
Solvent-Induced Cobalt(II) Cyanoguanidine Bromides: Syntheses, Crystal Structure, Optical and Magnetic Properties



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthetic Procedures: All Reagents Were Commercially Bought (Alfa, Kandel, Rheinland-Pfalz, Germany) and Used without Further Purification
2.2. PXRD Measurements
2.3. CHN Measurements and Thermogravimetric Analysis (TGA)
2.4. IR Spectra
2.5. UV-Vis Spectra
2.6. Single-Crystal Diffraction
2.7. Magnetic Measurement
3. Results and Discussion
3.1. Stability and Structure
3.2. UV−Vis and IR Optical Properties
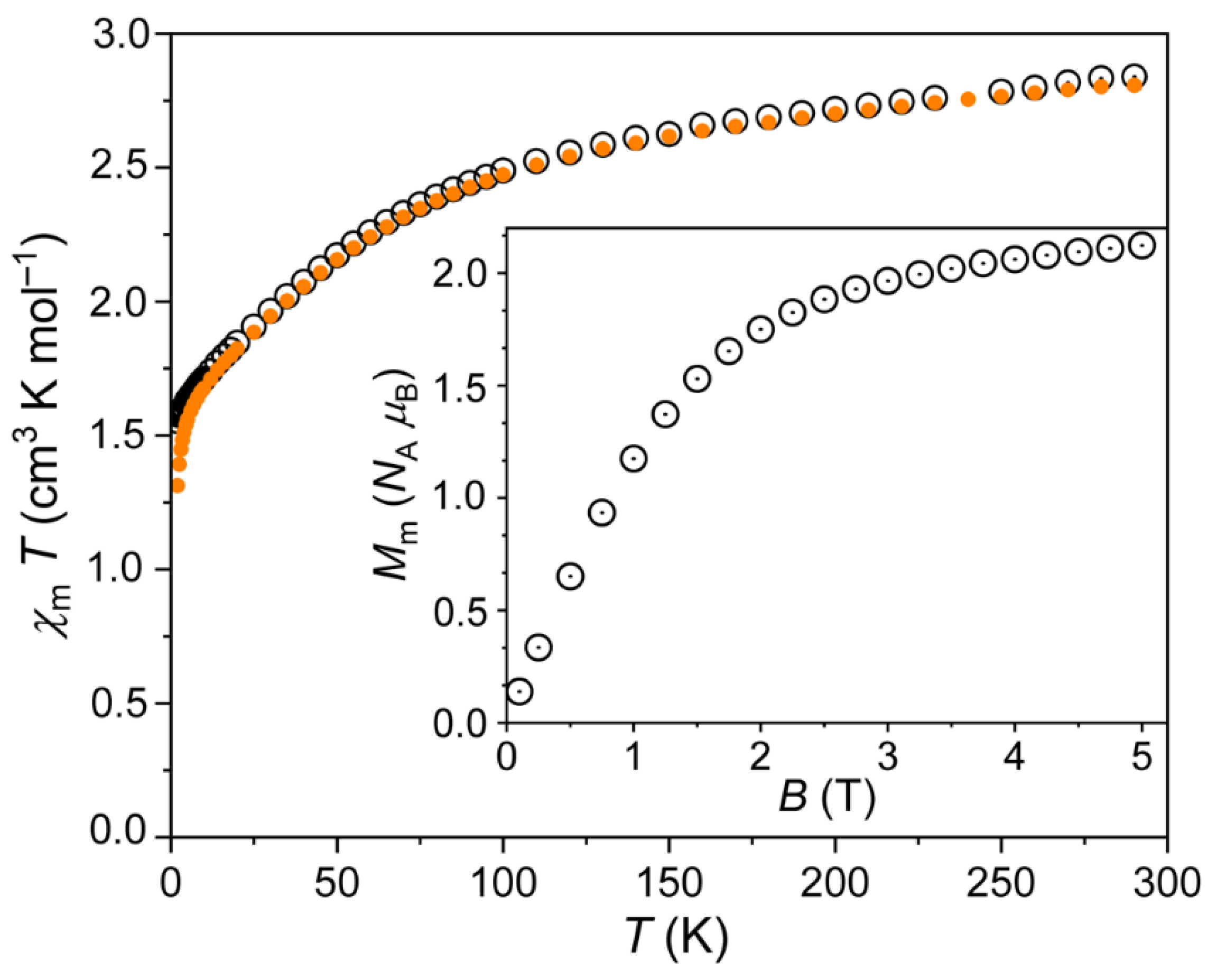
3.3. Magnetic Properties
4. Conclusions
- Synopsis Two solvent-induced Co(II) cyanoguanidine bromides (1 and 2) were synthesized in water and methanol, respectively. X-ray crystal structure, UV-vis absorption spectra and IR spectra analysis highlighted similarities and differences between these two compounds. Magnetic susceptibility measurements were consistent with the presence of cobalt(II) and evidence for weak antiferromagnetic interactions observed in 2.
- TOC Figure

Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pickardt, J.; Kühn, B. Crystal structure of dichlorobis(cyanoguanidine)zinc(II), Zn(C2N4H4)2Cl2. Z. Kristallogr. Cryst. Mater. 1995, 210, 901. [Google Scholar] [CrossRef]
- Fowkes, A.; Harrison, W.T.A. A monoclinic polymorph of dichlorobis-(cyanoguanidine)zinc(II). Acta Cryst. E 2005, 61, m2021–m2022. [Google Scholar] [CrossRef]
- Ritche, L.K.; Harrison, W.T.A. catena-poly[[dichlorozinc(II)]-μ-cyanoguanidine]. Acta Cryst. E 2007, 63, m617–m618. [Google Scholar] [CrossRef]
- Bessler, K.E.; de Sousa, A.T.; Deflon, V.M.; Niquet, E. Silver complexes with cyanoguanidine: Preparation and crystal structures of [Ag(cgn)2]F and [Ag(cgn)2][BF4]. Z. Anorg. Allg. Chem. 2003, 629, 1091–1095. [Google Scholar] [CrossRef]
- Zhang, J.; Corkett, A.J.; van Leusen, J.; Nelson, R.; Dronskowski, R. Cu(C2N4H4)2Br2·2H2O: An antiferromagnetic cyanoguanidine coordination compound and its characterization. Z. Naturforsch. B 2022, 77, 411–417. [Google Scholar] [CrossRef]
- Dronskowski, R.; Liu, X.-H. Bis(cyanoguanidine)silver(I) nitrate–cyanoguanidine (1/1). Acta Cryst. C 2003, 59, m243–m245. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Corkett, A.J.; Luo, D.; Dronskowski, R. Silver cyanoguanidine nitrate hydrate: Ag(C2N4H4)NO3·1⁄2H2O, a cyanoguanidine compound coordinating by an inner nitrogen atom. Inorganics 2020, 8, 64. [Google Scholar] [CrossRef]
- Pickardt, J.; Kühn, B. Metallkomplexe mit Cyanoguanidin als Liganden: Kristallstrukturen von [Cd(cnge)2F2]·3H2O, Cd(cnge)2Br2 und Hg(cnge)Cl2 (enge = Cyanoguanidin). Z. Naturforsch. B 1996, 51, 1701–1706. [Google Scholar] [CrossRef]
- Irzoqi, A.A.; Salih, M.M. Synthesis and characterization complexes of Ni(II) that contain cyanoguanidine and phosphines Ligands. Tikrit J. Pure Sci. 2018, 23, 93–101. [Google Scholar] [CrossRef]
- Beilstein, F.; Geuther, A. Ueber das Natriumamid. Liebigs Ann. Chem. 1858, 108, 88–102. [Google Scholar] [CrossRef]
- Barrio, J.; Volokh, M.; Shalom, M. Polymeric carbon nitrides and related metal-free materials for energy and environmental applications. J. Mater. Chem. A 2020, 8, 11075–11116. [Google Scholar] [CrossRef]
- Williams, P.A.; Ferrer, E.G.; Baeza, N.; Piro, O.E.; Castellano, E.E.; Baran, E.J. Transition metal promoted addition of methanol to cyanoguanidine. Molecular structure and properties of the generated Copper (II) and Nickel (II) complexes. Z. Anorg. Allg. Chem. 2005, 631, 1502–1506. [Google Scholar] [CrossRef]
- Kose, M.; Duman, S.E.; McKee, V.; Akyol, I.; Kurtoglu, M. Hydrogen bond directed 1D to 3D structures of square-planar Ni(II) complexes and their antimicrobial studies. Inorg. Chim. Acta 2017, 462, 281–288. [Google Scholar] [CrossRef]
- Bamberger, E.E. Ueber Dicyandiamid(I). Ber. Dtsch. Chem. Ges. 1883, 16, 1074–1078. [Google Scholar] [CrossRef]
- Pohl, F. Zur Kenntnis des Dicyandiamids. J. Prakt. Chem. 1908, 77, 533–548. [Google Scholar] [CrossRef]
- Bourosh, P.; Bologa, O.; Deseatnic-Ciloci, A.; Tiurina, J.; Bulhac, I. Synthesis, structure, and biological properties of mixed cobalt (III) dioximates with guanidine derivatives. Russ. J. Coord. Chem. 2017, 43, 591–599. [Google Scholar] [CrossRef]
- Aremu, J.A.; Durosinmi, L.M.; Oluyemi, E.A.; Ojo, I.A.O. Synthesis and Characterization of Guanidine derivatives of Benzothiazole and their Cobalt(II), Nickel(II), Zinc(II), Copper(II) and Iron(II) Complexes. IOSR J. Appl. Chem. 2018, 11, 53–71. [Google Scholar]
- Uzǎrević, K.; Halasz, I.; Frisčǐc, T. Real-time and in situ monitoring of mechanochemical reactions: A new playground for all chemists. J. Phys. Chem. Lett. 2015, 6, 4129–4140. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal gazing: Structure prediction and polymorphism. Science 1997, 278, 404–405. [Google Scholar] [CrossRef]
- Nimax, P.R.; Reimann, D.; Sünkel, K. Solvent effects on the crystal structure of silver pentacyanocyclopentadienide: Supramolecular isomerism and solvent coordination. Dalton Trans. 2018, 47, 8476–8482. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.K.; He, X.N.; Yan, H.B.; Lv, Z.W.; Chen, X.; Zhao, C.Y.; Tang, X.L. Synthesis, structural characterization and solvent effect of copper(II) complexes with a variational multidentate Salen-type ligand with bisoxime groups. Polyhedron 2009, 28, 1419–1428. [Google Scholar] [CrossRef]
- Bi, W.Y.; Lü, X.Q.; Chai, W.L.; Jin, W.J.; Song, J.R.; Wong, W.K. Synthesis, structure and near-infrared (NIR) luminescence of three solvent-induced pseudo-polymorphic complexes from a bimetallic Zn–Nd Schiff-base molecular unit. Inorg. Chem. Commun. 2008, 11, 1316–1319. [Google Scholar] [CrossRef]
- Hchicha, K.; Korb, M.; Kliuikov, A.; Čižmár, E.; Naïli, H. A cobalt(II)-based semiconductor complex with two-channel slow magnetic relaxation. J. Magn. Magn. Mater. 2021, 536, 168140. [Google Scholar] [CrossRef]
- Brader, M.L.; Kaarsholm, N.C.; Harnung, S.E.; Dunn, M.F. Ligand perturbation effects on a pseudotetrahedral Co(II)(His)3-Ligand Site: A Magnetic circular dichroism study of the Co(II)-substituted insulin hexamer. J. Biol. Chem. 1997, 272, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Rueff, J.M.; Masciocchi, N.; Rabu, P.; Sironi, A.; Skoulios, A. Structure and magnetism of a polycrystalline transition metal soap− CoII[OOC (CH2)10COO](H2O)2. Eur. J. Inorg. Chem. 2001, 11, 2843–2848. [Google Scholar] [CrossRef]
- Baker, W.A., Jr.; Daniels, M. Dicyandiamide-copper (II) complexes and the metal ion catalysed addition of alcohols to a nitrile. J. Inorg. Nucl. Chem. 1963, 25, 1194–1196. [Google Scholar] [CrossRef]
- Quinzani, O.V.; Tarulli, S.; Marcos, C.; Granda, S.G.; Baran, E.J. Crystal structure, spectroscopic and thermal behaviour of bis (saccharinato) tetrakis (pyridine) nickel (II) dipyridine. Z. Anorg. Allg. Chem. 1999, 625, 1848–1852. [Google Scholar] [CrossRef]
- Sawinski, P.K.; Meven, M.; Englert, U.; Dronskowski, R. Single-Crystal Neutron Diffraction Study on Guanidine, CN3H5. Cryst. Growth Des. 2013, 13, 1730–1735. [Google Scholar] [CrossRef]
- Ceccarelli, C.; Jeffrey, G.A.; Taylor, R. A survey of OH⋯O hydrogen bond geometries determined by neutron diffraction. J. Mol. Struct. 1981, 70, 255–271. [Google Scholar] [CrossRef]
- Gašparovič, L.; Koreňová, Z.; Jelemenský, Ľ. Kinetic study of wood chips decomposition by TGA. Chem. Pap. 2010, 64, 174–181. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectroscopy. In Studies in Physical and Theoretical Chemistry, 2nd ed.; Elsevier: New York, NY, USA, 1984; Volume 33, pp. 480–490. [Google Scholar]
- Papadopoulos, C.; Cristóvão, B.; Ferenc, W.; Hatzidimitriou, A.; Ciprioti, S.V.; Risoluti, R.; Lalia-Kantouri, M. Thermoanalytical, magnetic and structural investigation of neutral Co(II) complexes with 2,20-dipyridylamine and salicylaldehydes. J. Therm. Anal. Calorim. 2016, 123, 717–729. [Google Scholar] [CrossRef]
- Witschard, G.; Griffin, C.E. Infrared absorption characteristics of alkyl and aryl substituted phosphonium salts. Spectrochim. Acta 1963, 19, 1905–1910. [Google Scholar] [CrossRef]
- Sukhorukov, B.I.; Finkel’shtein, A.I. Optical Investigation of the Molecular Structure of Cyanamide and its Derivatives. 1. The Molecular Structure of Dicyandiamide. Opt. Spektrosk. 1959, 6, 637–641. [Google Scholar]
- Sheludyakova, L.A.; Sobolev, E.V.; Kozhevina, L.I. Concerning the nature of the doublet at 2200 cm−1 in the vibrational spectra of cyanoguanidine. J. Appl. Spectrosc. 1991, 55, 661–665. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared Spectra of Inorganic and Coordination Compounds; Wiley: New York, NY, USA, 1963. [Google Scholar]
- Begley, M.J.; Hubberstey, P.; Walton, P.H. Bis(μ-2-cyanoguanidine)-bis ((2-cyanoguanidine) copper (I)), a planar dimeric cation containing co-ordinatively unsaturated copper (I). J. Chem. Soc. Dalton Trans. 1995, 6, 957–962. [Google Scholar] [CrossRef]
- Lueken, H.; Magnetochemie, B. Teubner Stuttgart. Leipzig 1999. [Google Scholar] [CrossRef]
- Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 1959, 10, 87–98. [Google Scholar] [CrossRef]
- van Leeuwen, G.C.M. Investigation of the angular dependency of Co-O-Co superexchange by means of magnetic susceptibility measurements in dilute systems. Rec. Trav. Chim. Pays-Bas 1973, 92, 1249–1266. [Google Scholar] [CrossRef]
- Weihe, H.; Güdel, H.U. Angular and distance dependence of the magnetic properties of oxo-bridged iron (III) dimers. JACS 1997, 119, 6539–6543. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Corkett, A.J.; van Leusen, J.; Englert, U.; Dronskowski, R. Solvent-Induced Cobalt(II) Cyanoguanidine Bromides: Syntheses, Crystal Structure, Optical and Magnetic Properties. Crystals 2022, 12, 1377. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12101377
Zhang J, Corkett AJ, van Leusen J, Englert U, Dronskowski R. Solvent-Induced Cobalt(II) Cyanoguanidine Bromides: Syntheses, Crystal Structure, Optical and Magnetic Properties. Crystals. 2022; 12(10):1377. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12101377
Chicago/Turabian StyleZhang, Jing, Alex. J. Corkett, Jan van Leusen, Ulli Englert, and Richard Dronskowski. 2022. "Solvent-Induced Cobalt(II) Cyanoguanidine Bromides: Syntheses, Crystal Structure, Optical and Magnetic Properties" Crystals 12, no. 10: 1377. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12101377