
Screening of II-IV-V2 Materials for Photovoltaic Applications Based on Density Functional Theory Calculations


Abstract
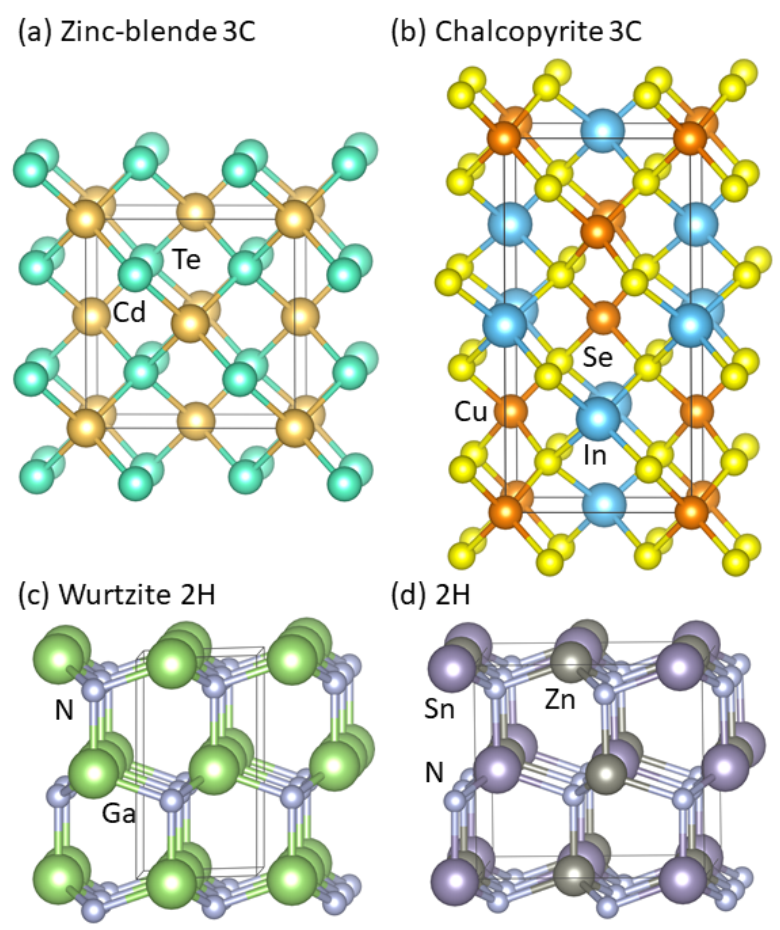
:1. Introduction
2. Calculation Method
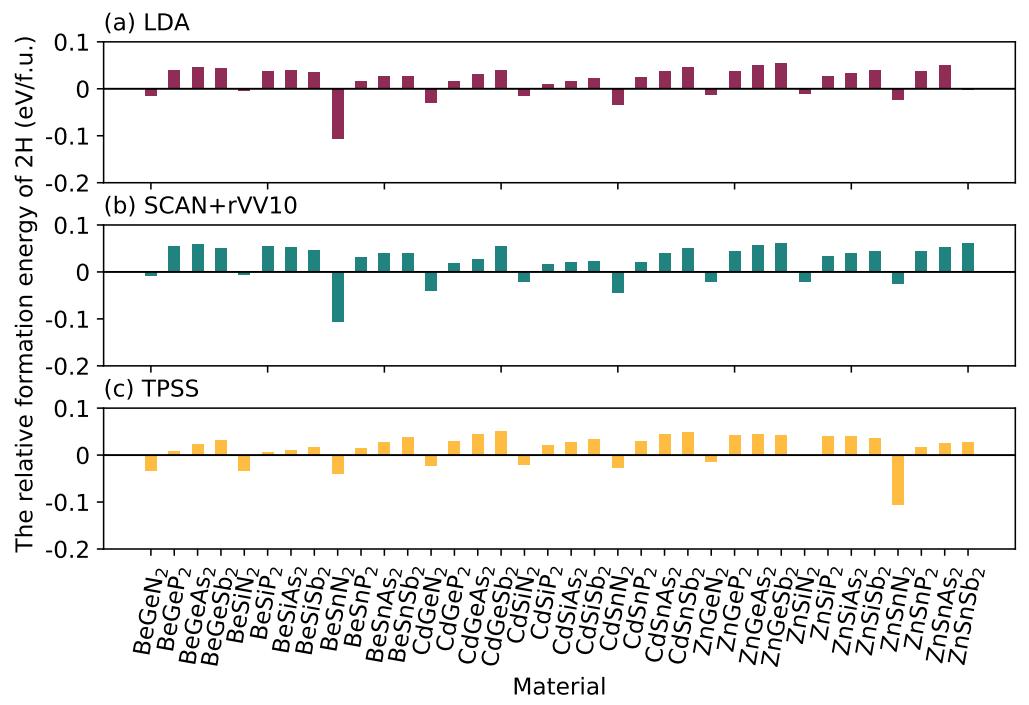
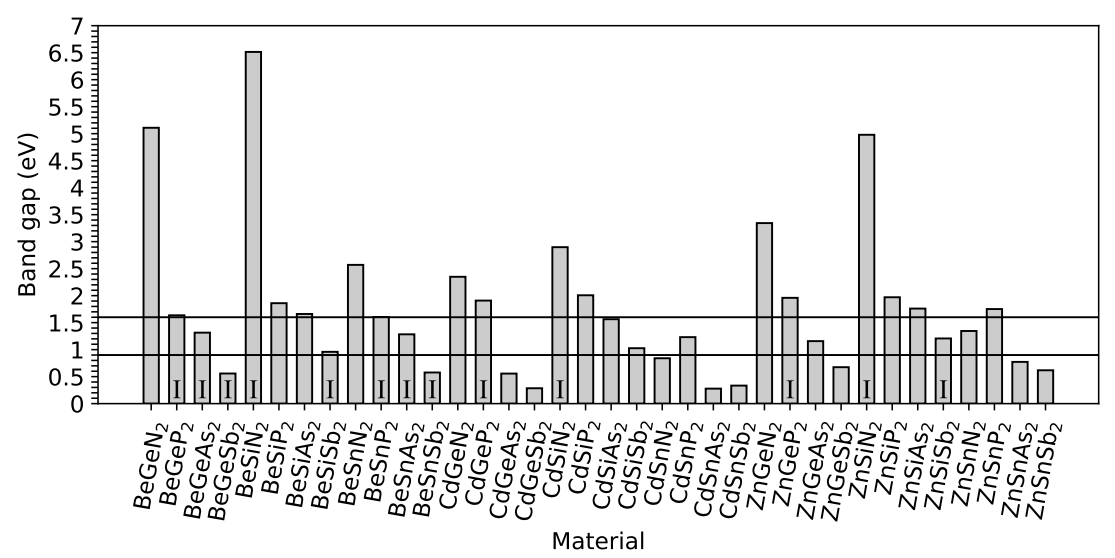
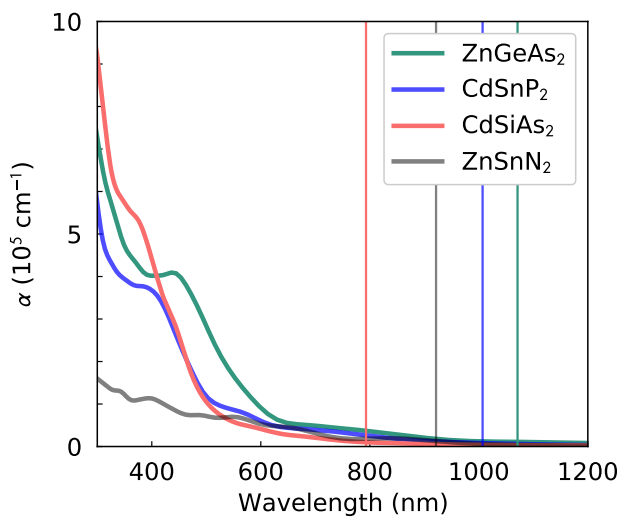
3. Results and Discussion
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okada, Y.; Ekins-Daukes, N.; Kita, T.; Tamaki, R.; Yoshida, M.; Pusch, A.; Hess, O.; Phillips, C.; Farrell, D.; Yoshida, K.; et al. Intermediate band solar cells: Recent progress and future directions. Appl. Phys. Rev. 2015, 2, 021302. [Google Scholar] [CrossRef] [Green Version]
- Haverkort, J.E.; Garnett, E.C.; Bakkers, E.P. Fundamentals of the nanowire solar cell: Optimization of the open circuit voltage. Appl. Phys. Rev. 2018, 5, 031106. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, C.Z.; Shi, M.; Chen, H. New phase for organic solar cell research: Emergence of Y-series electron acceptors and their perspectives. ACS Energy Lett. 2020, 5, 1554–1567. [Google Scholar] [CrossRef]
- Nayak, P.K.; Mahesh, S.; Snaith, H.J.; Cahen, D. Photovoltaic solar cell technologies: Analysing the state of the art. Nat. Rev. Mater. 2019, 4, 269–285. [Google Scholar] [CrossRef]
- Walsh, A.; Chen, S.; Wei, S.H.; Gong, X.G. Kesterite thin-film solar cells: Advances in materials modelling of Cu2ZnSnS4. Adv. Energy Mater. 2012, 2, 400–409. [Google Scholar] [CrossRef]
- Khan, I.S.; Heinselman, K.N.; Zakutayev, A. Review of ZnSnN2 semiconductor material. J. Phys. Energy 2020, 2, 032007. [Google Scholar] [CrossRef]
- Emery, A.A.; Wolverton, C. High-throughput dft calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci. Data 2017, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Voznyy, O.; Sargent, E.H. High-throughput screening of lead-free perovskite-like materials for optoelectronic applications. J. Phys. Chem. C 2017, 121, 7183–7187. [Google Scholar] [CrossRef]
- Huo, Z.; Wei, S.H.; Yin, W.J. High-throughput screening of chalcogenide single perovskites by first-principles calculations for photovoltaics. J. Phys. D Appl. Phys. 2018, 51, 474003. [Google Scholar] [CrossRef]
- Oganov, A.R.; Pickard, C.J.; Zhu, Q.; Needs, R.J. Structure prediction drives materials discovery. Nat. Rev. Mater. 2019, 4, 331–348. [Google Scholar] [CrossRef]
- Niki, S.; Contreras, M.; Repins, I.; Powalla, M.; Kushiya, K.; Ishizuka, S.; Matsubara, K. CIGS absorbers and processes. Prog. Photovolt. Res. Appl. 2010, 18, 453–466. [Google Scholar] [CrossRef]
- Crovetto, A.; Hansen, O. What is the band alignment of Cu2ZnSn(S,Se)4 solar cells? Sol. Energy Mater. Sol. Cells 2017, 169, 177–194. [Google Scholar] [CrossRef] [Green Version]
- Kanevce, A.; Reese, M.O.; Barnes, T.; Jensen, S.; Metzger, W. The roles of carrier concentration and interface, bulk, and grain-boundary recombination for 25% efficient CdTe solar cells. J. Appl. Phys. 2017, 121, 214506. [Google Scholar] [CrossRef]
- Kurtz, S.; Repins, I.; Metzger, W.K.; Verlinden, P.J.; Huang, S.; Bowden, S.; Tappan, I.; Emery, K.; Kazmerski, L.L.; Levi, D. Historical analysis of champion photovoltaic module efficiencies. IEEE J. Photovolt. 2018, 8, 363–372. [Google Scholar] [CrossRef]
- Shockley, W.; Queisser, H.J. Detailed balance limit of efficiency of p-n junction solar cells. J. Appl. Phys. 1961, 32, 510–519. [Google Scholar] [CrossRef]
- Shaposhnikov, V.; Krivosheeva, A.; Borisenko, V.; Lazzari, J.L.; d’Avitaya, F.A. Ab initio modeling of the structural, electronic, and optical properties of AIIBIVC2V semiconductors. Phys. Rev. B 2012, 85, 205201. [Google Scholar] [CrossRef]
- Pandey, M.; Kuhar, K.; Jacobsen, K.W. II–IV–V2 and III–III–V2 Polytypes as Light Absorbers for Single Junction and Tandem Photovoltaic Devices. J. Phys. Chem. C 2017, 121, 17780–17786. [Google Scholar] [CrossRef]
- Wellendorff, J.; Lundgaard, K.T.; Jacobsen, K.W.; Bligaard, T. mBEEF: An accurate semi-local Bayesian error estimation density functional. J. Chem. Phys. 2014, 140, 144107. [Google Scholar] [CrossRef] [Green Version]
- Gritsenko, O.; van Leeuwen, R.; van Lenthe, E.; Baerends, E.J. Self-consistent approximation to the Kohn-Sham exchange potential. Phys. Rev. A 1995, 51, 1944. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kitchaev, D.A.; Yang, J.; Chen, T.; Dacek, S.T.; Sarmiento-Pérez, R.A.; Marques, M.A.; Peng, H.; Ceder, G.; Perdew, J.P.; et al. Efficient first-principles prediction of solid stability: Towards chemical accuracy. Npj Comput. Mater. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Yang, J.H.; Kitchaev, D.A.; Ceder, G. Rationalizing accurate structure prediction in the meta-GGA SCAN functional. Phys. Rev. B 2019, 100, 035132. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S. Comparison study of exchange-correlation functionals on prediction of ground states and structural properties. Curr. Appl. Phys. 2021, 22, 61–64. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Crystal Structures; Interscience Publishers: New York, NY, USA, 1965; Volume 1. [Google Scholar]
- Park, J.S.; Walsh, A. Modeling Grain Boundaries in Polycrystalline Halide Perovskite Solar Cells. Annu. Rev. Condens. Matter Phys. 2020, 12, 95–109. [Google Scholar] [CrossRef]
- Park, J.S.; Li, Z.; Wilson, J.N.; Yin, W.J.; Walsh, A. Hexagonal Stacking Faults Act as Hole-Blocking Layers in Lead Halide Perovskites. ACS Energy Lett. 2020, 5, 2231–2233. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Yang, Z.H.; Perdew, J.P.; Sun, J. Versatile van der Waals density functional based on a meta-generalized gradient approximation. Phys. Rev. X 2016, 6, 041005. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, F.; Imura, M.; Murata, H.; Yamada, N.; Taniguchi, T. Synthesis of a Novel Rocksalt-Type Ternary Nitride Semiconductor MgSnN2 Using the Metathesis Reaction Under High Pressure. Eur. J. Inorg. Chem. 2020, 2020, 446–451. [Google Scholar] [CrossRef]
- Reddy, N.; Kistaiah, P.; Murthy, K. High-temperature thermal expansion study of cadmium stannidiphosphide by an X-ray method. J. Phys. D Appl. Phys. 1982, 15, 2247. [Google Scholar] [CrossRef]
- Avirović, M.; Lux-Steiner, M.; Elrod, U.; Hönigschmid, J.; Bucher, E. Single crystal growth of CdSiAs2 by chemical vapor transport; its structure and electrical properties. J. Cryst. Growth 1984, 67, 185–194. [Google Scholar] [CrossRef]
- Chelluri, B.; Chang, T.; Ourmazd, A.; Dayem, A.; Zyskind, J.; Srivastava, A. Molecular beam epitaxial growth of II–V semiconductor Zn3As2 and II–IV–V chalcopyrite ZnGeAs2. J. Cryst. Growth 1987, 81, 530–535. [Google Scholar] [CrossRef]
- Kimmel, M.; Lux-Steiner, M.; Vögt, M.; Bucher, E. n-ZnO/p-CdSiAs2 heterojunction solar cells. J. Appl. Phys. 1988, 64, 1560–1561. [Google Scholar] [CrossRef]
- Feldberg, N.; Aldous, J.; Linhart, W.; Phillips, L.; Durose, K.; Stampe, P.; Kennedy, R.; Scanlon, D.; Vardar, G.; Field, R., III; et al. Growth, disorder, and physical properties of ZnSnN2. Appl. Phys. Lett. 2013, 103, 042109. [Google Scholar] [CrossRef]
- Peshek, T.J.; Zhang, L.; Singh, R.K.; Tang, Z.; Vahidi, M.; To, B.; Coutts, T.J.; Gessert, T.A.; Newman, N.; Van Schilfgaarde, M. Criteria for improving the properties of ZnGeAs2 solar cells. Prog. Photovolt. Res. Appl. 2013, 21, 906–917. [Google Scholar] [CrossRef]
- Martinez, A.D.; Fioretti, A.N.; Toberer, E.S.; Tamboli, A.C. Synthesis, structure, and optoelectronic properties of II–IV–V2 materials. J. Mater. Chem. A 2017, 5, 11418–11435. [Google Scholar] [CrossRef]
- Nakatsuka, S.; Inoue, R.; Nose, Y. Fabrication of CdSnP2 Thin Films by Phosphidation for Photovoltaic Application. ACS Appl. Energy Mater. 2018, 1, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S. Examination of high-throughput hybrid calculations using coarser reciprocal space meshes. Curr. Appl. Phys. 2020, 20, 379–383. [Google Scholar] [CrossRef]
- Veal, T.D.; Feldberg, N.; Quackenbush, N.F.; Linhart, W.M.; Scanlon, D.O.; Piper, L.F.; Durbin, S.M. Band gap dependence on cation disorder in ZnSnN2 solar absorber. Adv. Energy Mater. 2015, 5, 1501462. [Google Scholar] [CrossRef] [Green Version]
- Quayle, P.C.; Blanton, E.W.; Punya, A.; Junno, G.T.; He, K.; Han, L.; Zhao, H.; Shan, J.; Lambrecht, W.R.; Kash, K. Charge-neutral disorder and polytypes in heterovalent wurtzite-based ternary semiconductors: The importance of the octet rule. Phys. Rev. B 2015, 91, 205207. [Google Scholar] [CrossRef] [Green Version]
- Lany, S.; Fioretti, A.N.; Zawadzki, P.P.; Schelhas, L.T.; Toberer, E.S.; Zakutayev, A.; Tamboli, A.C. Monte Carlo simulations of disorder in ZnSnN2 and the effects on the electronic structure. Phys. Rev. Mater. 2017, 1, 035401. [Google Scholar] [CrossRef]
- Sun, C.; Lu, N.; Wang, J.; Lee, J.; Peng, X.; Klie, R.F.; Kim, M.J. Creating a single twin boundary between two CdTe (111) wafers with controlled rotation angle by wafer bonding. Appl. Phys. Lett. 2013, 103, 252104. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, S.; Walsh, A. Opposing effects of stacking faults and antisite domain boundaries on the conduction band edge in kesterite quaternary semiconductors. Phys. Rev. Mater. 2018, 2, 014602. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Rothmann, M.U.; Zhu, Y.; Chen, W.; Yang, C.; Yuan, Y.; Choo, Y.Y.; Wen, X.; Cheng, Y.B.; Bach, U.; et al. The critical role of composition-dependent intragrain planar defects in the performance of MA1−xFAxPbI3 perovskite solar cells. Nat. Energy 2021, 6, 624–632. [Google Scholar] [CrossRef]
- Jeong, B.H.; Park, J.S. Stability and electronic structure of stacking faults and polytypes in ZnSnN2, ZnGeN2, and ZnSiN2. J. Korean Phys. Soc. 2021. [Google Scholar] [CrossRef]
- Pikhtin, A.; Razbegaev, V.; Goryunova, N.; Leonov, E.; Orlov, V.; Karavaev, G.; Poplavnoi, A.; Chaldyshev, V. Energy band structure and optical properties of CdSnP2. Phys. Status Solidi A 1971, 4, 311–318. [Google Scholar] [CrossRef]
- Shay, J.; Buehler, E. Electroreflectance Spectra of CdSiAs2 and CdGeAs2. Phys. Rev. B 1971, 3, 2598. [Google Scholar] [CrossRef]
- Shah, S.; Greene, J. Growth and physical properties of ZnGeAs2. Mater. Lett. 1983, 2, 115–118. [Google Scholar] [CrossRef]
- Peshek, T.; Tang, Z.; Zhang, L.; Singh, R.; To, B.; Gessert, T.; Coutts, T.; Newman, N.; Van Schilfgaarde, M. ZnGeAs2 thin films properties: A potentially useful semiconductor for photovoltaic applications. In Proceedings of the 2009 34th IEEE Photovoltaic Specialists Conference (PVSC), Philadelphia, PA, USA, 7–12 June 2009; pp. 001367–001369. [Google Scholar]
- Senabulya, N.; Feldberg, N.; Makin, R.A.; Yang, Y.; Shi, G.; Jones, C.M.; Kioupakis, E.; Mathis, J.; Clarke, R.; Durbin, S.M. Stabilization of orthorhombic phase in single-crystal ZnSnN2 films. AIP Adv. 2016, 6, 075019. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | a (Å) | b (Å) | c (Å) | (eV) | (eV/nm) | Ref. |
|---|---|---|---|---|---|---|
| CdSnP | 5.896 | 5.896 | 11.548 | 1.23 | 0.06 | |
| (5.9015) | (5.9015) | (11.5144) | (1.165) | [33,49] | ||
| CdSiAs | 5.878 | 5.878 | 10.896 | 1.56 | 0.15 | |
| (5.8848) | (5.8848) | (10.8820) | (1.55) | [34,50] | ||
| ZnGeAs | 5.634 | 5.634 | 11.133 | 1.16 | 0.47 | |
| (5.672) | (5.672) | (11.153) | (1.15) | [51,52] | ||
| ZnSnN | 6.699 | 5.822 | 5.464 | 1.35 | 0.28 | [48] |
| (6.8852) | (5.9557) | (5.5778) | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, B.-H.; Jeong, M.; Song, Y.; Park, K.; Park, J.-S. Screening of II-IV-V2 Materials for Photovoltaic Applications Based on Density Functional Theory Calculations. Crystals 2021, 11, 883. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080883
Jeong B-H, Jeong M, Song Y, Park K, Park J-S. Screening of II-IV-V2 Materials for Photovoltaic Applications Based on Density Functional Theory Calculations. Crystals. 2021; 11(8):883. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080883
Chicago/Turabian StyleJeong, Byeong-Hyeon, Minwoo Jeong, Youbin Song, Kanghyeon Park, and Ji-Sang Park. 2021. "Screening of II-IV-V2 Materials for Photovoltaic Applications Based on Density Functional Theory Calculations" Crystals 11, no. 8: 883. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080883