybridizations [5,6,7,8] and sp2 + sp3 hybridization BN [9], similar to carbon structures having many allotropes, including sp3 hybridizations [10,11,12,13,14], sp2 hybridizations [15,16,17,18] and sp2 + sp3 hybridizations [19,20,21]. Cubic boron nitride (c-BN) is potentially a third-generation semiconductor material with properties that make it suitable for electronic devices working under extreme circumstances, as well as deep UV luminescence emitters and detectors.
BN polymorphs are attracting increasing attention, not only because they have excellent wear resistance and are super hard, such as Pbca-BN [22], c-BN, P-BN [23], O-BN [24], BC8-BN [25], etc. A completely tetrahedral-bonded boron nitride within an orthorhombic system, namely Pbca-BN (space group: Pbca), was proposed by Zhang et al. [26] and investigated by Fan et al. using first-principles calculations [22]. According to Fan et al., the B, G and hardness of Pbca-BN are 344 GPa, 316 GPa and 60.1 GPa, respectively. Therefore, Pbca-BN presents super hard characteristics and may be used in important applications in technology and the industry. A novel polymorph of boron nitride with monoclinic symmetry, m-BN, was established by Ma et al. [27]. The B, G, E and hardness of m-BN are 329 GPa, 328 GPa, 729 GPa and 56.1 GPa, respectively, the bulk modulus of m-BN is slightly smaller than that of Pbca-BN and the G is slightly greater than that of Pbca-BN. BN polymorphs have attracted increased attention from researchers, because they are essentially broad-bandgap semiconductors, and they have excellent physical properties, such as high thermal conductivity, great resistivity, high mobility, a small dielectric constant and strong breakdown electric field. They are recommended for the production of electronic equipment employed under extreme circumstances. Regarding Pnma-BN, the bandgap is 7.18 eV [28], which is larger than that of c-BN (6.1–6.4 eV [29,30]); thus, it may be the largest bandgap BN polymorph in theory. In contrast, the bandgaps of Pbca-BN, m-BN and P4/mbm BN are 5.399 eV, 4.629 eV and 4.8 eV [31], respectively. In addition to its super hard properties and wide bandgap, the BN polymorph has many other interesting properties, such as ductility in the case of P213 BN [32]. The B/G ratio of P213 BN is 2.220, which is close to that of dz4-BN [6] and larger than that of lz2-BN [6], while it is lower than that of cT8-BN [5].
In this work, we propose an all sp2 + sp3 hybridization BN polymorph, P4/m BN, with dynamical stability and mechanical stability, and the physical characteristics (including crystal structure, mechanical anisotropy, elastic properties and electronic characteristics) of P4/m BN are analyzed by means of density functional theory [33,34].
2. Materials and Methods
As in the majority of crystal structure and physical characteristics predictions, the Perdew–Burke–Ernzerhof (PBE) functional of the exchange correlation potential and the generalized gradient approximation (GGA) [35] are used in this work, and the density functional theory (DFT) calculations are performed by utilizing the ultrasoft pseudopotentials [36] under the Cambridge Sequential Total Energy Package (CASTEP) code [37]. The valence electron structures of N and B are 2s22p3 and 2s22p1, respectively. A high k-point separation, smaller than or approximately equal to 0.025 Å−1 × 2π, is used for P4/m BN, and 6 × 6 × 10 Monkhorst-Pack meshes [38] are employed for a conventional cell of P4/m BN. The Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization scheme [39] is employed for crystal geometric majorization. The plane wave cut-off energy for structural majorization is 500 eV, and this was adopted for the crystal property calculations for P4/m BN. The electronic bands were investigated utilizing the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional [40]. All the phonon spectra of P4/m BN were investigated based on the density functional perturbation theory (DFPT) method [41].
3. Results and Discussion
3.1. Crystal Structure
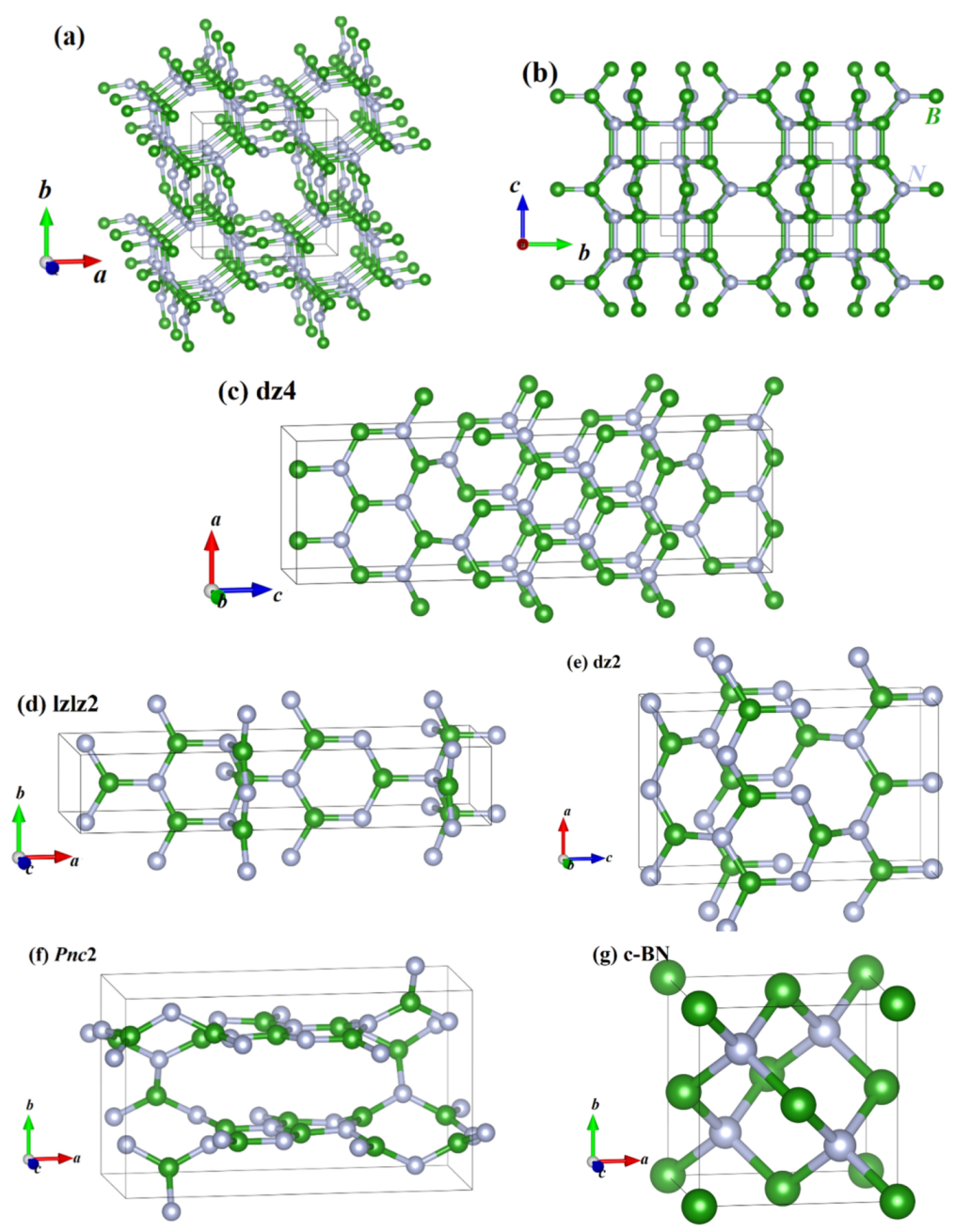
The crystal structures of the BN polymorphs are shown in Figure 1. The green and gray spheres represent B atoms and N atoms, respectively. According to Figure 1, in the crystal structure of P4/m BN, one conventional cell contains 12 N and 12 B atoms, and these N and B atoms are connected by sp2 and sp3 hybridizations, including eight nitrogen atoms and eight boron atoms connected by sp3 hybridizations and four nitrogen atoms and four boron atoms connected by sp2 hybridizations. The P4/m BN involves a 4-membered BN ring, 6-membered BN ring, 8-membered BN ring and 16-membered BN ring. These ring structures are composed of alternating connections of N atoms and B atoms, and there is only one 4-, 6- and 16-membered BN ring each, as well as two eight-membered rings. The lattice parameters of the crystal structures of P4/m BN are presented in Table 1, together with the lattice constants of other BN polymorphs. The theoretical results regarding the GGA level for c-BN are in better agreement with the experimental results [42] than those of the LDA method. Thus, the results presented later in this work are based on the GGA level. The volumes per BN unit of BN in the P4/m, Pnc2, dz4, dz2 and lzlz2 phases and c-BN are also shown in Table 1. All the BN polymorphs with sp2 + sp3 hybridizations and sp2 hybridizations are larger than the sp3 hybridizations (c-BN), so the bulk modulus of the BN polymorphs with sp2 + sp3 hybridizations and sp2 hybridizations is less than that of c-BN.
3.2. Stability
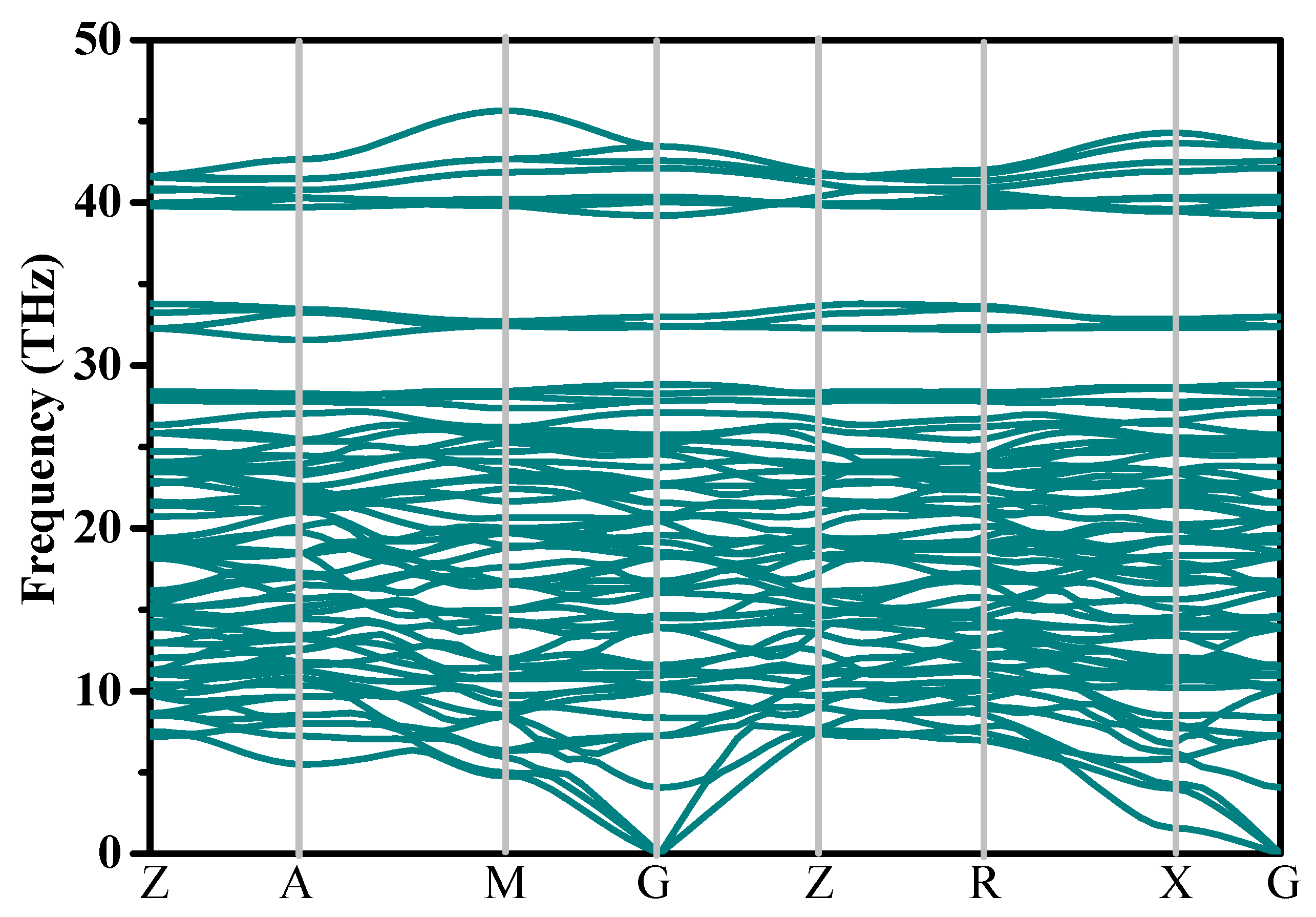
Stability is a significant physical property of new phases. In order to investigate the dynamical stability of P4/m BN, the phonon spectra of P4/m BN were generated, as presented in Figure 2. There was no hypothetical frequency observed in the entire Brillouin zone of P4/m BN, which proves that P4/m BN is dynamically stable. To study the mechanical stability of P4/m BN, the elastic constants of P4/m BN and other BN polymorphs were consulted, and these are shown in Table 2. The theoretical results of the elastic parameter Cij of c-BN are closer to the experimental results [43]; this also indicates that our prediction of the theoretical results of the elastic constants of P4/m BN is also reliable. The space group of P4/m BN is P4/m; it can be classified as having tetragonal symmetry, and the mechanical stability can be verified by [44]
The elastic constants (see Table 2) of P4/m BN satisfy the above Born criteria, thus proving that P4/m BN are mechanically stable.
3.3. Mechanical and Mechanical Anisotropy Properties
The elastic moduli and the B/G of P4/m BN are also shown in Table 2. The Voigt–Reuss-Hill approximations [45,46,47] are estimated for the values of B and G. For the tetragonal structure, BV, BR, GV and GR can be taken as [48]
For the B, G and E of P4/m BN, the following equations are used:
According to Table 2, the B of the BN polymorphs with sp2 + sp3 hybridizations and sp2 hybridizations, P4/m BN, Pnc2 BN, dz4 BN, dz2 BN and lzlz2 BN, are all smaller than that of c-BN; this is consistent with our previous conclusions in the crystal structure section. In addition, the B of P4/m BN are greater than those of dz4 BN but less than those of Pnc2 BN, dz2 BN, lzlz2 BN and c-BN. In contrast, the G and E of P4/m BN are greater than those of Pnc2 BN and dz4 BN and smaller than those of dz2 BN, lzlz2 BN and c-BN. The B/G values of the BN materials are also shown in Table 2. It is well-known that B/G < 1.75 indicates ductility; otherwise, the material is brittle [49]. According to Table 2, the values for P4/m BN, dz2 BN, dz4 BN, lzlz2 BN and c-BN are smaller than 1.75, i.e., their brittleness is confirmed; moreover, the B/G of Pnc2 BN exceeds 1.75, which means that the ductility for Pnc2 BN is verified. Additionally, the ratio of the B/G of P4/m BN indicates its greater ductility compared to the other BN polymorphs mentioned in this work.
The mechanical anisotropy of crystals is very important in the study of their physical characteristics [50]. A directional dependence can visually show the anisotropic characteristics due to the varying Young’s moduli generated in different planes. The three-dimensional (3D) surface is a regular sphere, which means that the material is isotropic; otherwise, the material shows anisotropic characteristics [8,51,52,53,54,55], and the smaller the resemblance to a sphere, the larger the anisotropy, to evaluate the mechanical anisotropy properties of E.
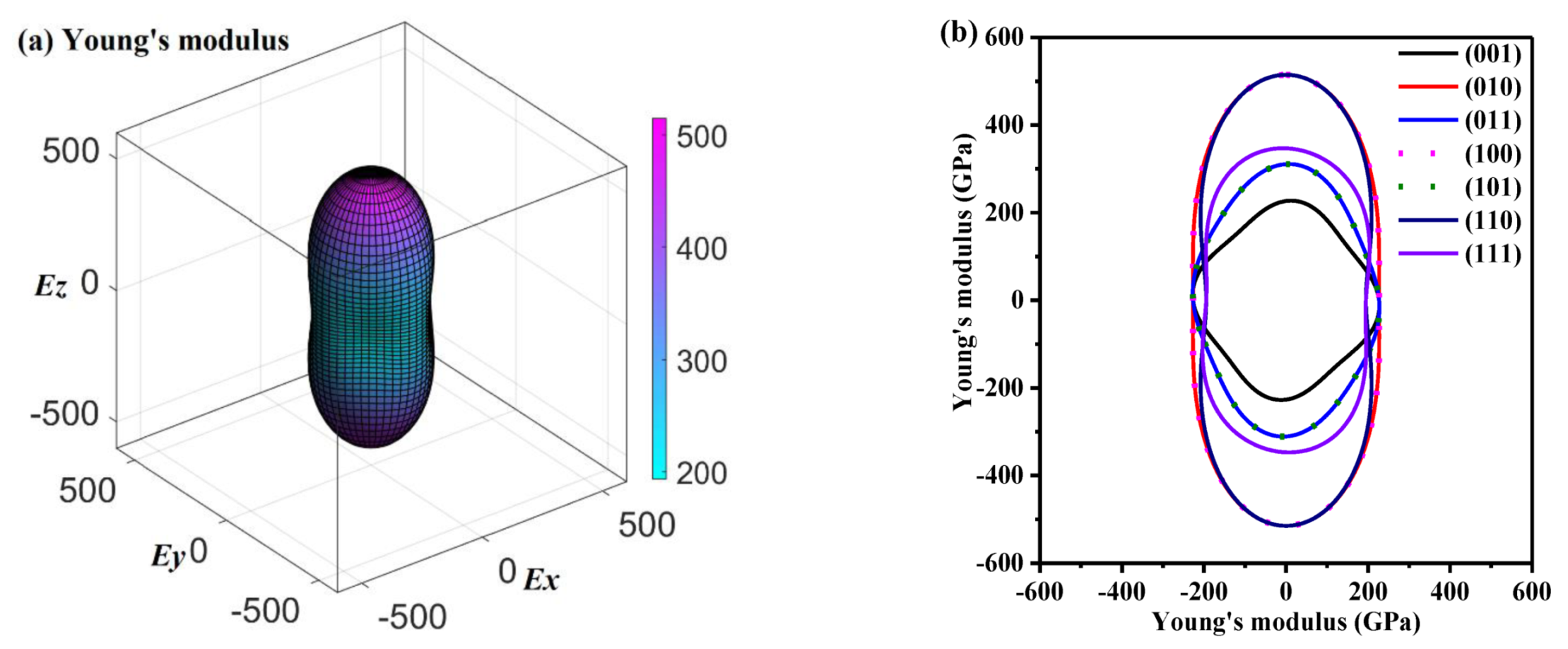
For P4/m BN in distinct directions, the variations of E of P4/m BN in different planes were plotted, as shown in Figure 3a. In Figure 3a, different colors represent different value ranges: the minimum value and maximum value of E in P4/m BN are indicated by the curved surface of the solid cyan line and the solid purple line, respectively. The ratio between the maximum value and the minimum value (Emax/Emin = 514.72/193.57 = 2.66) of E is greater than that of the Pbca phase [22], P42/mnm phase [50] and Pm-3m phase [56], while it is much weaker than that of P4/mbm BN, B7N7, B11N11 and B15N15 [57]. The anisotropy in E of P4/mbm BN, B7N7, B11N11 and B15N15 is approximately 4, 6, 8 and 24 times that of P4/m BN, respectively. Therefore, the anisotropy in E of P4/m BN is much lower than that of P4/mbm BN, B7N7, B11N11 and B15N15. The two-dimensional (2D) extreme values of E in the (001), (101), (100), (110), (010) and (111) planes for P4/m BN are presented in Figure 3b, respectively. The distribution of E in the (100) plane and (010) plane is the same, including the Emax and Emin, and the same situation also occurs in the (101) plane and (011) plane. The anisotropy in E of P4/m BN at the (001) plane (Emax/Emin = 228.00/193.57 = 1.18) is the lowest, and the anisotropy in E at the (110) plane (Emax/Emin = 514.72/194.54 = 2.65) is the strongest.
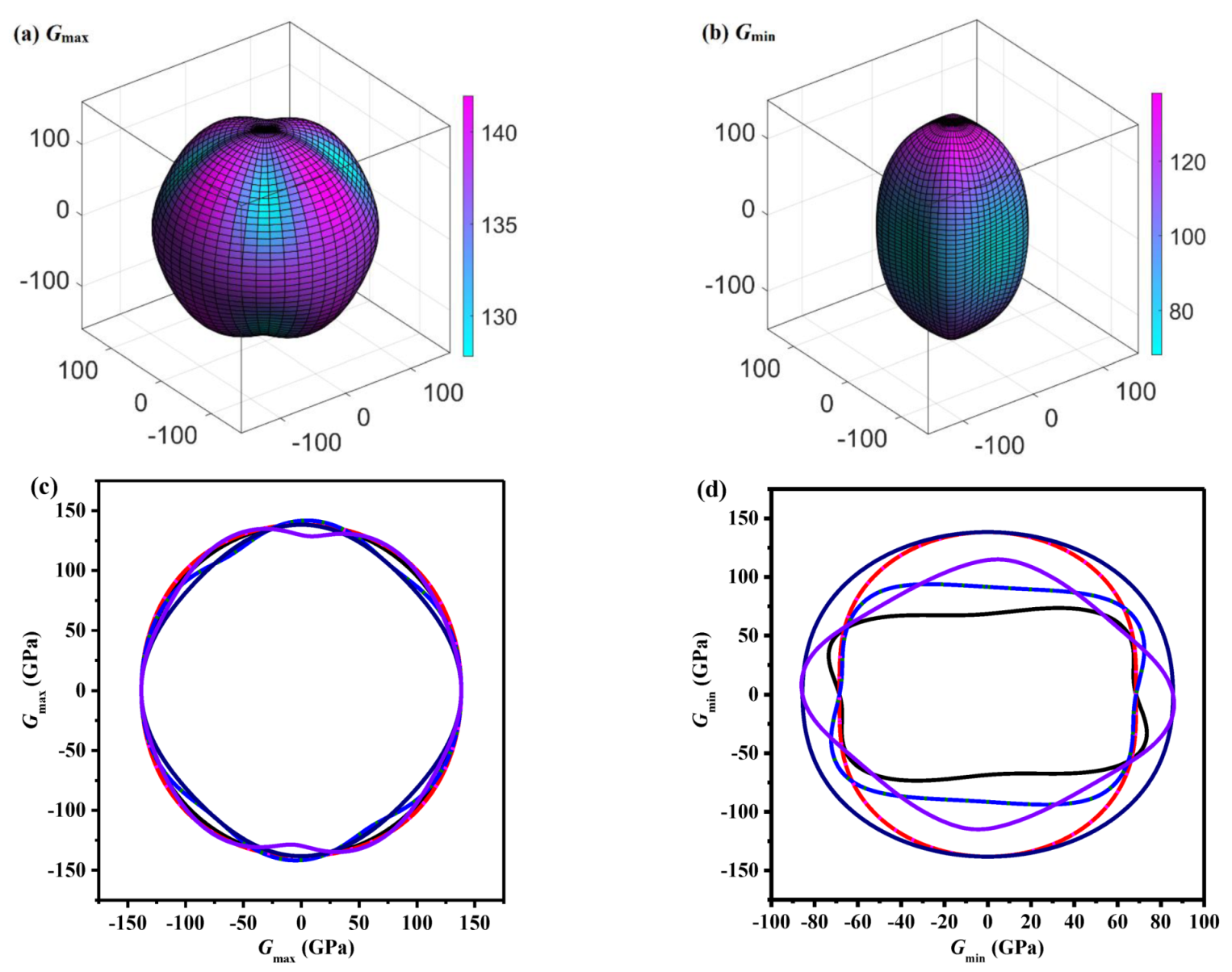
The variations of the Gmax and Gmin of P4/m BN in different directions are displayed in Figure 4a,b, which indicate the mechanical anisotropy properties of G for P4/m BN in distinct directions. The shape of the 3D-structured surface of G for P4/m BN is a very close sphere; the Gmax/Gmin for P4/m BN is 2.08, while it is larger than that for the Pbca phase [22], P42/mnm phase [50] and Pm-3m phase [56]. Regarding the Young’s modulus, the anisotropy of the shear modulus of P4/m BN is still weaker than that of P4/mbm BN, B7N7, B11N11 and B15N15. The Gmax/Gmin of P4/mbm BN, B7N7, B11N11 and B15N15 is still many times that of P4/m BN, and the anisotropy of G of P4/mbm BN, B7N7, B11N11 and B15N15 is approximately 7, 8, 14 and 44 times that of P4/m BN, respectively. The 2D representations of the shear modulus in the major planes for P4/m BN are provided in Figure 4c,d, respectively. As demonstrated in Figure 4c,d, as in the case of the Young’s modulus, the distribution of G in the (100) and (010) planes is identical, and the distribution of G in the (101) plane and (011) plane is the same. The anisotropy of G of P4/m BN in the (110) plane (Gmax/Gmin = 138.27/85.62 = 1.61) is the lowest, and the anisotropy of the shear modulus in the (101) and (011) planes (Gmax/Gmin = 141.94/68.22 = 2.08) is the greatest.
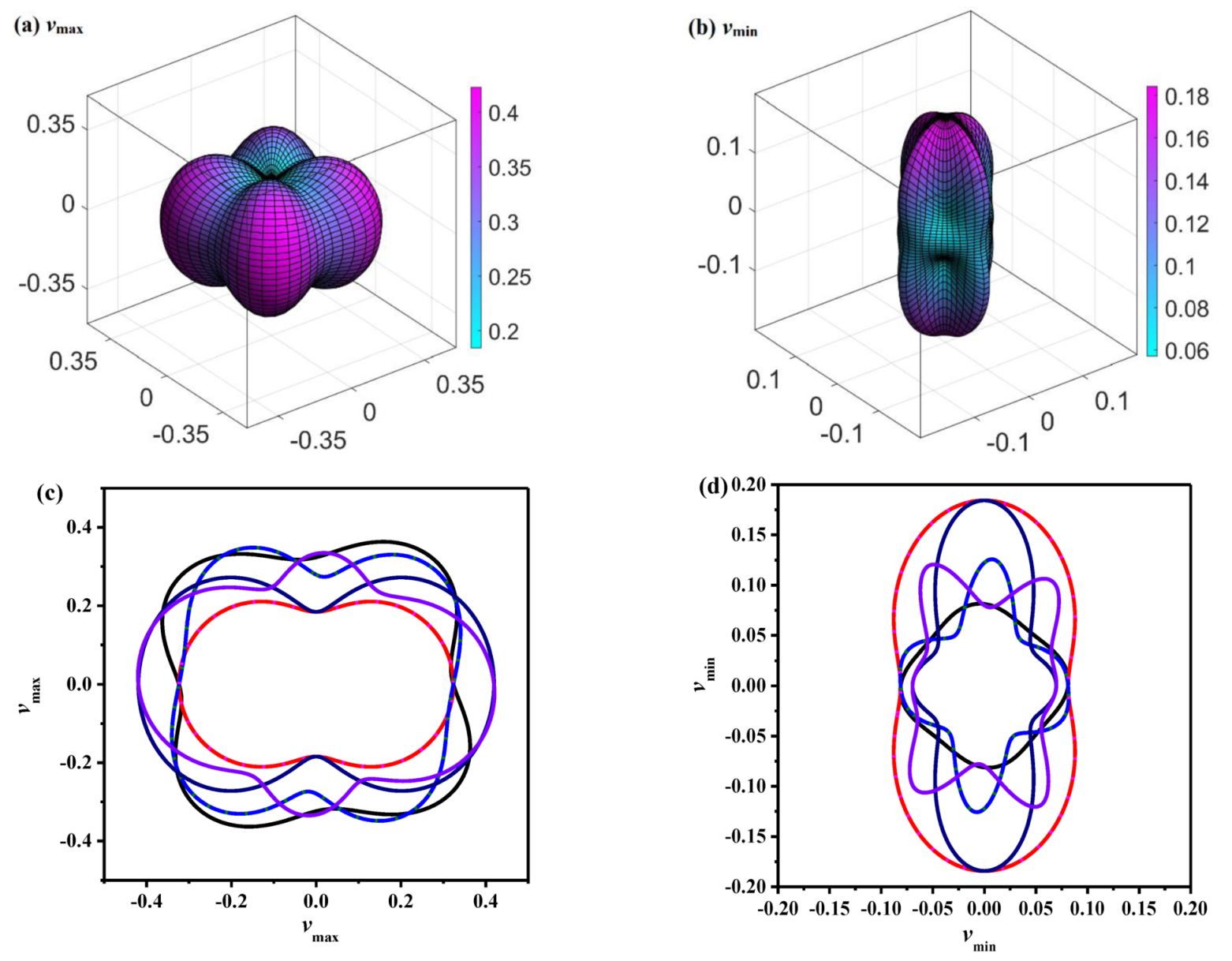
The variations of the extremum value of v for P4/m BN in different directions are demonstrated in Figure 5a,b, and the 2D representations of the Poisson’s ratio in the (001), (101), (100), (110), (010) and (111) planes for P4/m BN are displayed in Figure 5c,d, respectively. The shape of the three-dimensional (3D) contour map of the P4/m BN shear modulus deviates far from the sphere; the vmax/vmin for P4/m BN is 7.00, while it is larger than that of the Pbca phase [22], P42/mnm phase [50] and Pm-3m phase [56]. Since all these materials are BN polymorphs, the reason for the differences in their anisotropy is not the different constituent elements. The anisotropy of the Young’s modulus, Poisson’s ratio and shear modulus is due to the different stacking modes of these BN polymorphs. Different stacking modes form different element rings. The larger the element ring, the larger the number of pore structures in these BN polymorphs. The more pore structures present, the greater the variations in the Young’s modulus, shear modulus and Poisson’s ratio in different directions. According to Figure 5c,d, for E and the shear modulus, the distribution of G in the (100) plane and (010) plane is the same, and the distribution of the Young’s modulus and shear modulus in the (101) plane and (011) plane is the same. The anisotropy of the Poisson’s ratio of P4/m BN in the (110) plane and (111) plane (vmax/vmin = 0.42/0.06 = 7.00) is the largest, and the anisotropy in the v ratio in the (100) and (010) planes (vmax/vmin = 0.32/0.08 = 4.00) is the smallest.
3.4. Electronic Band Structures
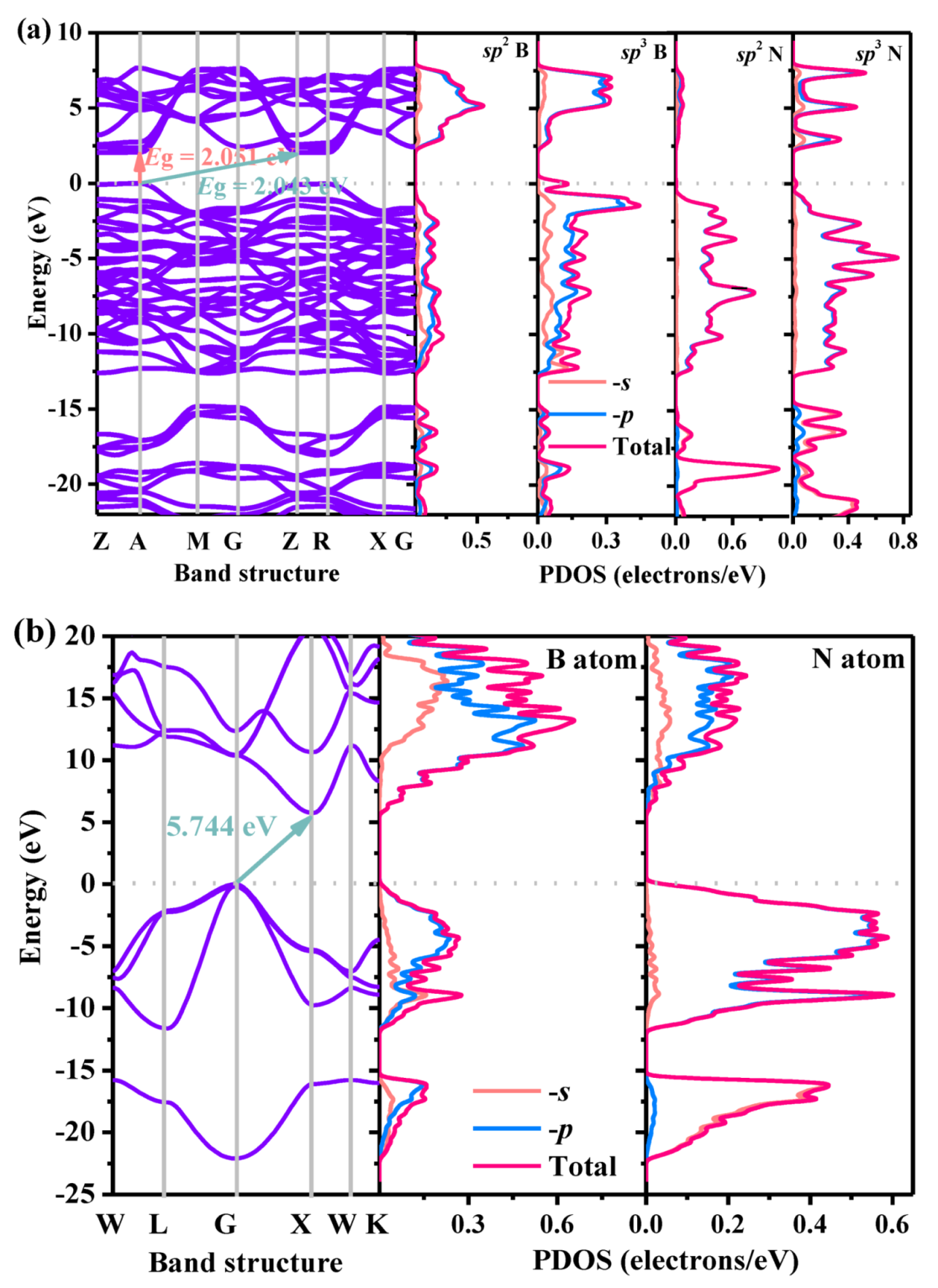
The electronic band structures and partial density of states (PDOS) using the HSE06 hybrid functional of P4/m BN and c-BN are plotted in Figure 6a,b, respectively. From Figure 6, one can observe that the P4/m BN is a semiconductor material with a quasi-direct bandgap, as it has a direct bandgap of 2.043 eV; while the direct bandgap of P4/m BN is 2.051 eV, the conduction band minimum (CBM) is located at the Z point, and the valence band maximum (VBM) is located at the A point. For other BN polymorphs, most of them are semiconductor materials and have a wide bandgap of more than 4 eV, such as P42/mnm BN (6.13 eV [50]), Pbca BN (6.81 eV [22] and 6.79 eV [3]), Pm-3m BN (5.87 eV [56]), m-BN (4.629 eV [27]), Pnma BN (7.18 eV [28]), B4N4-II (5.32 eV [3]) and B4N4-I (4.86 eV [3]). Although the bandgap of P4/m BN is smaller than that of the above BN polymorphs, it is still larger than that of P213 BN (1.826 eV [32]). For P4/m BN, there are two hybridization modes of nitrogen atoms and boron atoms: sp2 hybridization and sp3 hybridization. The PDOS of atoms with different hybridization modes were studied, and the results are shown in Figure 6a. According to the partial density of the states in Figure 6a, the contribution of 2p orbitals from B atoms with sp2 hybrids is larger than that of B atoms with sp3 hybrids in the lower energy range (−8 to −4 eV), while the contribution of 2p orbitals from B atoms with sp2 hybrids is less than that of B atoms with sp3 hybrids in the range near the Fermi level (0 to 2 eV). For N atoms, the contribution from N 2p orbitals is greater than that of B 2p orbitals from 0 to 12 eV. In the range of 18–20 eV, the contribution of 2p orbitals from N atoms with sp2 hybrids is greater than that of N atoms with sp3 hybrids, while the contribution of 2p orbitals from N atoms with sp2 hybrids is less than that of N atoms with sp3 hybrids at 15–18 eV and 20–22 eV. While, in c-BN, boron atoms and nitrogen atoms are all combined by sp3 hybridization, in the energy range of −12 eV to the Fermi level, 6–20 eV, the contributions of the N 2p orbitals and B 2p orbitals are greater than those of the B 2s orbitals and N 2s orbitals; in the energy range from −22 eV to −15 eV, the contribution of B 2p orbitals is greater than that of B 2s orbitals, while the contribution of N 2s orbitals is greater than that of N 2p orbitals.
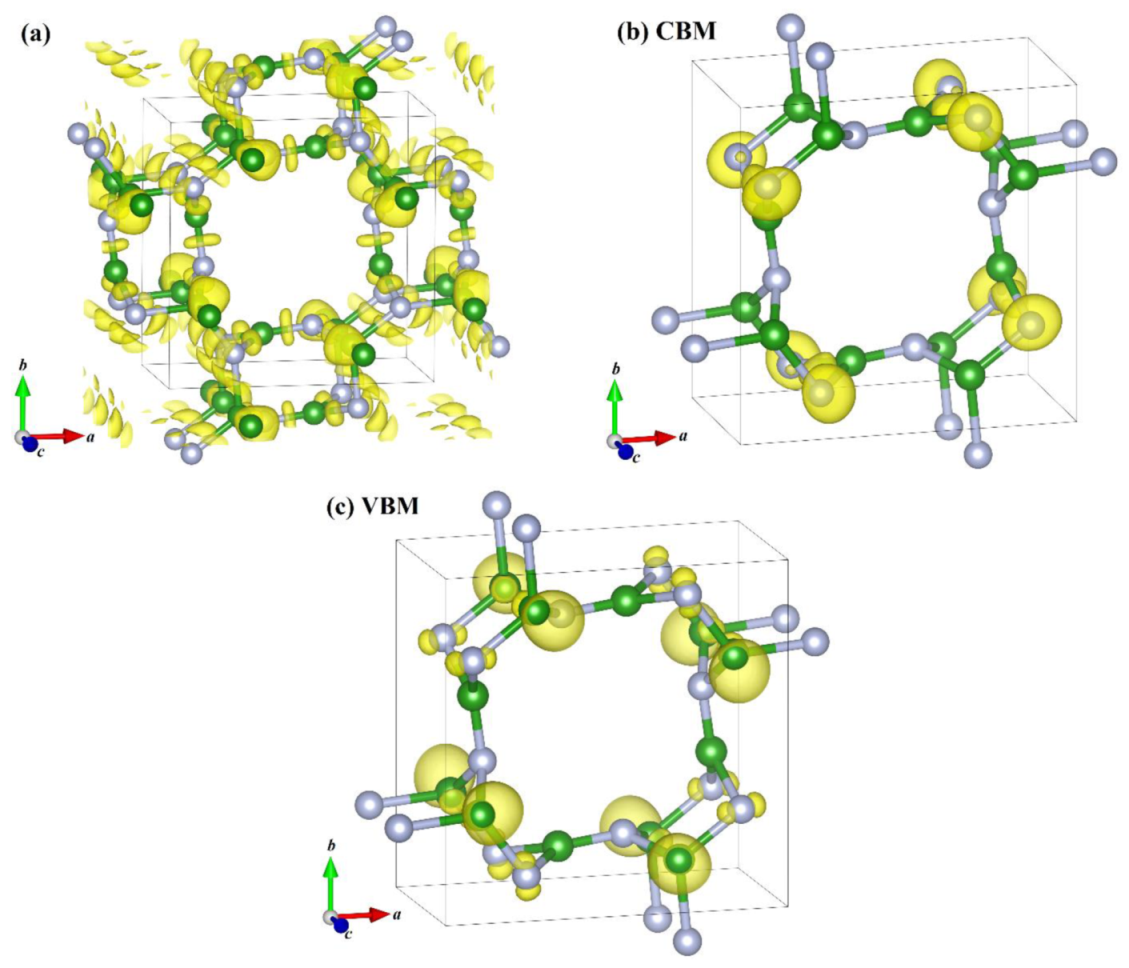
In addition, the electron localization functions (ELF) of P4/m BN and the band decomposed charge densities (BDCD) of P4/m BN are shown in Figure 7; the ELF with an isosurface level was set to 0.88, and the isosurface of the BDCD at the CBM and VBM was set to 0.005 eV/Å3, respectively. The electron localization at the center of the sp3 B–B bonds was stronger than that of the sp2 + sp2 BN bonds and stronger than that of the sp3 − sp3 BN bonds. The band decomposed charge densities (BDCD) of the CBM and VBM for P4/m BN are shown in Figure 7b,c, respectively. As shown in Figure 6 and Figure 7b, the CBM is composed mainly of sp3 N 2p orbitals and sp2 B atom 2p orbitals to a lesser extent, while the VBM is composed mainly of sp3 B 2p orbitals and, to a lesser extent, sp3 N atom 2p orbitals. Due to the quasi-direct bandgap of the P4/m BN, P4/m BN is a good candidate for photoelectric devices.
3.5. X-ray Diffraction
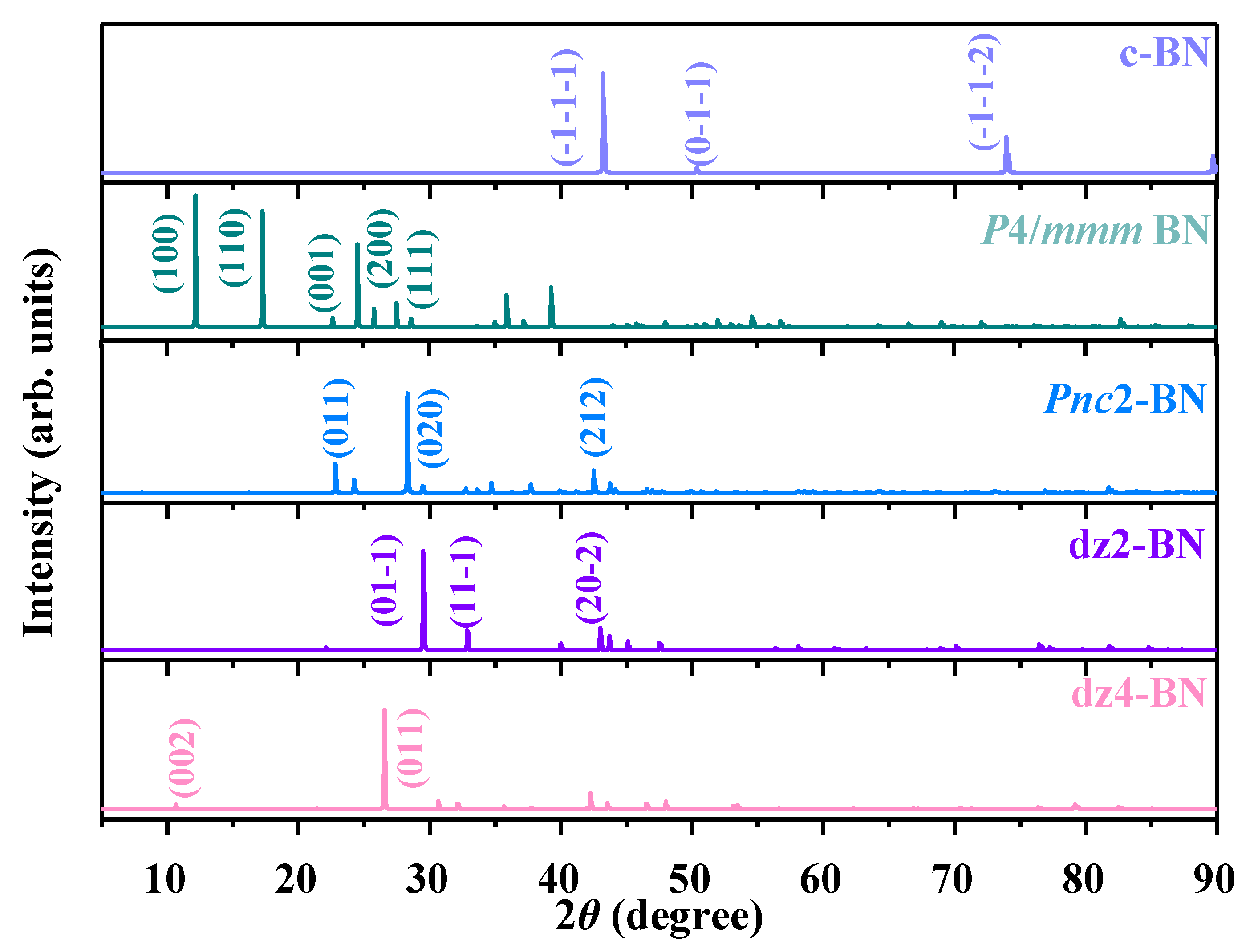
In order to obtain further characteristics of P4/m BN for future synthetic verification, the simulation of X-ray diffraction (XRD) patterns for P4/m BN and other BN polymorphs was performed, as illustrated in Figure 8. Strong peaks of c-BN appeared between 40° and 90°, while strong peaks of P4/m BN appeared between 5° and 40°, respectively. The strongest peak of c-BN was (-1-1-1), which was located at around 43.21°, while the strongest peak of P4/m BN was (100), (110) and (200) for P4/m BN. In addition to the main diffraction peaks in Figure 7, the XRD spectrum also included several other weak diffraction peaks. For the (100) peak, the diffraction angles were 12.21° and 8.06° for P4/m BN and Pnc2 BN, respectively; for the (110) peak, the diffraction angles were 17.29°, 8.06° and 32.98° for P4/m BN, Pnc2 BN and dz2 BN, respectively. For the (011) peak, the diffraction angles were 22.81°, 29.62° and 26.79° for Pnc2 BN, dz2 BN and dz4 BN, respectively. For the (111) peak, the diffraction angles were 28.62°, 24.23° and 32.34° for P4/m BN, Pnc2 BN and dz4 BN, respectively. These XRD characteristics are crucial for distinguishing the structures of P4/m BN in future experiments.
4. Conclusions
In summary, three novel boron nitride polymorphs, P4/m BN, with wide bandgap properties are proposed theoretically through first-principles calculations. The structural characteristics, dynamical stability, mechanical stability, mechanical anisotropy, mechanical properties and electronic characteristics of P4/m BN are investigated in this work. P4/m BN has the strongest brittleness compared to that of dz2 BN, lzlz2 BN and dz4 BN. The B of P4/m BN is greater than that of dz4 BN but less than that of Pnc2 BN, dz2 BN, lzlz2 BN and c-BN. The G and E of P4/m BN are larger than those of Pnc2 BN and dz4 BN but smaller than those of dz2 BN, lzlz2 BN and c-BN. The anisotropy of the shear and Young’s modulus of P4/m BN is greater than that of Pbca BN, P42/mnm BN and Pm-3m BN, and it is less than that of P4/mbm BN, B7N7, B11N11 and B15N15. The Gmax/Gmin and Emax/Emin of P4/mbm BN, B7N7, B11N11 and B15N15 are still many times that of P4/m BN. In addition, P4/m BN is a quasi-direct and wide bandgap semiconductor material. P4/m BN may play an important role in the manufacture of photoelectric devices. Finally, it is possible that the XRD characteristics will be very helpful in determining the crystal structure of P4/m BN in future experiments.
Author Contributions
Methodology, X.Y.; investigation, X.Y., R.S., B.H. and B.M.; data curation, X.Y. and R.S.; writing—original draft preparation, X.Y.; project administration, X.Y. and R.S. and funding acquisition, X.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China, grant number 61901162, the Research Project in Inner Mongolia Higher Education Institutions, grant number NJZY20222, and the Science and Technology Research Project of Hetao College, grant number HYZZ201930.
Data Availability Statement
Data are contained within the article.
Acknowledgments
Y. Liu (School of Microelectronics, Xidian University) is thanked for allowing us to use the CASTEP code in Materials Studio.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xiong, M.; Yuan, Z.; Mao, F.; Wang, X.; Jin, D.; Zhang, Q.; Yu, D.; Wang, C.; Wei, S. Superhard B28N32 with three-dimensional metallicity: First-principles prediction. Comput. Mater. Sci. 2021, 188, 110121. [Google Scholar] [CrossRef]
- Tian, Y.; Kou, C.; Lu, M.; Yan, Y.; Zhang, D.; Li, W.; Cui, X.; Zhang, S.; Zhao, M.; Gao, L. Superhard monoclinic BN allotrope in M-carbon structure. Phys. Lett. A 2020, 384, 126518. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, P.; Yan, F.; Shi, C.; Tian, Y. Physical properties of B4N4-I and B4N4-II: First-principles study. Chin. Phys. B 2019, 28, 036101. [Google Scholar] [CrossRef]
- He, C.; Sun, L.; Zhang, C.; Peng, X.; Zhang, K.; Zhong, J. Z-BN: A novel superhard boron nitride phase. Phys. Chem. Chem. Phys. 2012, 14, 10967–10971. [Google Scholar] [CrossRef]
- Niu, C.; Wang, J. Three-dimensional three-connected tetragonal BN: Ab initio calculations. Phys. Lett. A 2014, 378, 2303–2307. [Google Scholar] [CrossRef]
- Dai, J.; Wu, X.; Yang, J.; Zeng, X. Porous boron nitride with tunable pore size. J. Phys. Chem. Lett. 2014, 5, 393–398. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, W.; Huai, P. Novel 3D metallic boron nitride containing only sp2 bonds. J. Phys. D Appl. Phys. 2017, 50, 385302. [Google Scholar] [CrossRef]
- Ma, Z.; Zuo, J.; Wang, P.; Shi, C. Physical properties of Ima2-BN under pressure: First principles calculations. Chin. J. Phys. 2019, 59, 317–324. [Google Scholar] [CrossRef]
- Yu, X.; Su, R.; He, B. A novel BN Polymorph with ductile manner. J. Solid State Chem. 2022, in press. [CrossRef]
- Yang, X.; Lv, C.; Liu, S.; Zang, J.; Qin, J.; Du, M.; Yang, D.; Li, X.; Liu, B.; Shan, C. Orthorhombic C14 carbon: A novel superhard sp3 carbon allotrope. Carbon 2020, 156, 309–312. [Google Scholar] [CrossRef]
- Fan, Q.; Peng, H.; Zhang, W.; Yu, X.; Yun, S. Physical properties of group 14 elements in P2/m phase. J. Solid State Chem. 2022, 305, 122641. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, X.; Liu, S.; Gan, L. Three tetragonal superhard sp3 carbon allotropes. Solid State Commun. 2021, 323, 114095. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Wang, J.; Guan, S.; Li, Y. Dense as diamond: Pn-C10, a superhard sp3 carbon allotrope. Appl. Phys. Lett. 2021, 118, 012107. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, H.; Jiang, L.; Yu, X.; Zhang, W.; Yun, S. Two orthorhombic superhard carbon allotropes: C16 and C24. Diam. Relat. Mater. 2021, 116, 108426. [Google Scholar] [CrossRef]
- Fu, W.; Zhang, Y.; Shang, J.; Zeng, L.; Cai, Y. Lattice thermal conductivity and bandgap engineering of a three-dimensional sp2-hybridized Dirac carbon material: HS-C48. Comput. Mater. Sci. 2018, 155, 293–297. [Google Scholar] [CrossRef]
- Su, H.; Lai, Z.; Kan, E.; Zhu, X. CP-C20, a new metallic cubic carbon allotrope with an sp2 network. J. Solid State Chem. 2020, 283, 121136. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, X.; Wang, M. A prediction of a new porous metallic carbon allotrope with an sp2 hybridized network: cP-C24. Solid State Sci. 2020, 105, 106247. [Google Scholar] [CrossRef]
- Zhao, C.; Yang, Y.; Niu, C.; Wang, J.; Jia, Y. C-57 carbon: A two-dimensional metallic carbon allotrope with pentagonal and heptagonal rings. Comp. Mater. Sci. 2019, 160, 115–119. [Google Scholar] [CrossRef]
- Ram, B.; Mizuseki, H. C568: A new two-dimensional sp2-sp3 hybridized allotrope of carbon. Carbon 2020, 158, 827–835. [Google Scholar] [CrossRef]
- Liu, L.; Hu, M.; Zhao, Z.; Pan, Y.; Dong, H. Superhard conductive orthorhombic carbon polymorphs. Carbon 2020, 158, 546–552. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, H.; Jiang, L.; Zhang, W.; Song, Y.; Wei, Q.; Yu, X.; Yun, S. Three-dimensional metallic carbon allotropes with superhardness. Nanotech. Rev. 2021, 10, 1266–1276. [Google Scholar] [CrossRef]
- Fan, Q.; Wei, Q.; Yan, H.; Zhang, M.; Zhang, Z.; Zhang, J.; Zhang, D. Elastic and electronic properties of Pbca-BN: First-principles calculations. Comput. Mater Sci. 2014, 85, 80–87. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, D.; Zhao, Z.; Fu, S.; Xiong, M.; Wang, Q.; Gao, Y.; Luo, K.; He, J.; Tian, Y. First-principles study of O-BN: A sp3-bonding boron nitride allotrope. J. Appl. Phys. 2012, 112, 053518. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, J.; Ahuja, R. A novel superhard BN allotrope under cold compression of h-BN. J. Phys-Condens. Mat. 2013, 25, 122204. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhao, C.; Niu, C.; Wang, J.; Jia, Y.; Cho, J. First-principles study of the crystal structures and physical properties of H18-BN and Rh6-BN. Phys. Lett. A 2016, 380, 3891. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Lv, J.; Zhu, C.; Li, Q.; Zhang, M.; Li, Q.; Ma, Y. First-principles structural design of superhard materials. J. Chem. Phys. 2013, 138, 114101. [Google Scholar] [CrossRef]
- Ma, Z.; Zuo, J.; Tang, C.; Wang, P.; Shi, C. Physical properties of a novel phase of boron nitride and its potential applications. Mater. Chem. Phys. 2020, 252, 123245. [Google Scholar] [CrossRef]
- Ma, Z.; Han, Z.; Liu, X.; Yu, X.; Wang, D.; Tian, Y. Pnma-BN: Another boron nitride polymorph with interesting physical properties. Nanomaterials 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Hernandez, P.; Gonzales-Diaz, M.; Munoz, A. Electronic and structural properties of cubic BN and BP. Phys. Rev. B 1995, 51, 14705–14708. [Google Scholar] [CrossRef]
- Ferhat, M.; Zaoui, A.; Certier, M.; Aourag, H. Electronic structure of BN, BP and Bas—The art of scientifique computing. Phys. B 1998, 252, 229–236. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, W.; Wu, W.; Li, B. A novel BN polymorph in P4/mbm phase with a (4,4) nanotube. Phys. Status Solidi B 2021, 2100333. [Google Scholar] [CrossRef]
- Fan, Q.; Wu, N.; Chen, S.; Jiang, L.; Zhang, W.; Yun, S. P213 BN: A novel large-cell boron nitride polymorph. Commun. Theor. Phys. 2021, 73, 125701. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892R–7895R. [Google Scholar] [CrossRef]
- Clark, S.; Segall, M.; Pickard, C.; Hasnip, P.; Probert, M.I.; Refson, K.; Payne, M. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pfrommer, B.; Côté, M.; Louie, S.; Cohen, M. Relaxation of crystals with the Quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef] [Green Version]
- Petrescu, M. Boron nitride theoretical hardness compared to carbon polymorphs. Diam. Relat. Mater. 2004, 13, 1848. [Google Scholar] [CrossRef]
- Grimsditch, M.; Zouboulis, E.S.; Polian, A. Elastic constants of boron nitride. J. Appl. Phys. 1994, 76, 832. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Voigt, W. Lehrburch der Kristallphysik; Teubner, B.G., Ed.; Johnson Reprint Corp: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. J. Appl. Math. Mech. 1929, 9, 49–58. (In German) [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Phys. Soc. Lond. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Wu, Z.; Zhao, E.; Xiang, H.; Hao, X.; Liu, X.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philosop. Mag. J. Sci. Ser. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Fan, Q.; Ai, X.; Zhou, J.; Yu, X.; Zhang, W.; Yun, S. Novel III-V nitride polymorphs in the P42/mnm and Pbca phases. Materials 2020, 13, 3743. [Google Scholar] [CrossRef]
- Duan, Y.; Sun, Y.; Peng, M.; Zhou, S. Anisotropic elastic properties of the Ca–Pb compounds. J. Alloys Compd. 2014, 595, 14–21. [Google Scholar] [CrossRef]
- Fan, Q.; Hao, B.; Jiang, L.; Yu, X.; Zhang, W.; Song, Y.; Yun, S. Group 14 semiconductor alloys in the P41212 phase: A comprehensive study. Res. Phys. 2021, 25, 104254. [Google Scholar] [CrossRef]
- Qiao, L.; Jin, Z. Two B-C-O compounds: Structural, mechanical anisotropy and electronic properties under pressure. Materials 2017, 10, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Liu, H.; Yang, R.; Yu, X.; Zhang, W.; Yun, S. An orthorhombic superhard carbon allotrope: Pmma C24. J. Solid State Chem. 2021, 300, 122260. [Google Scholar] [CrossRef]
- Fan, Q.; Li, C.; Yang, R.; Yu, X.; Zhang, W.; Yun, S. Stability, mechanical, anisotropic and electronic properties of oP8 carbon: A superhard carbon allotrope in orthorhombic phase. J. Solid State Chem. 2021, 294, 121894. [Google Scholar] [CrossRef]
- Zhang, Q.; Zou, Y.; Fan, Q.; Yang, Y. Physical properties of XN (X = B, Al, Ga, In) in the Pm-3n phase: First-principles calculations. Materials 2020, 13, 1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, Q.; Kawazoe, Y.; Jena, P. Three-dimensional metallic boron nitride. J. Am. Chem. Soc. 2013, 135, 18216–18221. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The crystal construction of P4/m BN (a), and the crystal construction of P4/m BN along the a-axis (b), dz4 BN (c), lzlz2 BN (d), dz2 BN (e), Pnc2 BN (f) and c-BN (g).
Figure 1.
The crystal construction of P4/m BN (a), and the crystal construction of P4/m BN along the a-axis (b), dz4 BN (c), lzlz2 BN (d), dz2 BN (e), Pnc2 BN (f) and c-BN (g).

Figure 2.
Phonon spectra of P4/m BN.

Figure 3.
The directional dependence of the Young’s modulus (in GPa) for P4/m BN, (a) and the 2D representation of Young’s modulus (in GPa) for P4/m BN (b).
Figure 3.
The directional dependence of the Young’s modulus (in GPa) for P4/m BN, (a) and the 2D representation of Young’s modulus (in GPa) for P4/m BN (b).

Figure 4.
The directional dependence of the maximum values for the shear modulus (in GPa) for P4/m BN (a), the minimum values for the shear modulus (in GPa) for P4/m BN (b), the 2D representation of the maximum values for the shear modulus (in GPa) for P4/m BN (c) and the 2D representation of the minimum values for the shear modulus (in GPa) for P4/m BN (d).
Figure 4.
The directional dependence of the maximum values for the shear modulus (in GPa) for P4/m BN (a), the minimum values for the shear modulus (in GPa) for P4/m BN (b), the 2D representation of the maximum values for the shear modulus (in GPa) for P4/m BN (c) and the 2D representation of the minimum values for the shear modulus (in GPa) for P4/m BN (d).

Figure 5.
The directional dependence of the maximum values for Poisson’s ratio for P4/m BN (a), the minimum values for Poisson’s ratio for P4/m BN (b), the 2D representation of the maximum values for Poisson’s ratio for P4/m BN (c) and the 2D representation of the minimum values for Poisson’s ratio for P4/m BN (d).
Figure 5.
The directional dependence of the maximum values for Poisson’s ratio for P4/m BN (a), the minimum values for Poisson’s ratio for P4/m BN (b), the 2D representation of the maximum values for Poisson’s ratio for P4/m BN (c) and the 2D representation of the minimum values for Poisson’s ratio for P4/m BN (d).

Figure 6.
Electronic band structure and partial density of state for P4/m BN (a) and c-BN (b).

Figure 7.
Electron localization functions with an isosurface level set to 0.88 (a), and band decomposed charge density (BDCD) for CBM (b) and VBM (c) of P4/m BN, the isosurface of the BDCD at the CBM and VBM was set to 0.005 eV/Å3.
Figure 7.
Electron localization functions with an isosurface level set to 0.88 (a), and band decomposed charge density (BDCD) for CBM (b) and VBM (c) of P4/m BN, the isosurface of the BDCD at the CBM and VBM was set to 0.005 eV/Å3.

Figure 8.
Simulated XRD patterns of P4/m BN using an X-ray wavelength (1.5406 Å) with a copper source.
Figure 8.
Simulated XRD patterns of P4/m BN using an X-ray wavelength (1.5406 Å) with a copper source.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Lattice constants (Å) and volumes per BN unit (Å3) in the P4/m, Pnc2, dz4, dz2 and lzlz2 phases and c-BN.
Table 1.
Lattice constants (Å) and volumes per BN unit (Å3) in the P4/m, Pnc2, dz4, dz2 and lzlz2 phases and c-BN.
| Materials | Methods | a | b | c | β | V | ρ |
|---|---|---|---|---|---|---|---|
| P4/m | GGA | 7.2455 | 3.9257 | 17.1740 | 2.3995 | ||
| LDA | 7.1143 | 3.8736 | 16.3376 | 2.5334 | |||
| Pnc2 | GGA 1 | 10.9536 | 6.3020 | 4.9544 | 17.1001 | 2.4099 | |
| LDA 1 | 10.8877 | 6.0603 | 4.9076 | 16.1899 | 2.5454 | ||
| dz4 | GGA | 4.9850 | 3.4268 | 16.6081 | 17.7321 | 2.3240 | |
| dz2 | GGA | 4.9349 | 3.2640 | 8.0329 | 16.1736 | 2.5480 | |
| lzlz2 | GGA | 13.0946 | 2.5104 | 4.3953 | 89.3 | 18.0596 | 2.2819 |
| c-BN | GGA | 3.6217 | 11.8762 | 3.4699 | |||
| LDA | 3.5779 | 11.4505 | 3.5989 | ||||
| Experimental 2 | 3.6200 | 11.8595 | 3.4748 |
Table 2.
The elastic parameters (GPa) and E (Young’s modulus), B (bulk modulus) and G (shear modulus) (GPa) of BN polymorphs with the GGA functional.
Table 2.
The elastic parameters (GPa) and E (Young’s modulus), B (bulk modulus) and G (shear modulus) (GPa) of BN polymorphs with the GGA functional.
| Space Group | C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 | C16 | C26 | B | G | E | B/G |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P4/m | 261 | 261 | 539 | 138 | 138 | 69 | 90 | 65 | 65 | 3 | −3 | 160 | 117 | 282 | 1.368 |
| Pnc2 | 659 1 | 122 | 661 | 36 | 136 | 56 | 117 | 149 | 68 | 176 | 93 | 237 | 1.892 | ||
| dz4 | 810 | 60 | 672 | 20 | 261 | 28 | 26 | 197 | 79 | 148 | 92 | 229 | 1.609 | ||
| dz2 | 786 | 107 | 629 | 86 | 290 | 72 | 64 | 229 | 131 | 183 | 127 | 309 | 1.441 | ||
| lzlz2 | 548 | 835 | 335 | 132 | 32 | 214 | 97 | 47 | 51 | 207 | 133 | 329 | 1.556 | ||
| c-BN | 779 | 447 | 165 | 370 | 384 | 856 | 0.964 | ||||||||
| 820 2 | 480 | 190 | 400 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yu, X.; Su, R.; He, B.; Ma, B. Theoretical Investigations of a BN Polymorph with sp2 + sp3 Hybridizations. Crystals 2021, 11, 1574. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121574
AMA Style
Yu X, Su R, He B, Ma B. Theoretical Investigations of a BN Polymorph with sp2 + sp3 Hybridizations. Crystals. 2021; 11(12):1574. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121574
Chicago/Turabian StyleYu, Xinhai, Riguge Su, Bei He, and Binchang Ma. 2021. "Theoretical Investigations of a BN Polymorph with sp2 + sp3 Hybridizations" Crystals 11, no. 12: 1574. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11121574
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.