
Synthesis, Cytotoxic Activity, Crystal Structure, DFT Studies and Molecular Docking of 3-Amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
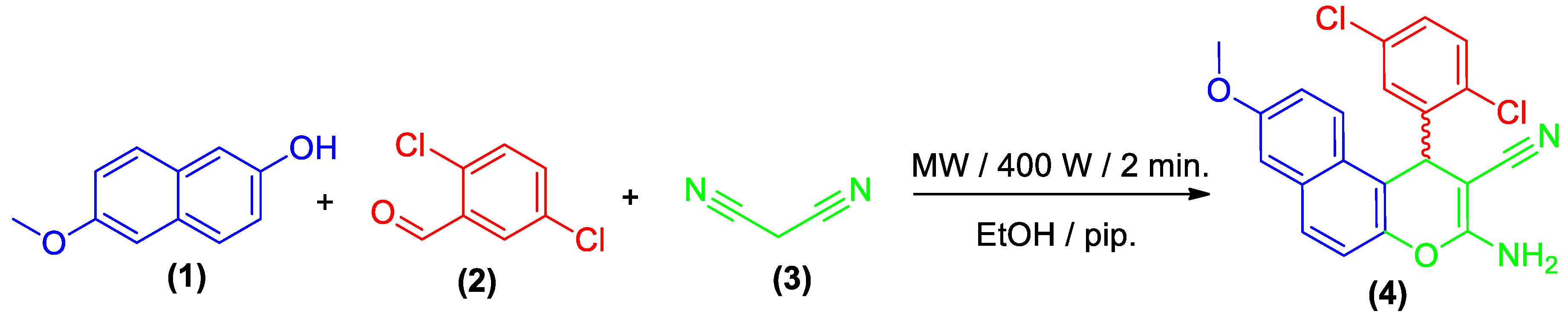
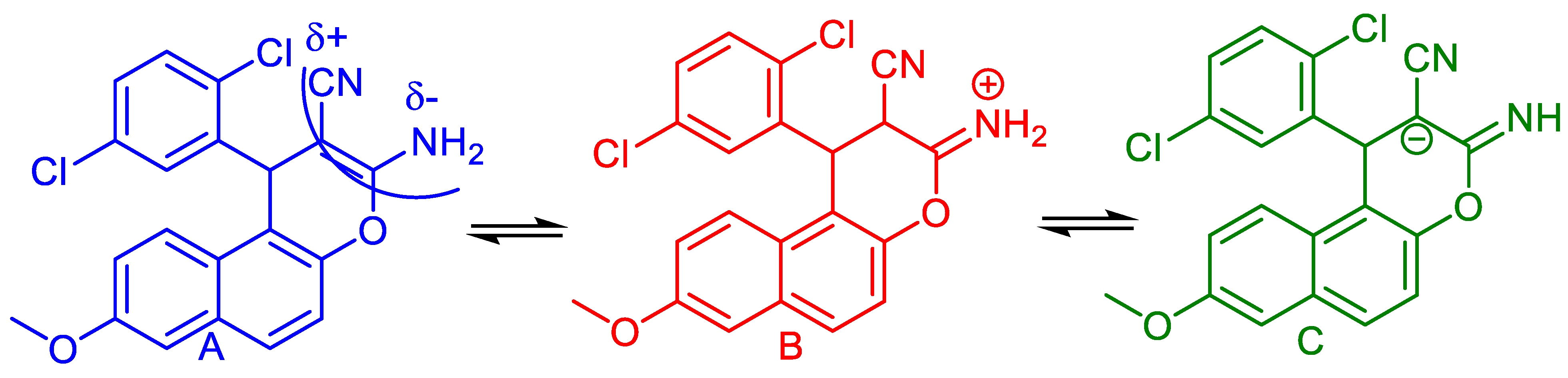
2.1. Chemistry
2.2. Spectroscopic Data
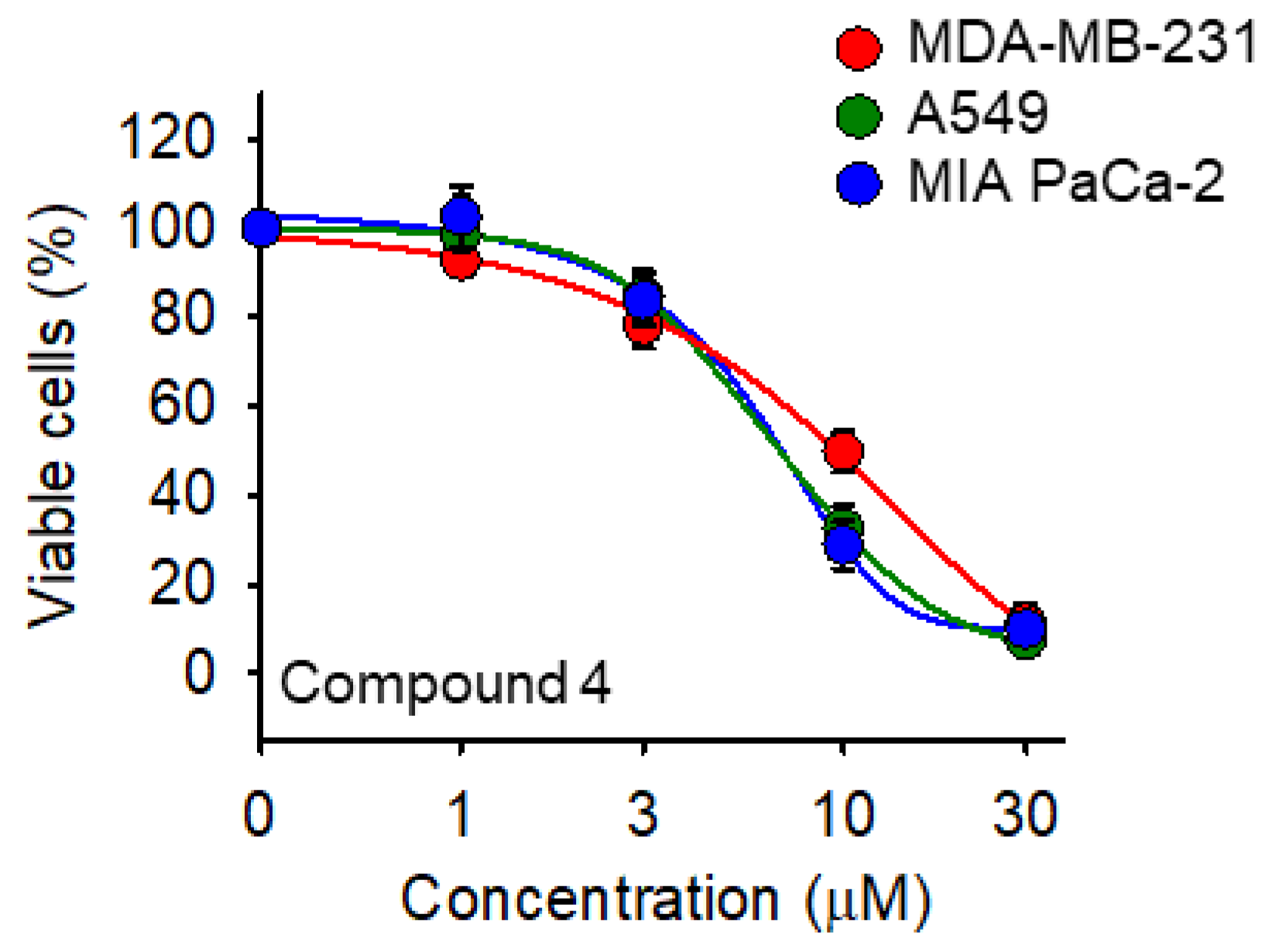
2.3. Cell Viability Assay
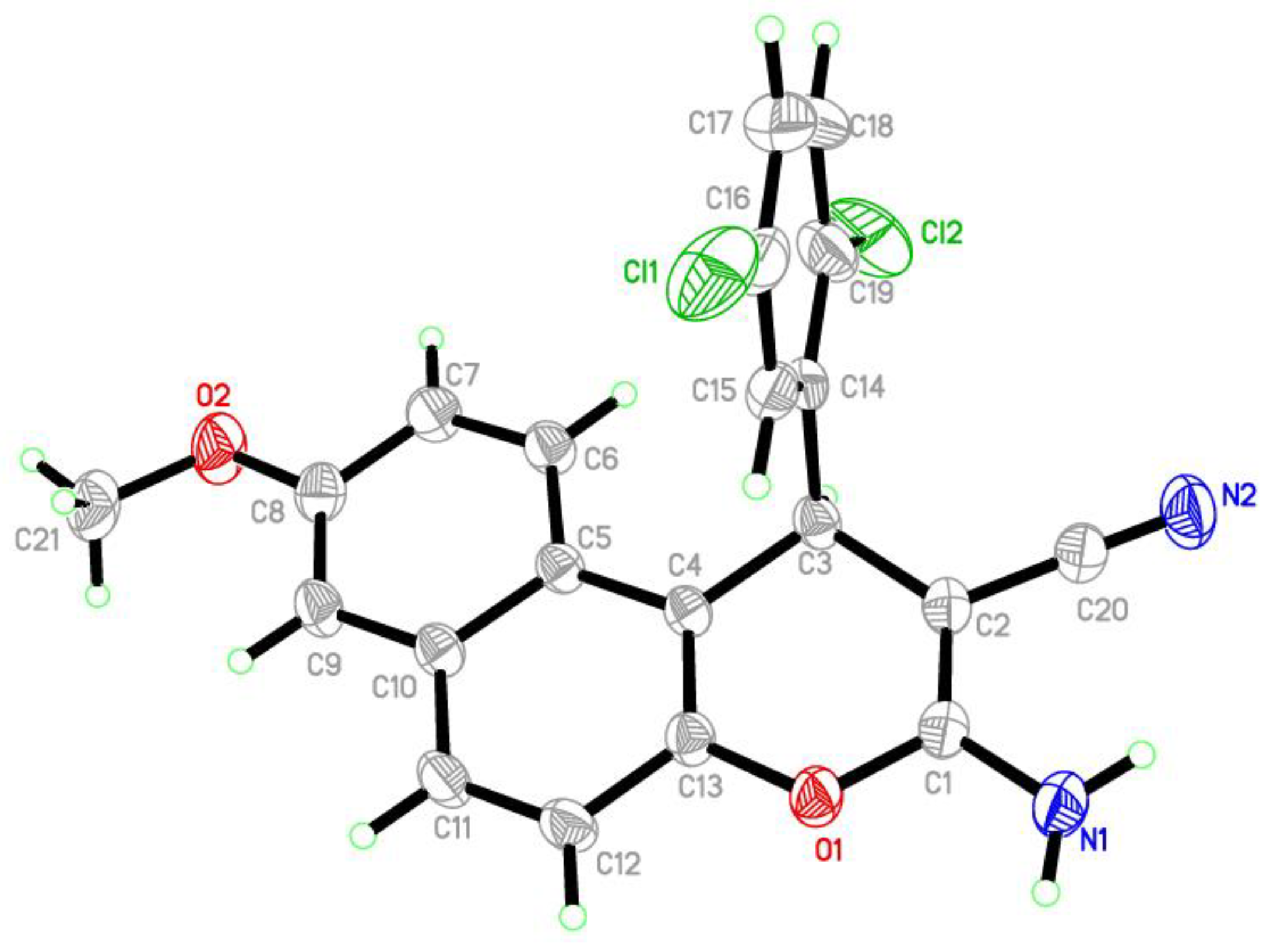
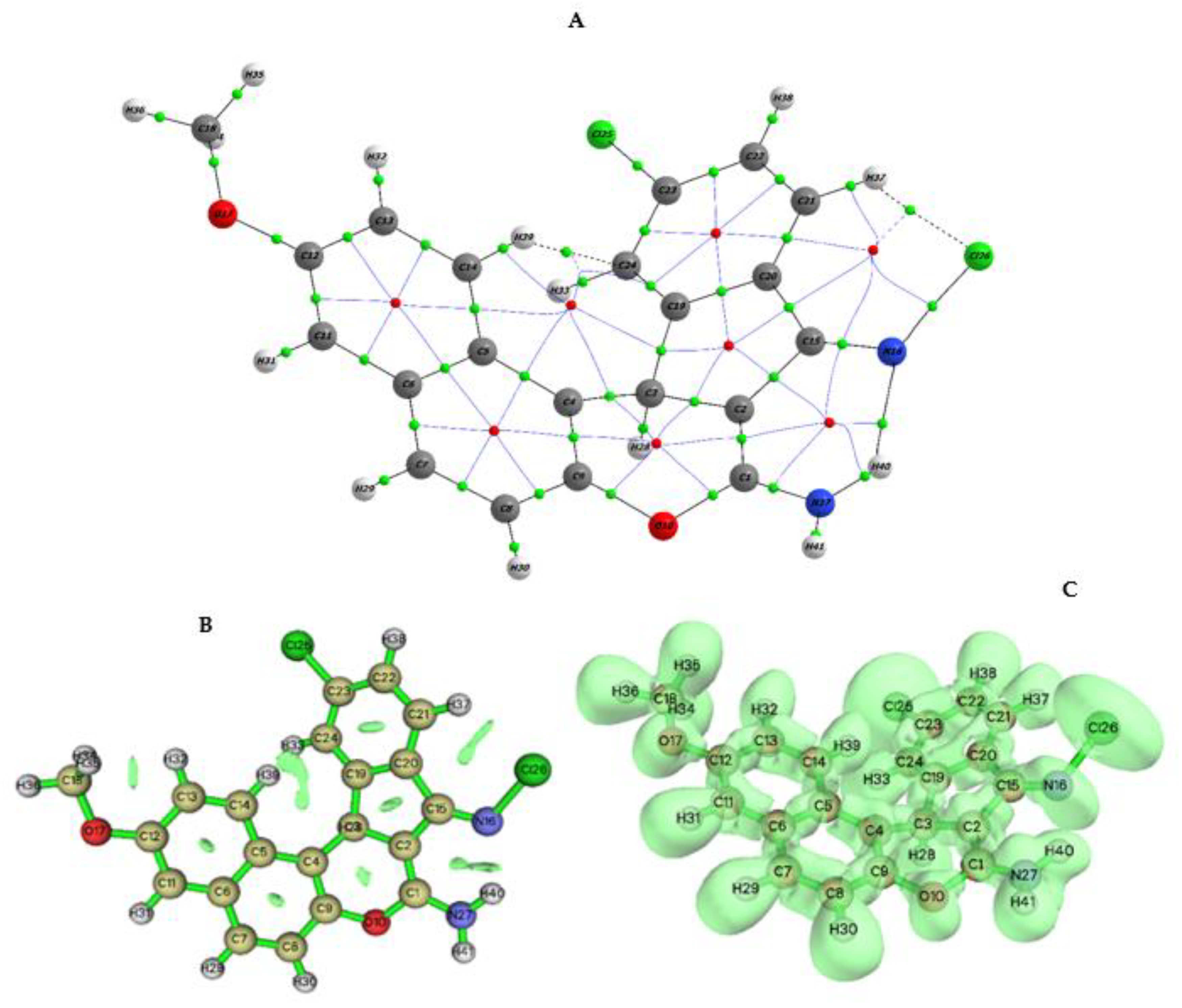
2.4. Crystal Data
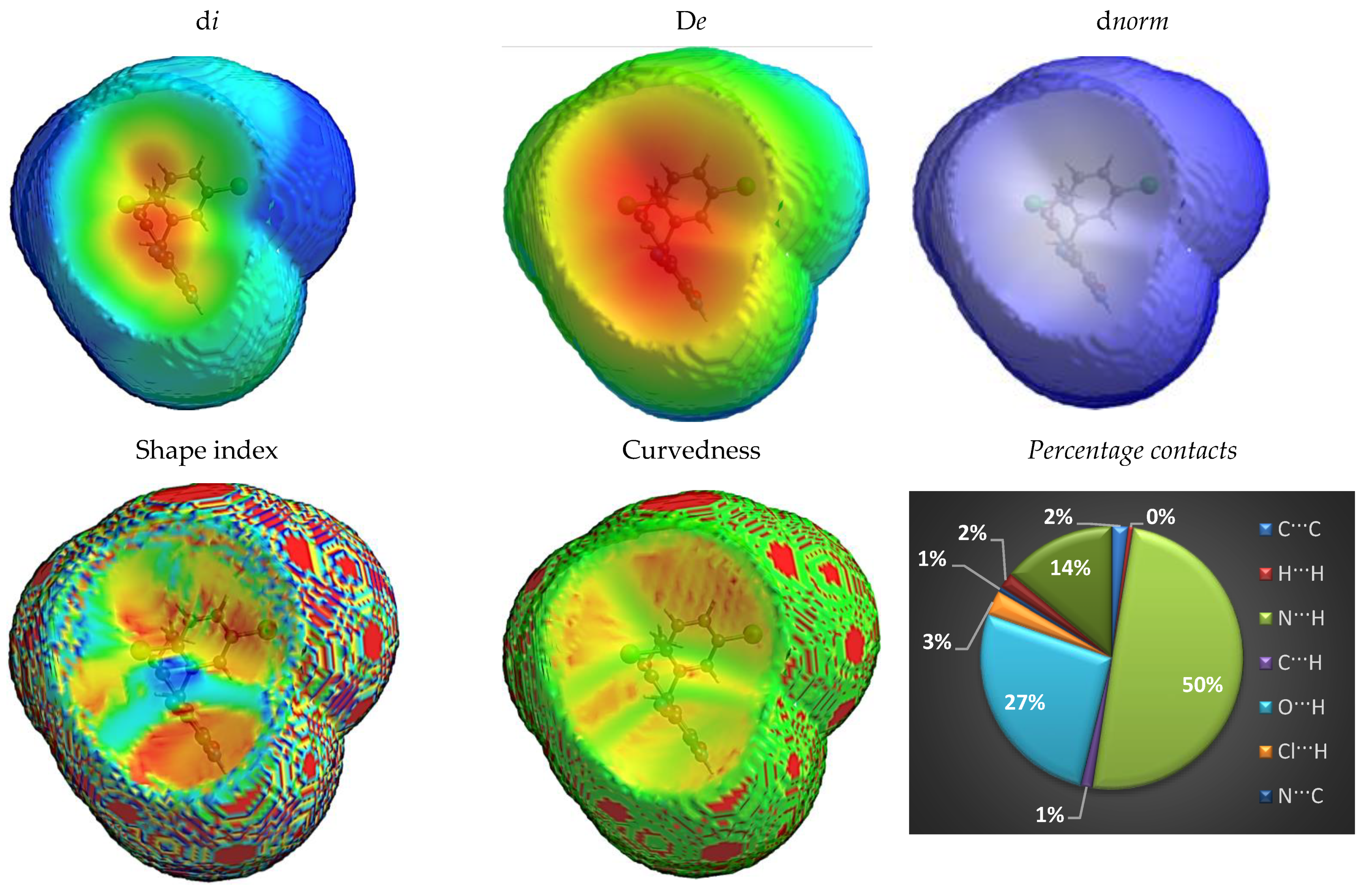
Fingerprint Plots for the Hirshfeld Surface
2.5. Molecular DFT Studies
2.5.1. Geometry Optimization
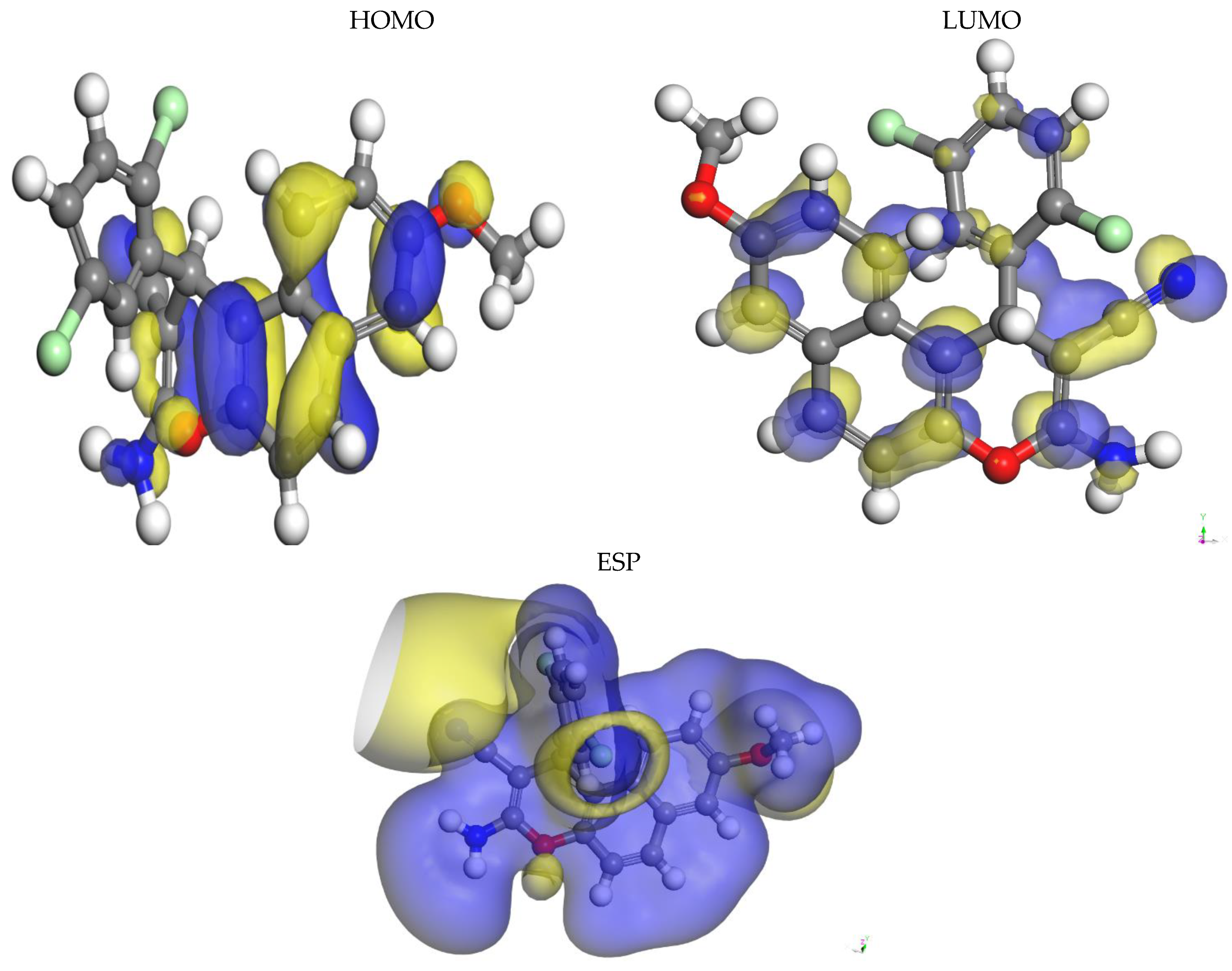
2.5.2. Analysis of Frontier Molecular Orbital
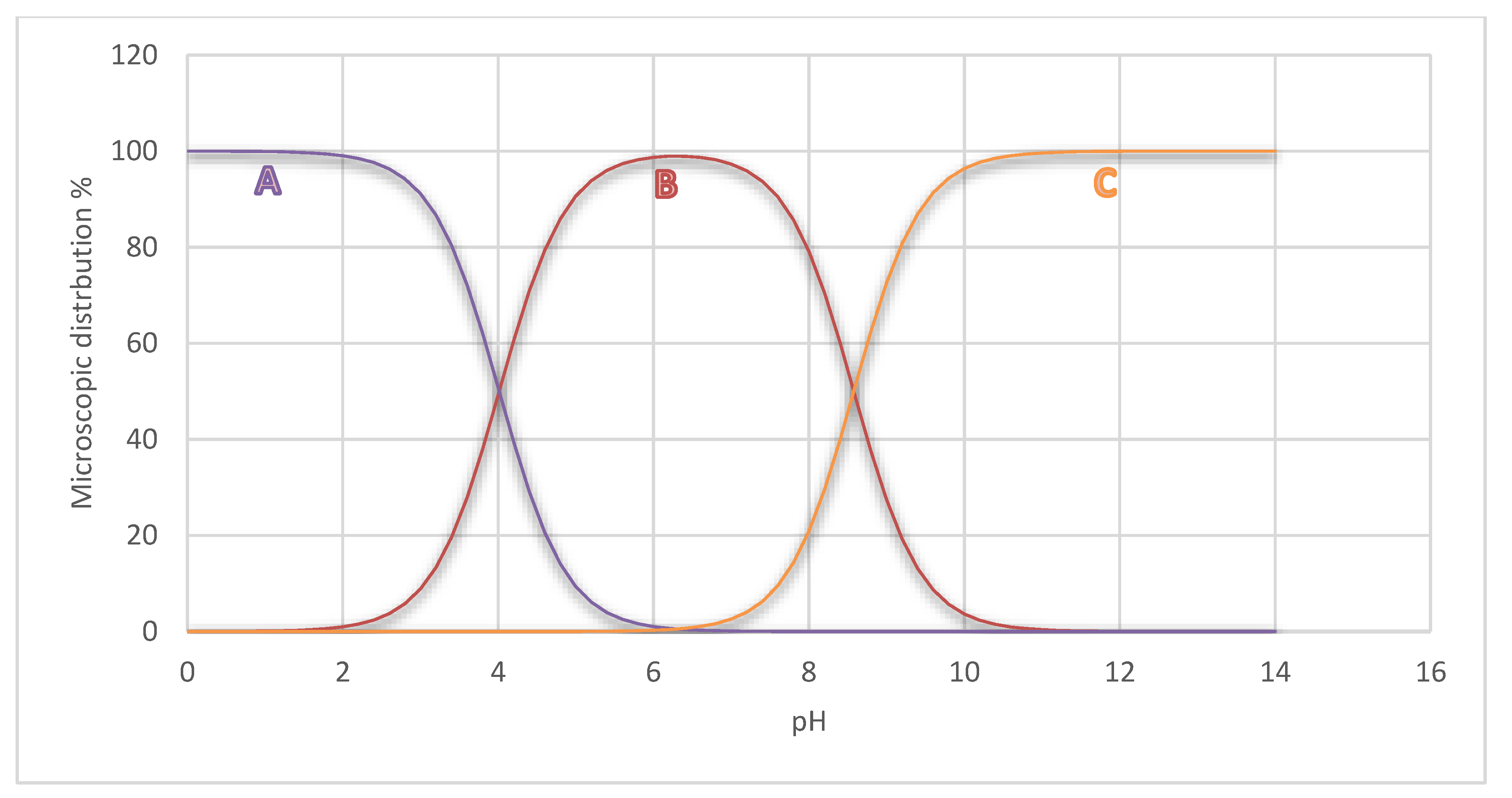
2.5.3. Chemical Reactivity
2.5.4. Analysis of the MEP Surface
2.5.5. QTAIM, NCI and ELF Approaches
2.5.6. NMR Spectra
2.5.7. ADMET and Bioactivity Profile in Silico
2.6. Pharmacokinetic-Descriptors (Table S2, Figure S5; Supplementary Materials)
Bioactivity and Toxicity Risk Prediction
- No observed carcinogenic effect with ranged values for the synthesized compound 4 (~0.8–0.9 mg/kg bw/d).
- The positive result against Caco-2 indicates a good permeability for the tested compound 4 (Caco-2 is a human colon epithelial cancer cell line, which is applied as a model for human intestinal absorption for medication).
- Compound 4 exhibits no acute oral toxicity observed with low acute lethal dose values for rat (LD50 = ~2.8–2.9 mol/kg).
- A positivity acceptance with high permeability value through the BBB (blood brain barrier) indicates that compound 4 may be effective in treating tuberculosis meningitis [72].
- It has no mutagenic effect on the DNA of microorganisms when employing the Ames test (a wide examination of mutagenic effects for bioactive molecules against DNA of microorganisms [73].
- Compound 4 does not exhibit skin sensitivity (utilizing 254 compounds able to induce skin sensitization) and hepatotoxicity effects (using 531 compounds that have a liver-associated side effect in humans).
- This compound was not biodegradable when compared with 1604 compounds [74].
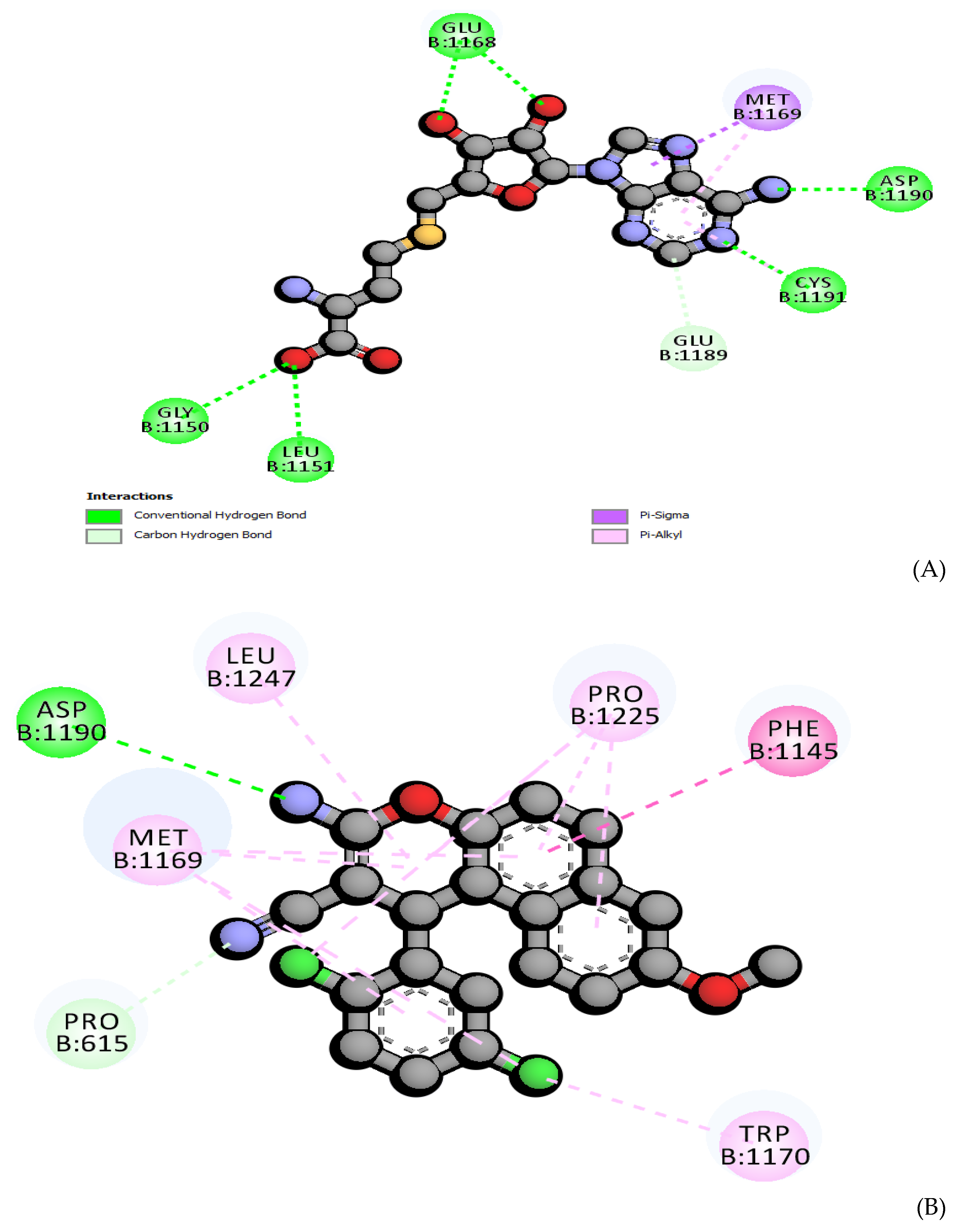
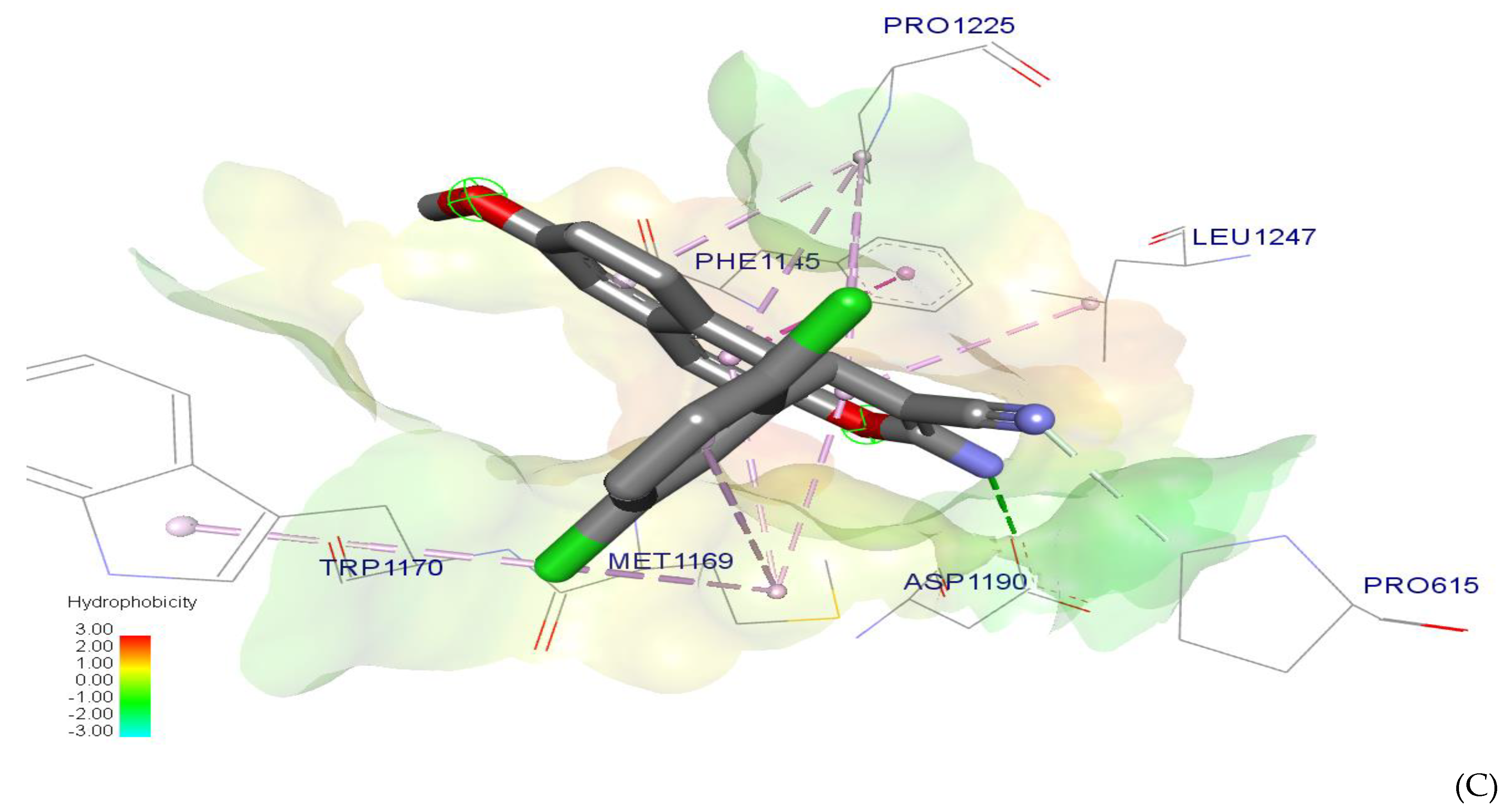
2.7. Molecular Docking
Efficiency of Ligand based on Binding Energy
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 3-Amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile (4)
3.3. Cytotoxic Activity
3.3.1. Cell Culture
3.3.2. Cytotoxicity Evaluation Using Cell Viability Assay
3.4. X-ray Crystallography Analysis
3.5. Computational Model
3.6. Protein Preparation
3.7. Molecular Docking
3.8. ADMET Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Alenazy, R.; Gu, X.; Polyak, S.W.; Zhang, P.; Sykes, M.J.; Zhang, N.; Venter, H.; Ma, S. Design and structural optimization of novel 2H-benzo[h]chromene derivatives that target AcrB and reverse bacterial multidrug resistance. Eur. J. Med. Chem. 2020, 113049. [Google Scholar] [CrossRef]
- Radwan, H.A.M.; El-Mawgoud, H.K.A.; El-Mariah, F.; El-Agrody, A.M.; Amr, A.E.; Al-Omar, M.A.; Ghabbour, H.A. Single-Crystal Structure and Antimicrobial Activity of Ethyl 3-Amino-1-(4-chlorophenyl)-9-hydroxy-1H-benzo[f] chromene-2- carboxylate Combined with Ethyl α-Cyano-4-chlorocinnamate. Russ. J. Gen. Chem. 2020, 90, 299–304. [Google Scholar] [CrossRef]
- Fouda, A.M.; Hassan, A.H.; Eliwa, E.M.; Ahmed, H.E.A.; Al-Dies, A.A.M.; Omar, A.M.; Nassar, H.S.; Halawa, A.H.; Aljuhani, N.; El-Agrody, A.M. Targeted potent antimicrobial benzochromene-based analogues: Synthesis, computational studies, and inhibitory effect against 14α-Demethylase and DNA Gyrase. Bioorg. Chem. 2020, 105, 104387. [Google Scholar] [CrossRef]
- Abd El-Mawgoud, H.K.; Radwan, H.A.M.; El-Mariah, F.; El-Agrody, A.M. Synthesis, Characterization, Biological Activity of Novel 1H-benzo[f]chromene and 12H-benzo[f]chromeno[2,3-d]pyrimidine Derivatives. Lett. Drug Des. Discov. 2018, 15, 857–865. [Google Scholar] [CrossRef]
- Abdella, A.M.; Moatasim, Y.; Ali, M.A.; Elwahy, A.H.M.; Abdelhamid, I.A. Synthesis and Anti-influenza Virus Activity of Novel bis(4H-chromene-3-carbonitrile) Derivatives. J. Heterocycl. Chem. 2017, 54, 1854–1862. [Google Scholar] [CrossRef]
- Jayaprakash Rao, Y.; Yadaiah Goud, E.; Hemasri, Y.; Jain, N.; Gabriella, S. Synthesis and antiproliferative activity of 6,7-aryl/hetaryl coumarins. Russ. J. Gen. Chem. 2016, 86, 184–189. [Google Scholar] [CrossRef]
- Parthiban, A.; Kumaravel, M.; Muthukumaran, J.; Rukkumani, R.; Krishna, R.; Rao, H.S.P. Synthesis, in vitro and in silico anti-proliferative activity of 4-aryl-4H-chromene derivatives. Med. Chem. Res. 2016, 25, 1308–1315. [Google Scholar] [CrossRef]
- Ahagh, M.H.; Dehghan, G.; Mehdipour, M.; Teimuri-Mofrad, R.; Payami, E.; Sheibani, N.; Ghaffari, M.; Asadid, M. Synthesis, characterization, anti-proliferative properties and DNA binding of benzochromene derivatives: Increased Bax/Bcl-2 ratio and caspasedependent apoptosis in colorectal cancer cell. Bioorg. Chem. 2019, 93, 103329. [Google Scholar] [CrossRef]
- Fu, Z.-Y.; Jin, Q.-H.; Qu, Y.-L.; Guan, L.-P. Chalcone derivatives bearing chromen or benzo[f]chromen moieties: Design, synthesis, and evaluations of anti-inflammatory, analgesic, selective COX-2 inhibitory activities. Bioorg. Med. Chem. Lett. 2019, 29, 1909–1912. [Google Scholar] [CrossRef]
- Dehkordi, M.F.; Dehghan, G.; Mahdavi, M.; Feizi, M.A.H. Multispectral studies of DNA binding, antioxidant and cytotoxic activities of a new pyranochromene derivative. Spectrochim. Acta Part A 2015, 145, 353–359. [Google Scholar] [CrossRef]
- Schmitt, F.; Gold, M.; Rothemund, M.; Andronache, I.; Biersack, B.; Schobert, R.; Mueller, T. New naphthopyran analogues of LY290181 as potential tumor vascular-disrupting agents. Eur. J. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Alblewi, F.F.; Okasha, R.M.; Eskandrani, A.A.; Afifi, T.H.; Mohamed, H.M.; Halawa, A.H.; Fouda, A.M.; Al-Dies, A.A.M.; Mora, A.; El-Agrody, A.M. Design and Synthesis of Novel Heterocyclic-Based 4H-benzo[h]chromene Moieties: Targeting antitumor caspase 3/7 activities and cell cycle analysis. Molecules 2019, 24, 1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiba, M.E.; Al-Abdullah, E.S.; Ghabbour, H.A.; Riyadh, S.M.; Abdel-Kader, R.M. Inhibitory activity of benzo[h]quinoline and benzo[h]chromene in human glioblastoma cells. Trop. J. Pharm. Res. 2016, 15, 2337–2343. [Google Scholar] [CrossRef] [Green Version]
- Haiba, M.E.; Al-Abdullah, E.S.; Ahmed, N.S.; Ghabbour, H.A.; Awad, H.M. Efficient and easy synthesis of new Benzo[h]chromene and Benzo[h]quinoline derivatives as a new class of cytotoxic agents. J. Mol. Struct. 2019, 1195, 702–711. [Google Scholar] [CrossRef]
- Kheirollahi, A.; Pordeli, M.; Safavi, M.; Mashkouri, S.; Naimi-Jamal, M.R.; Ardestani, S.K. Cytotoxic and apoptotic effects of synthetic benzochromene derivatives on human cancer cell lines. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 1199–1208. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; Abd El-Mawgoud, H.K.; Fouda, A.M.; Khattab, E.S.A.E.H. Synthesis, in-vitro cytotoxicity of 4H-benzo[h]chromene derivatives and structure-activity relationships of 4-aryl group and 3-, 7-positions. Chem. Pap. 2016, 70, 1279–1292. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; Fouda, A.M.; Khattab, E.S.A.E.H. Synthesis, antitumor activity of 2-amino-4H-benzo[h]chromene derivatives, and structure-activity relationships of the 3- and 4-positions. Med. Chem. Res. 2013, 22, 6105–6120. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; Fouda, A.M.; Khattab, E.S.A.E.H. Halogenated 2-amino-4H-benzo[h]chromene derivatives as antitumor agents and the relationship between lipophilicity and antitumor activity. Med. Chem. Res. 2017, 26, 691–700. [Google Scholar] [CrossRef]
- Okasha, R.M.; Alblewi, F.F.; Afifi, T.H.; Naqvi, A.; Fouda, A.M.; Al-Dies, A.A.M.; El-Agrody, A.M.; Belmont, P.; Bunce, R.A. Design of new Benzo[h]chromene derivatives: Antitumor activities and structure-activity relationships of the 2,3-Positions and fused rings at the 2,3-positions. Molecules 2017, 22, 479. [Google Scholar] [CrossRef]
- Halawa, A.H.; Fouda, A.M.; Al-Dies, A.-A.M.; El-Agrody, A.M. Synthesis, Biological Evaluation and Molecular Docking Studies of 4H-benzo[h]chromenes, 7H-benzo[h]chromeno[2,3-d]pyrimidines as Antitumor Agents. Lett. Drug Des. Discov. 2015, 13, 77–88. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; Fouda, A.M.; Al-Dies, A.A.M. Studies on the synthesis, in vitro antitumor activity of 4H-benzo[h]chromene, 7H-benzo[h]chromene[2,3-d]pyrimidine derivatives and structure-Activity relationships of the 2-,3- and 2,3-positions. Med. Chem. Res. 2014, 23. [Google Scholar] [CrossRef]
- Ahmed, H.E.A.; El-Nassag, M.A.A.; Hassan, A.H.; Okasha, R.M.; Ihmaid, S.; Fouda, A.M.; Afifi, T.H.; Aljuhani, A.; El-Agrody, A.M. Introducing novel potent anticancer agents of 1H-benzo[f]chromene scaffolds, targeting c-Src kinase enzyme with MDA-MB-231 cell line anti-invasion effect. J. Enzyme Inhib. Med. Chem. 2018, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazzi, L.; Cavalli, A.; Belluti, F.; Bisi, A.; Gobbi, S.; Rizzo, S.; Bartolini, M.; Andrisano, V.; Recanatini, M.; Rampa, A. Extensive SAR and computational studies of 3-{4-[(benzylmethylamino)methyl] phenyl}-6,7-dimethoxy-2H-2-chromenone (AP2238) derivatives. J. Med. Chem. 2007, 50. [Google Scholar] [CrossRef] [PubMed]
- Gorle, S.; Maddila, S.; Maddila, S.; Naicker, K.; Singh, M.; Singh, P.; Jonnalagadda, S. Synthesis, Molecular Docking Study and in vitro Anticancer Activity of Tetrazole Linked Benzochromene Derivatives. Anticancer Agents Med. Chem. 2017, 17. [Google Scholar] [CrossRef]
- Afifi, T.H.; Okasha, R.M.; Ahmed, H.E.A.; Ilaš, J.; Saleh, T.; Abd-El-aziz, A.S. Structure-activity relationships and molecular docking studies of chromene and chromene based azo chromophores: A novel series of potent antimicrobial and anticancer agents. EXCLI J. 2017, 16, 868. [Google Scholar] [CrossRef]
- Fouda, A.M.; Assiri, M.A.; Mora, A.; Ali, T.E.; Afifi, T.H.; El-Agrody, A.M. Microwave synthesis of novel halogenated β-enaminonitriles linked 9-bromo-1H-benzo[f]chromene moieties: Induces cell cycle arrest and apoptosis in human cancer cells via dual inhibition of topoisomerase I and II. Bioorg. Chem. 2019, 93. [Google Scholar] [CrossRef]
- Fouda, A.M.; Okasha, R.M.; Alblewi, F.F.; Mora, A.; Afifi, T.H.; El-Agrody, A.M. A proficient microwave synthesis with structure elucidation and the exploitation of the biological behavior of the newly halogenated 3-amino-1H-benzo[f]chromene molecules, targeting dual inhibition of topoisomerase II and microtubules. Bioorg. Chem. 2020, 95. [Google Scholar] [CrossRef]
- Okasha, R.M.; Amr, A.E.; El-Agrody, A.M.; Al-Omar, M.A.; Ghabbour, H.A. Crystal structure of 3-amino-8-methoxy-1-phenyl-1H-benzo[f]chromene-2-carbonitrile, C21H16N2O2. Z. Kristallogr. NCS 2017, 232. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, H.M.; Amr, A.E.; El-Agrody, A.M.; Al-Omar, M.A.; Ghabbour, H.A. Crystal structure of 3-amino-1-(4-bromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile, C21H15BrN2O2. Z. Kristallogr. NCS 2017, 232. [Google Scholar] [CrossRef] [Green Version]
- Fouda, A.M.; Amr, A.E.; El-Agrody, A.M.; Al-Omar, M.A.; Ghabbour, H.A. Crystal structure of 3-amino-8-methoxy-1-(4-methoxy phenyl)-1H-benzo[f]chromene-2-carbonitrile, C22H18N2O3. Z. Kristallogr. NCS 2017, 232. [Google Scholar] [CrossRef] [Green Version]
- Okasha, R.M.; Alblewi, F.F.; Assiri, M.A.; Amr, A.E.; Ghabbour, H.A.; Afifi, T.H.; El-Agrody, A.M. Crystal Structure and Spectral Studies of 3-Amino-9-Methoxy-1-(4-methoxyphenyl)-1H-Benzo[f]Chromene-2-Carbonitrile. J. Comput. Theor. Nanosci. 2018, 15. [Google Scholar] [CrossRef]
- Okasha, R.M.; El-Agrody, A.M.; Amr, A.E.; Al-Omar, M.A.; Ghabbour, H.A. Synthesis and X-ray Single Crystals Characterizations of 2-Amino-4-(2-chlorophenyl)-6-Chloro-4H-Benzo[h]Chromene-3-Carbonitrile. J. Comput. Theor. Nanosci. 2017, 14, 5286–5291. [Google Scholar] [CrossRef]
- Amr, A.E.; Abd El-Mawgoud, H.K.; El-Agrody, A.M.; Al-Omar, A.E.; Alsultan, M.S. X-ray, Microwave Assisted Synthesis and Spectral Data of 3-Amino-1-(3,5-dibromo-2-methoxyphenyl)-8-methoxy-1H-benzo[f]-chromene-2-carbonitrile. J. Comput. Theor. Nanosci. 2017, 14, 3930–3935. [Google Scholar] [CrossRef]
- Afifi, T.H.; El-Agrody, A.M.; Amr, A.E.; Al-Omar, A.E.; Ghabbour, H.A. X-ray Characterization and Antimicrobial Activity of Synthesized New 3-Amino-8-Bromo-1-(3,4-dimethoxyphenyl)-1H-Benzo[f]-Chromene-2-Carbonitrile. J. Comput. Theor. Nanosci. 2017, 14, 3924–3929. [Google Scholar] [CrossRef]
- Paull, K.D.; Shoemaker, R.H.; Boyd, M.R.; Parsons, J.L.; Risbood, P.A.; Barbera, W.A.; Sharma, M.N.; Baker, D.C.; Hand, E.; Scudiero, D.A.; et al. The synthesis of XTT: A new tetrazolium reagent that is bioreducible to a water-soluble formazan. J. Heterocycl. Chem. 1988, 25, 911–914. [Google Scholar] [CrossRef]
- Scudiere, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a Soluble Tetrazolium/Formazan Assay for Cell Growth and Drug Sensitivity in Culture Using Human and Other Tumor Cell Lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by x-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Fabbiani, F.P.A.; Spackman, M.A. Comparison of polymorphic molecular crystal structures through hirshfeld surface analysis. Cryst. Growth Des. 2007, 7. [Google Scholar] [CrossRef]
- Dalal, J.; Sinha, N.; Yadav, H.; Kumar, B. Structural, electrical, ferroelectric and mechanical properties with Hirshfeld surface analysis of novel NLO semiorganic sodium p-nitrophenolate dihydrate piezoelectric single crystal. RSC Adv. 2015, 5. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, K. Role of frontier orbitals in chemical reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, P. Arvi Rauk:“Orbital Interaction Theory of Organic Chemistry” Wiley & Sons: New York, NY, USA, 1994. ISBN 0-471-59389-3. 307 Seiten, mit HMO-Programmdiskette, Preis: $45.50. Ber. Bunsenges. Phys. Chem. 1995, 99, 997–999. [Google Scholar]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ayers, P.W.; Parr, R.G. Variational principles for describing chemical reactions: The Fukui function and chemical hardness revisited. J. Am. Chem. Soc. 2000, 122, 2010–2018. [Google Scholar] [CrossRef]
- Lamaka, S.V.; Zheludkevich, M.L.; Yasakau, K.A.; Serra, R.; Poznyak, S.K.; Ferreira, M.G.S. Nanoporous titania interlayer as reservoir of corrosion inhibitors for coatings with self-healing ability. Prog. Org. Coat. 2007, 58, 127–135. [Google Scholar] [CrossRef]
- Komorowski, L.; Lipinski, J.; Szarek, P.; Ordon, P. Polarization justified Fukui functions: The theory and applications for molecules. J. Chem. Phys. 2011, 135, 14109. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Huizar, L.H.; Rios-Reyes, C.H.; Álvarez-Romero, G.A.; Palomar-Pardavé, M.E.; Ramírez-Silva, M.T. Electrophilic and nucleophilic chemical reactivity of neutral and anionic forms of 4-cpa, 24d-cpa, 34-cpa and 245t-cpa through conceptual dft reactivity descriptors. J. Chil. Chem. Soc. 2017, 62, 3411–3416. [Google Scholar] [CrossRef]
- Elhenawy, A.A.; Al-Harbi, L.M.; El-Gazzar, M.A.; Khowdiary, M.M.; Moustfa, A. Synthesis, molecular properties and comparative docking and QSAR of new 2-(7-hydroxy-2-oxo-2H-chromen-4-yl)acetic acid derivatives as possible anticancer agents. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 218, 248–262. [Google Scholar] [CrossRef]
- Luque, F.J.; López, J.M.; Orozco, M. Perspective on Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. In Theoretical Chemistry Accounts; Springer: Berlin, Germany, 2000; pp. 343–345. [Google Scholar]
- Mendoza-Huizar, L.H.; Rios-Reyes, C.H. Chemical reactivity of atrazine employing the Fukui function. J. Mex. Chem. Soc. 2011, 55, 142–147. [Google Scholar]
- Sureshkumar, B.; Mary, Y.S.; Panicker, C.Y.; Suma, S.; Armaković, S.; Armaković, S.J.; Van Alsenoy, C.; Narayana, B. Quinoline derivatives as possible lead compounds for anti-malarial drugs: Spectroscopic, DFT and MD study. Arab. J. Chem. 2017, 13, 632–648. [Google Scholar] [CrossRef]
- War, J.A.; Jalaja, K.; Mary, Y.S.; Panicker, C.Y.; Armaković, S.; Armaković, S.J.; Srivastava, S.K.; Van Alsenoy, C. Spectroscopic characterization of 1-[3-(1H-imidazol-1-yl) propyl]-3-phenylthiourea and assessment of reactive and optoelectronic properties employing DFT calculations and molecular dynamics simulations. J. Mol. Struct. 2017, 1129, 72–85. [Google Scholar] [CrossRef]
- Elhenawy, A.A.; Al-Harbi, L.M.; Moustafa, G.O.; El-Gazzar, M.A.; Abdel-Rahman, R.F.; Salim, A.E. Synthesis, comparative docking, and pharmacological activity of naproxen amino acid derivatives as possible anti-inflammatory and analgesic agents. Drug Des. Dev. Ther. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, I.O.; Nassar, H.S.; El-Nasser, K.S.; Bougarech, A.; Abid, M.; Elhenawy, A.A. Synthesis and characterization of MnII and CoII complexes with poly (vinyl alcohol-nicotinic acid) for photocatalytic degradation of Indigo carmine dye. Inorg. Chem. Commun. 2021, 124, 108360. [Google Scholar] [CrossRef]
- Theophilou, A.K. A novel density functional theory for atoms, molecules, and solids. J. Chem. Phys. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory, International Series of Monographs on Chemistry 22. Oxford University Press. Oxford Henkelman G, Arnaldsson A, Jónsson H A fast robust algorithm Bader decomposition of charge density. Comput. Mater. Sci. 1990, 88, 899. [Google Scholar]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.P.; Miller, B.J.; Kjaergaard, H.G. Are bond critical points really critical for hydrogen bonding? J. Chem. Theory Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of hydrogen-bond interaction potentials from the electron density: Integration of noncovalent interaction regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 397–409. [Google Scholar] [CrossRef]
- Sheikh, J.; Hatzade, K.; Bader, A.; Shaheen, U.; Sander, T.; Ben Hadda, T. Computational evaluation and experimental verification of antibacterial and antioxidant activity of 7-hydroxy-3-pyrazolyl-4H-chromen-4-ones and their o-glucosides: Identification of pharmacophore sites. Med. Chem. Res. 2014, 23, 243–251. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Govindasamy, J.; Khalilullah, H.; Mohan, G.; Stables, J.P.; Pannecouque, C.; Clercq, E. De POMA analyses as new efficient bioinformatics’ platform to predict and optimise bioactivity of synthesized 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. Bioorg. Med. Chem. Lett. 2012, 22, 7029–7035. [Google Scholar] [CrossRef] [PubMed]
- Mabkhot, Y.N.; Arfan, M.; Zgou, H.; Genc, Z.K.; Genc, M.; Rauf, A.; Bawazeer, S.; Ben Hadda, T. How to improve antifungal bioactivity: POM and DFT study of some chiral amides derivatives of diacetyl-L-tartaric acid and amines. Res. Chem. Intermed. 2016, 42, 8055–8068. [Google Scholar] [CrossRef]
- Rachedi, K.O.; Ouk, T.-S.; Bahadi, R.; Bouzina, A.; Djouad, S.-E.; Bechlem, K.; Zerrouki, R.; Ben Hadda, T.; Almalki, F.; Berredjem, M. Synthesis, DFT and POM analyses of cytotoxicity activity of α-amidophosphonates derivatives: Identification of potential antiviral O,O-pharmacophore site. J. Mol. Struct. 2019, 1197, 196–203. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Liu, X.; Xu, Y.; Li, H.; Luo, C.; Jiang, H. Computational methods for drug design and discovery: Focus on China. Trends Pharmacol. Sci. 2013, 34, 549–559. [Google Scholar] [CrossRef]
- Husain, A.; Ahmad, A.; Khan, S.A.; Asif, M.; Bhutani, R.; Al-Abbasi, F.A. Synthesis, molecular properties, toxicity and biological evaluation of some new substituted imidazolidine derivatives in search of potent anti-inflammatory agents. J Saudi Pharm. J. 2016, 24, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. J Mutat. Res. Mol. Mech. Mutagen. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Dueñas-González, A.; Jesús Naveja, J.; Medina-Franco, J.L. Introduction of Epigenetic Targets in Drug Discovery and Current Status of Epi-Drugs and Epi-Probes. In Epi-Informatics: Discovery and Development of Small Molecule Epigenetic Drugs and Probes; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780128028094. [Google Scholar]
- Fahy, J.; Jeltsch, A.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: A chemical and therapeutic patent overview and selected clinical studies. Expert Opin. Ther. Pat. 2012, 22, 1427–1442. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and Function of Mammalian DNA Methyltransferases. ChemBioChem 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenetics Chromatin 2017, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. 2010, 42, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, T. The BLAST Sequence Analysis Tool; National Center for Biotechnology Information (US): Bethesda, MA, USA, 2013.
- Ishiyama, S.; Nishiyama, A.; Saeki, Y.; Moritsugu, K.; Morimoto, D.; Yamaguchi, L.; Arai, N.; Matsumura, R.; Kawakami, T.; Mishima, Y.; et al. Structure of the Dnmt1 Reader Module Complexed with a Unique Two-Mono-Ubiquitin Mark on Histone H3 Reveals the Basis for DNA Methylation Maintenance. Mol. Cell 2017, 68, 350–360.e7. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Wang, L.; Du, Y.; Xie, S.; Yang, X.; Lian, F.; Zhou, Z.; Qian, C. Structural and mechanistic insights into UHRF1-mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res. 2018, 46, 3218–3231. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Maestro|Schrödinger. 2018. Available online: https://www.Schrödinger.com/materials-science (accessed on 27 January 2021).
- Siu, S.W.I.; Pluhackova, K.; Böckmann, R.A. Optimization of the OPLS-AA force field for long hydrocarbons. J. Chem. Theory Comput. 2012, 39–40. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. Procheck: Validation of Protein-Structure Coordinates; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assumpção, J.H.M.; Takeda, A.A.S.; Sforcin, J.M.; Rainho, C.A. Abstract A12: Brazilian Propolis as a Source of Novel DNA Methyltransferase Inhibitors: A Computer-Aided Discovery and in Vitro Approaches. Available online: https://clincancerres.aacrjournals.org/content/24/1_Supplement/A12 (accessed on 27 January 2021).
- Tarcsay, Á.; Nyíri, K.; Keserú, G.M. Impact of lipophilic efficiency on compound quality. J. Med. Chem. 2012, 55, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Thapa, R.; Chattopadhyay, K.K. Bandgap widening in highly conducting CdO thin film by Ti incorporation through radio frequency magnetron sputtering technique. Solid State Commun. 2008, 145, 33–37. [Google Scholar] [CrossRef]
- Siemens Analytical X-Ray Instruments, Inc. Anal. Chem. 1990. [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Tapp, H.S.; Kemsley, E.K. Notes on the practical utility of OPLS. TrAC Trends Anal. Chem. 2009, 28, 1322–1327. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | Ar | Cell Lines IC50(µM) a 24h | ||
|---|---|---|---|---|
| MDA-MB-231 | A549 | MIA PaCa-2 | ||
| 4 | 2,5-Cl2C6H3 | 10.7 ± 1.5 | 7.7 ± 0.7 | 7.3 ± 0.7 |
| Etoposide | - | >30 | >30 | >30 |
| Camptothecin | - | 22.4 ± 20.6 | 20.0 ± 10.2 | 0.9 ± 0.02 |
| Crystal Data | |
|---|---|
| Chemical formula | C21H14Cl2N2O2 |
| Formula weight | 397.24 |
| Crystal system, space group | Orthorhombic, Pbca |
| Temperature (K) | 293 |
| a, b, c (Å) | 9.3186 (5), 19.0894 (10), 20.7892 (12) |
| V (Å3) | 3698.1 (3) |
| Z | 8 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.37 |
| Crystal size (mm) | 0.32 × 0.27 × 0.25 |
| Data collection | |
| Diffractometer | Bruker APEX-II D8 venture diffractometer |
| Absorption correction | Multi-scan, SADABS Bruker 2018 |
| Tmin, Tmax | 0.932, 0.944 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 62944, 4248, 3181 |
| Rint | 0.049 |
| Refinement | |
| R[F2 > 2σ( F2)], wR( F2), S | 0.059, 0.170, 1.05 |
| No. of reflections | 4248 |
| No. of parameters | 253 |
| No. of restraints | 0 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.91, −0.73 |
| Bond Lengths | X-ray Crystal | DFT* 6-311++G(d,p) | Bond Lengths | X-ray Crystal | DFT* 6-311++G(d,p) |
|---|---|---|---|---|---|
| Cl1—C16 | 1.740 (3) | 1.832 | O2—C8 | 1.373 (3) | 1.411 |
| Cl2—C19 | 1.734 (3) | 1.845 | O2—C21 | 1.411 (4) | 1.470 |
| O1—C1 | 1.357 (3) | 1.384 | N1—C1 | 1.336 (4) | 1.361 |
| O1—C13 | 1.395 (3) | 1.419 | N2—C20 | 1.140 (4) | 1.173 |
| Bond Angles | X-ray Crystal | DFT* 6-311++G(d,p) | Bond Angles | X-ray Crystal | DFT* 6-311++G(d,p) |
| C1—O1—C13 | 118.33 (19) | 119.38 | O1—C13—C4 | 123.1 (2) | 122.36 |
| C8—O2—C21 | 117.7 (2) | 114.80 | O1—C13—C12 | 113.8 (2) | 11.6.97 |
| O1—C1—N1 | 110.7 (2) | 110.8 | Cl1—C16—C15 | 118.7 (2) | 1180.66 |
| O1—C1—C2 | 122.4 (2) | 122.23 | Cl1—C16—C17 | 120.0 (2) | 119.10 |
| N1—C1—C2 | 126.9 (3) | 127.21 | Cl2—C19—C14 | 120.8 (3) | 120.77 |
| O2—C8—C7 | 114.1 (2) | 119.91 | Cl2—C19—C18 | 118.2 (2) | 11631 |
| O2—C8—C9 | 125.3 (2) | 119.67 | N2—C20—C2 | 179.3 (3) | 178.66 |
| D—H···A | D—H | H···A | D···A | D—H···A |
|---|---|---|---|---|
| N1—H2N1···O2 i | 0.84(3) | 2.52(3) | 3.247(4) | 146(3) |
| N1—H1N1···N2 ii | 0.85(4) | 2.31(4) | 3.145(4) | 165(3) |
| C7—H7A···N2 iii | 0.93 | 2.60 | 3.369(4) | 141.0 |
| HOMO | LUMO | ΔG | I | A | η | S | χ | ωi | µ+ | µ− | ΔNmax | ΔEmax | ΔEmin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −5.947 | −2.389 | −3.558 | 5.947 | 2.389 | 1.779 | 0.562 | −4.168 | 4.882 | −3.278 | −5.057 | −1.171 | −4.685 | −2.342 |
| Cpd. | 4 | Reference Inhibitor |
|---|---|---|
| ΔG | −5.458 | −6.508 |
| RMSD | 0.802 | 1.637 |
| Int. | −46.7218 | −30.9447 |
| H.B. | −4.30374 | −4.55283 |
| E ele | −87.417 | −100.927 |
| Evdw | −104.168 | −72.201 |
| L.E. | −0.246 ± 0.08 * | −0.217 ± 0.11 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Gaafary, M.; Syrovets, T.; M. Mohamed, H.; A. Elhenawy, A.; M. El-Agrody, A.; El-Galil E. Amr, A.; Ghabbour, H.A.; Almehizia, A.A. Synthesis, Cytotoxic Activity, Crystal Structure, DFT Studies and Molecular Docking of 3-Amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile. Crystals 2021, 11, 184. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11020184
El Gaafary M, Syrovets T, M. Mohamed H, A. Elhenawy A, M. El-Agrody A, El-Galil E. Amr A, Ghabbour HA, Almehizia AA. Synthesis, Cytotoxic Activity, Crystal Structure, DFT Studies and Molecular Docking of 3-Amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile. Crystals. 2021; 11(2):184. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11020184
Chicago/Turabian StyleEl Gaafary, Menna, Tatiana Syrovets, Hany M. Mohamed, Ahmed A. Elhenawy, Ahmed M. El-Agrody, Abd El-Galil E. Amr, Hazem A. Ghabbour, and Abdulrahman A. Almehizia. 2021. "Synthesis, Cytotoxic Activity, Crystal Structure, DFT Studies and Molecular Docking of 3-Amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile" Crystals 11, no. 2: 184. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11020184