
Transfer of a Rational Crystal Contact Engineering Strategy between Diverse Alcohol Dehydrogenases

 , , ,
, , ,
Abstract
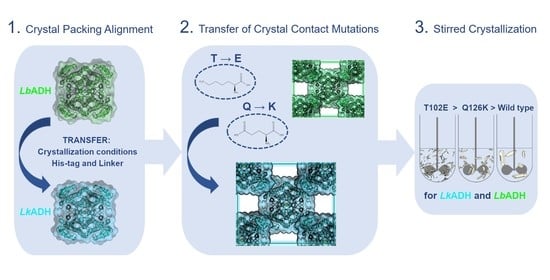
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mutagenesis
2.2. Protein production and Purification
2.3. Protein Crystallization
2.4. Protein Analytics and Analysis of Crystal Photomicrographs
2.5. X-ray Diffraction and Data Refinement
3. Results and Discussion
3.1. Transfer of Crystal Packing from LbADH to LkADH
3.2. Transfer of Mutations on Crystal Contacts from LbADH to LkADH
3.3. Stirred Crystallization of Dialyzed and Clarified E. coli Cell Lysate
3.4. Evaluation of Crystal Size Differences between LkADH Variants
3.5. Stirred Crystallization of Clarified E. coli Cell Lysate
3.6. Comparison of LkADH and LbADH Crystal Contacts at Position Q126 on a Molecular Level
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, J.; Fazio, V.J.; Lawson, B.; Peat, T.S. The C6 Web Tool: A Resource for the Rational Selection of Crystallization Conditions. Cryst. Growth Des. 2010, 10, 2785–2792. [Google Scholar] [CrossRef]
- Weber, P.; Pissis, C.; Navaza, R.; Mechaly, A.E.; Saul, F.; Alzari, P.M.; Haouz, A. High-Throughput Crystallization Pipeline at the Crystallography Core Facility of the Institut Pasteur. Molecules 2019, 24, 4451. [Google Scholar] [CrossRef] [Green Version]
- Longenecker, K.L.; Garrard, S.M.; Sheffield, P.J.; Derewenda, Z.S. Protein crystallization by rational mutagenesis of surface residues: Lys to Ala mutations promote crystallization of RhoGDI. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.M.; Artymiuk, P.J.; Yewdall, S.J.; Smith, J.M.; Livingstone, J.C.; Treffry, A.; Luzzago, A.; Levi, S.; Arosio, P.; Cesareni, G.; et al. Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 1991, 349, 541–544. [Google Scholar] [CrossRef]
- Braig, K.; Otwinowski, Z.; Hegde, R.; Boisvert, D.C.; Joachimiak, A.; Horwich, A.L.; Sigler, P.B. The crystal structure of the bacterial chaperonln GroEL at 2.8 Å. Nature 1994, 371, 578–586. [Google Scholar] [CrossRef]
- Horwich, A. Working with Paul Sigler. Nat. Struct. Biol. 2000, 7, 269–270. [Google Scholar] [CrossRef] [PubMed]
- McElroy, H.E.; Sisson, G.W.; Schoettlin, W.E.; Aust, R.M.; Villafranca, J.E. Studies on engineering crystallizability by mutation of surface residues of human thymidylate synthase. J. Cryst. Growth 1992, 122, 265–272. [Google Scholar] [CrossRef]
- Derewenda, Z.S. The use of recombinant methods and molecular engineering in protein crystallization. Methods 2004, 34, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S.; Vekilov, P.G. Entropy and surface engineering in protein crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Boczek, T.; Grelewska, K.; Pinkowska, M.; Sikorska, M.; Zawadzki, M.; Derewenda, Z. Protein crystallization by surface entropy reduction: Optimization of the SER strategy. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 636–645. [Google Scholar] [CrossRef]
- Derewenda, Z.S.; Godzik, A. The “Sticky Patch” Model of Crystallization and Modification of Proteins for Enhanced Crystallizability. Methods Mol. Biol. 2017, 1607, 77–115. [Google Scholar] [CrossRef] [Green Version]
- Derewenda, Z.S. Protein crystallization in drug design: Towards a rational approach. Expert Opin. Drug Discov. 2007, 2, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S. Rational protein crystallization by mutational surface engineering. Structure 2004, 12, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Laganowsky, A.; Zhao, M.; Soriaga, A.B.; Sawaya, M.R.; Cascio, D.; Yeates, T.O. An approach to crystallizing proteins by metal-mediated synthetic symmetrization. Protein Sci 2011, 20, 1876–1890. [Google Scholar] [CrossRef] [Green Version]
- Devedjiev, Y.; Surendranath, Y.; Derewenda, U.; Gabrys, A.; Cooper, D.R.; Zhang, R.-g.; Lezondra, L.; Joachimiak, A.; Derewenda, Z.S. The Structure and Ligand Binding Properties of the B.subtilis YkoF Gene Product, a Member of a Novel Family of Thiamin/HMP-binding Proteins. J. Mol. Biol. 2004, 343, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Roosen-Runge, F.; Zhang, F.; Schreiber, F.; Roth, R. Ion-activated attractive patches as a mechanism for controlled protein interactions. Sci. Rep. 2014, 4, 7016. [Google Scholar] [CrossRef] [Green Version]
- Banatao, D.R.; Cascio, D.; Crowley, C.S.; Fleissner, M.R.; Tienson, H.L.; Yeates, T.O. An approach to crystallizing proteins by synthetic symmetrization. Proc. Natl. Acad. Sci. USA 2006, 103, 16230–16235. [Google Scholar] [CrossRef] [Green Version]
- Hekmat, D. Large-scale crystallization of proteins for purification and formulation. Bioprocess Biosyst. Eng. 2015, 38, 1209–1231. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.; Carvalho, A.L.; Roque, A.C.A. Renaissance of protein crystallization and precipitation in biopharmaceuticals purification. Biotechnol. Adv. 2017, 35, 41–50. [Google Scholar] [CrossRef]
- Shenoy, B.; Wang, Y.; Shan, W.; Margolin, A.L. Stability of crystalline proteins. Biotechnol. Bioeng. 2001, 73, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Brange, J.; Vølund, A. Insulin analogs with improved pharmacokinetic profiles. Adv. Drug Deliv. Rev. 1999, 35, 307–335. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, H.; Li, J.; Cheng, Q.-D.; Zhang, X.; Zeng, X.-B.; Fiaz, A.; Wang, B.; Zhang, C.-Y.; Lu, Q.-Q.; et al. Direct Crystallization of Proteins from Impure Sources. Cryst. Growth Des. 2020, 20, 1694–1705. [Google Scholar] [CrossRef]
- Harrison, R.G.; Todd, P.W.; Rudge, S.R.; Petrides, D.P. Bioseparations Science and Engineering, 2nd ed.; Oxford University Press: New York, NY, USA, 2003. [Google Scholar]
- Grob, P.; Huber, M.; Walla, B.; Hermann, J.; Janowski, R.; Niessing, D.; Hekmat, D.; Weuster-Botz, D. Crystal Contact Engineering Enables Efficient Capture and Purification of an Oxidoreductase by Technical Crystallization. Biotechnol. J. 2020, e2000010. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, P.; Hermann, J.; Li, J.; Krautenbacher, A.; Klöpfer, K.; Hekmat, D.; Weuster-Botz, D. Rational Crystal Contact Engineering of Lactobacillus brevis Alcohol Dehydrogenase To Promote Technical Protein Crystallization. Cryst. Growth Des. 2019, 19, 2380–2387. [Google Scholar] [CrossRef]
- Hermann, J.; Bischoff, D.; Grob, P.; Janowski, R.; Hekmat, D.; Niessing, D.; Zacharias, M.; Weuster-Botz, D. Controlling Protein Crystallization by Free Energy Guided Design of Interactions at Crystal Contacts. Crystals 2021, 11, 588. [Google Scholar] [CrossRef]
- Hermann, J.; Nowotny, P.; Schrader, T.E.; Biggel, P.; Hekmat, D.; Weuster-Botz, D. Neutron and X-ray crystal structures of Lactobacillus brevis alcohol dehydrogenase reveal new insights into hydrogen-bonding pathways. Acta Crystallogr. Sect. F 2018, 74, 754–764. [Google Scholar] [CrossRef]
- Noey, E.L.; Tibrewal, N.; Jiménez-Osés, G.; Osuna, S.; Park, J.; Bond, C.M.; Cascio, D.; Liang, J.; Zhang, X.; Huisman, G.W.; et al. Origins of stereoselectivity in evolved ketoreductases. Proc. Natl. Acad. Sci. USA 2015, 112, E7065–E7072. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Baumann, U.; Reymond, J.L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004, 32, e115. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.H.C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Smejkal, B.; Agrawal, N.J.; Helk, B.; Schulz, H.; Giffard, M.; Mechelke, M.; Ortner, F.; Heckmeier, P.; Trout, B.L.; Hekmat, D. Fast and scalable purification of a therapeutic full-length antibody based on process crystallization. Biotechnol. Bioeng. 2013, 110, 2452–2461. [Google Scholar] [CrossRef]
- Hebel, D.; Ürdingen, M.; Hekmat, D.; Weuster-Botz, D. Development and Scale up of High-Yield Crystallization Processes of Lysozyme and Lipase Using Additives. Cryst. Growth Des. 2013, 13, 2499–2506. [Google Scholar] [CrossRef]
- Teng, C.C.; Man, H.T. Simple reflection technique for measuring the electro-optic coefficient of poled polymers. Appl. Phys. Lett. 1990, 56, 1734–1736. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Schmidt, S.; Havekost, D.; Kaiser, K.; Kauling, J.; Henzler, H.-J. Crystallization for the Downstream Processing of Proteins. Eng. Life Sci. 2005, 5, 273–276. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walla, B.; Bischoff, D.; Janowski, R.; von den Eichen, N.; Niessing, D.; Weuster-Botz, D. Transfer of a Rational Crystal Contact Engineering Strategy between Diverse Alcohol Dehydrogenases. Crystals 2021, 11, 975. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080975
Walla B, Bischoff D, Janowski R, von den Eichen N, Niessing D, Weuster-Botz D. Transfer of a Rational Crystal Contact Engineering Strategy between Diverse Alcohol Dehydrogenases. Crystals. 2021; 11(8):975. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080975
Chicago/Turabian StyleWalla, Brigitte, Daniel Bischoff, Robert Janowski, Nikolas von den Eichen, Dierk Niessing, and Dirk Weuster-Botz. 2021. "Transfer of a Rational Crystal Contact Engineering Strategy between Diverse Alcohol Dehydrogenases" Crystals 11, no. 8: 975. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11080975