
An Enhanced Oxidation of Formate on PtNi/Ni Foam Catalyst in an Alkaline Medium


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
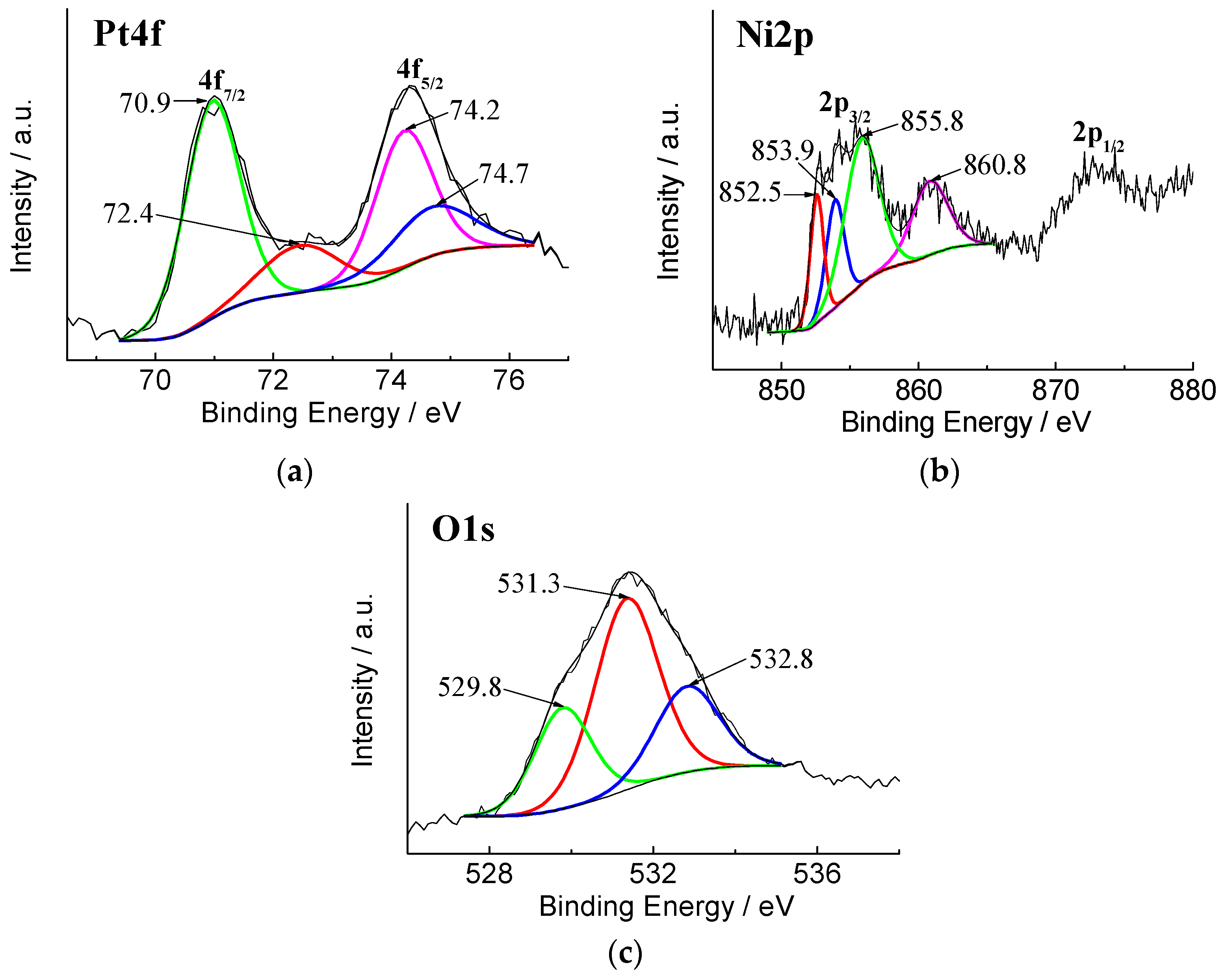
3.1. Characterization of the Electrodes
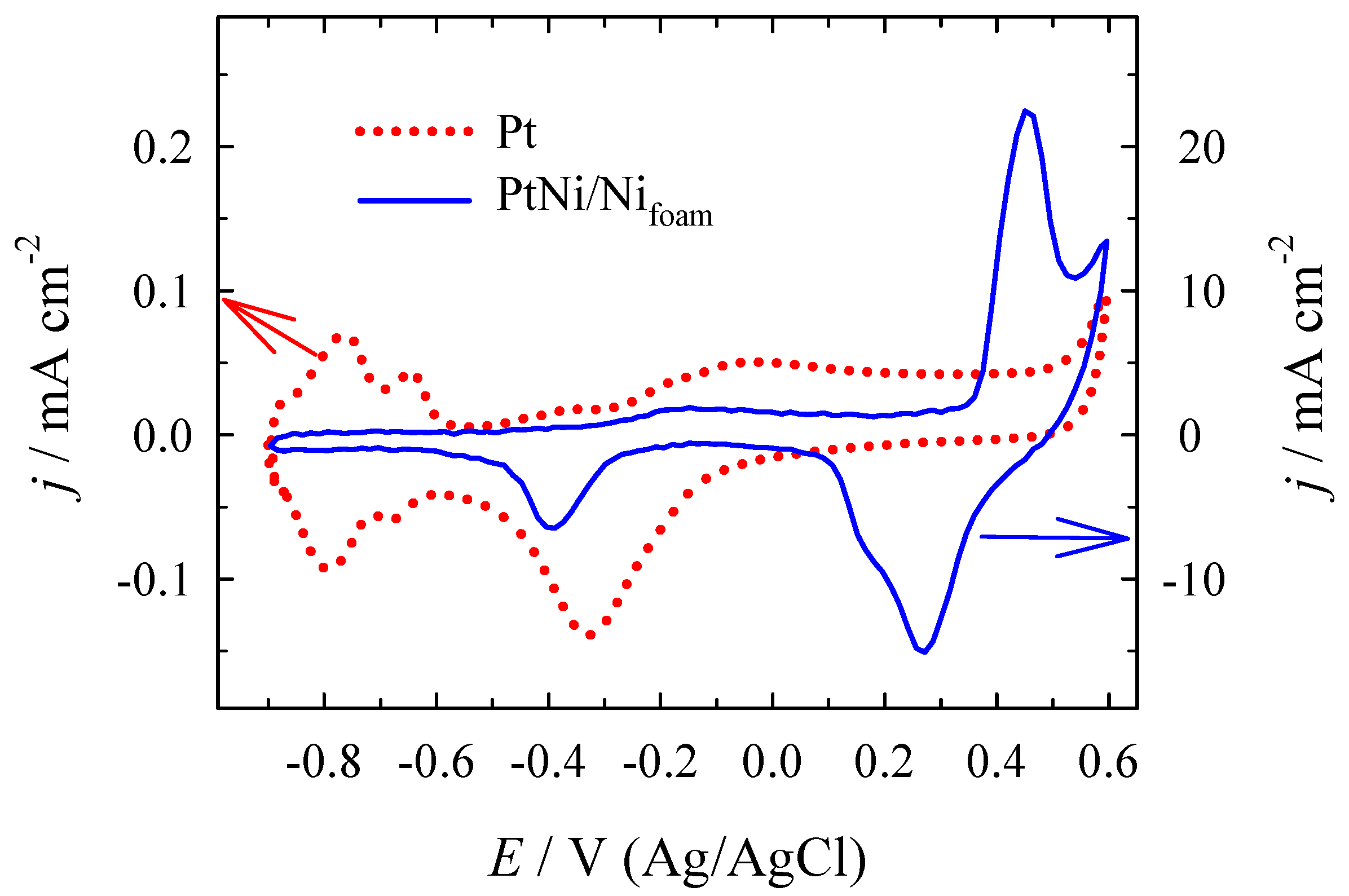
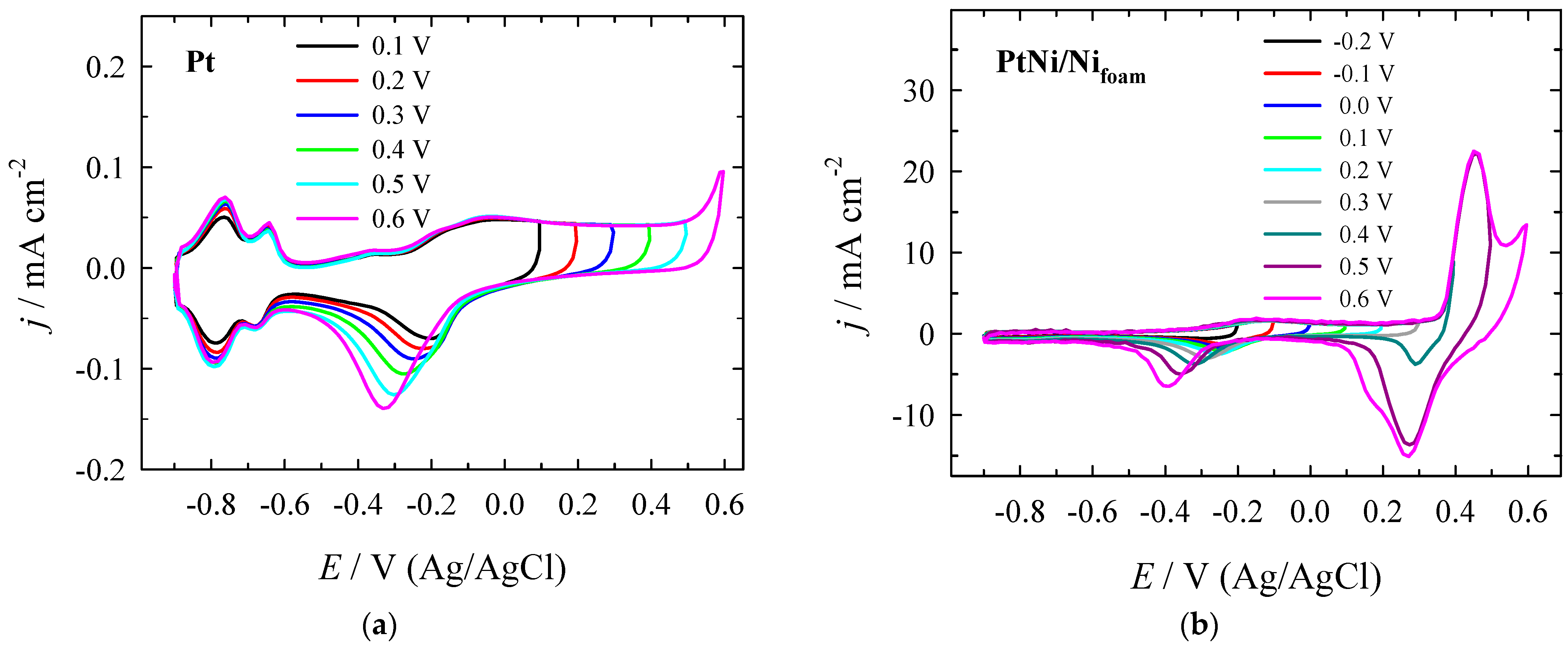
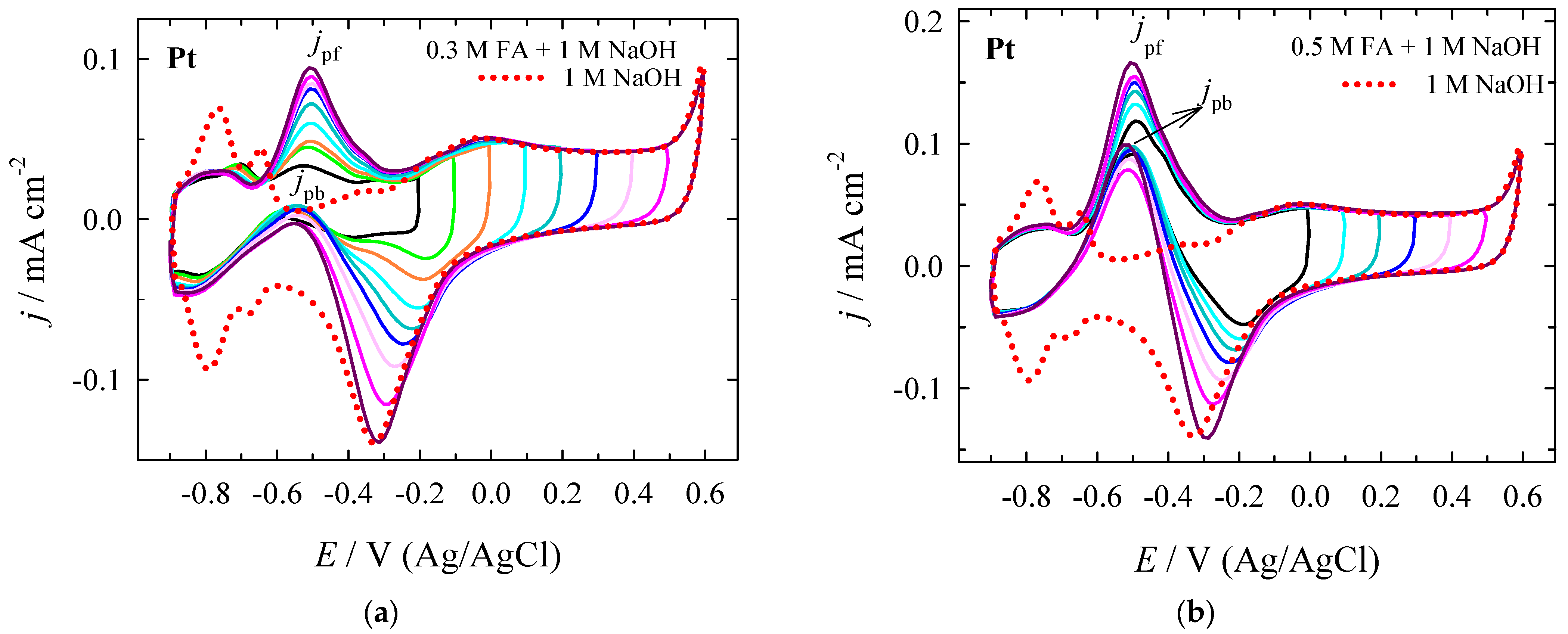
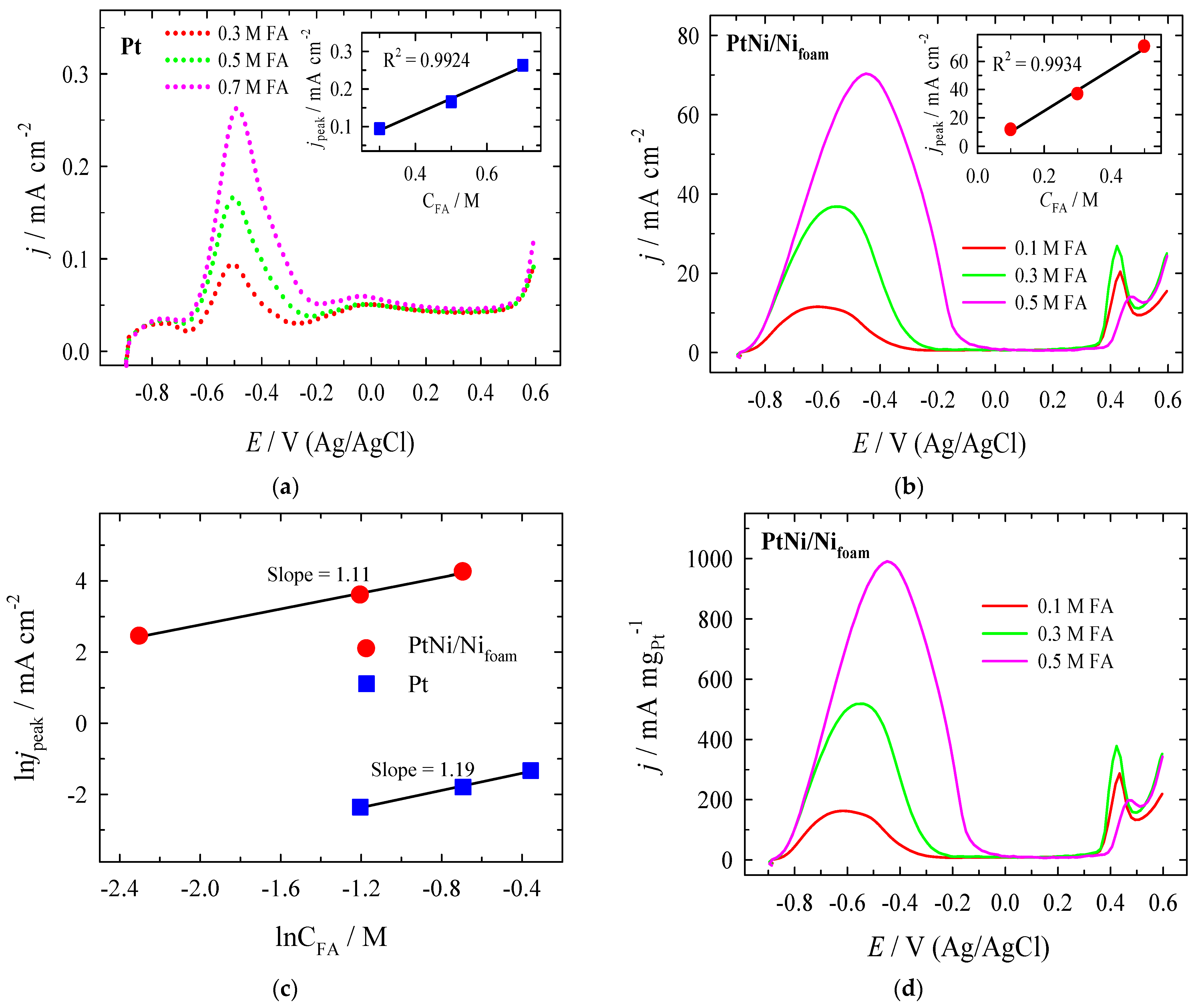
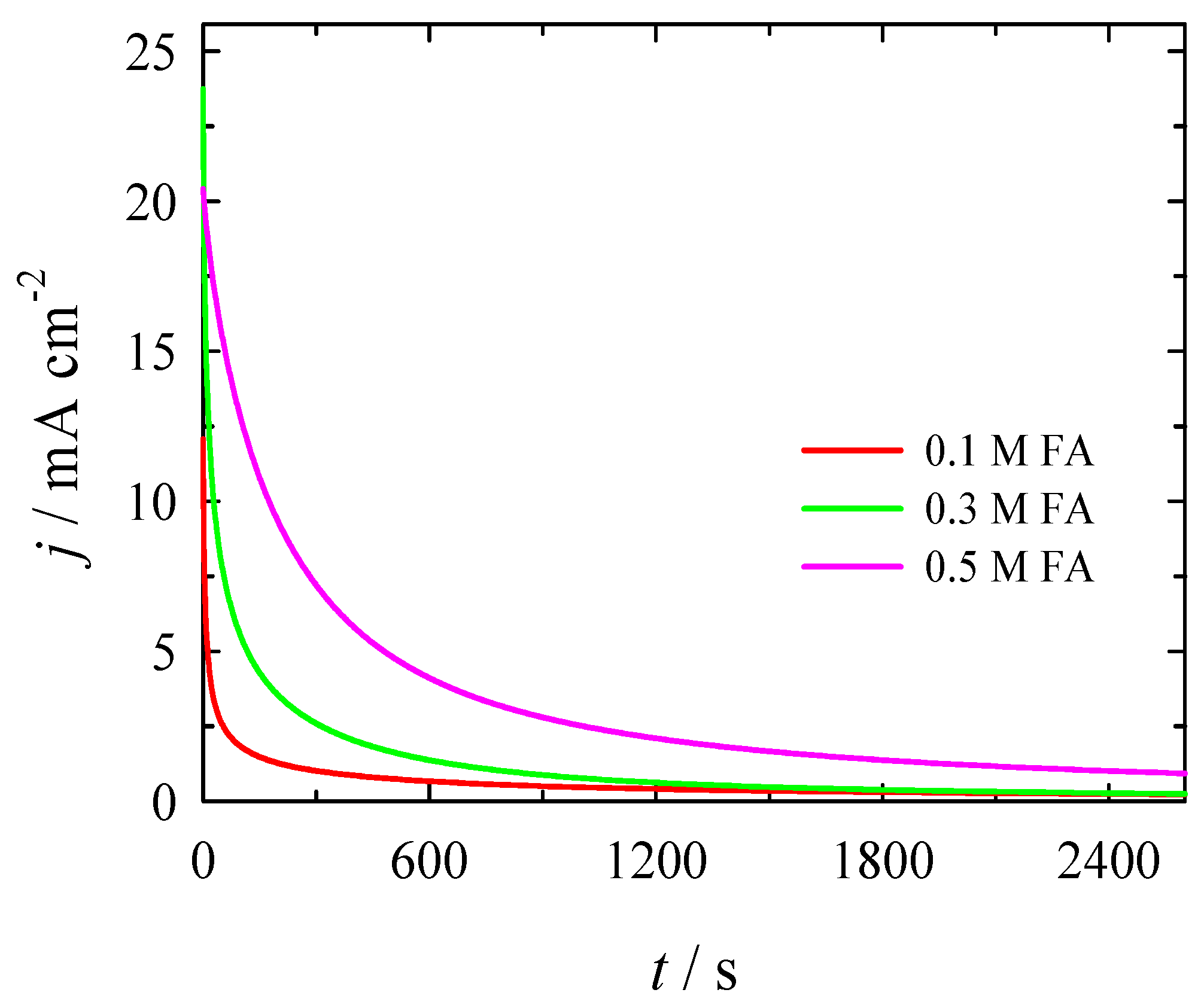
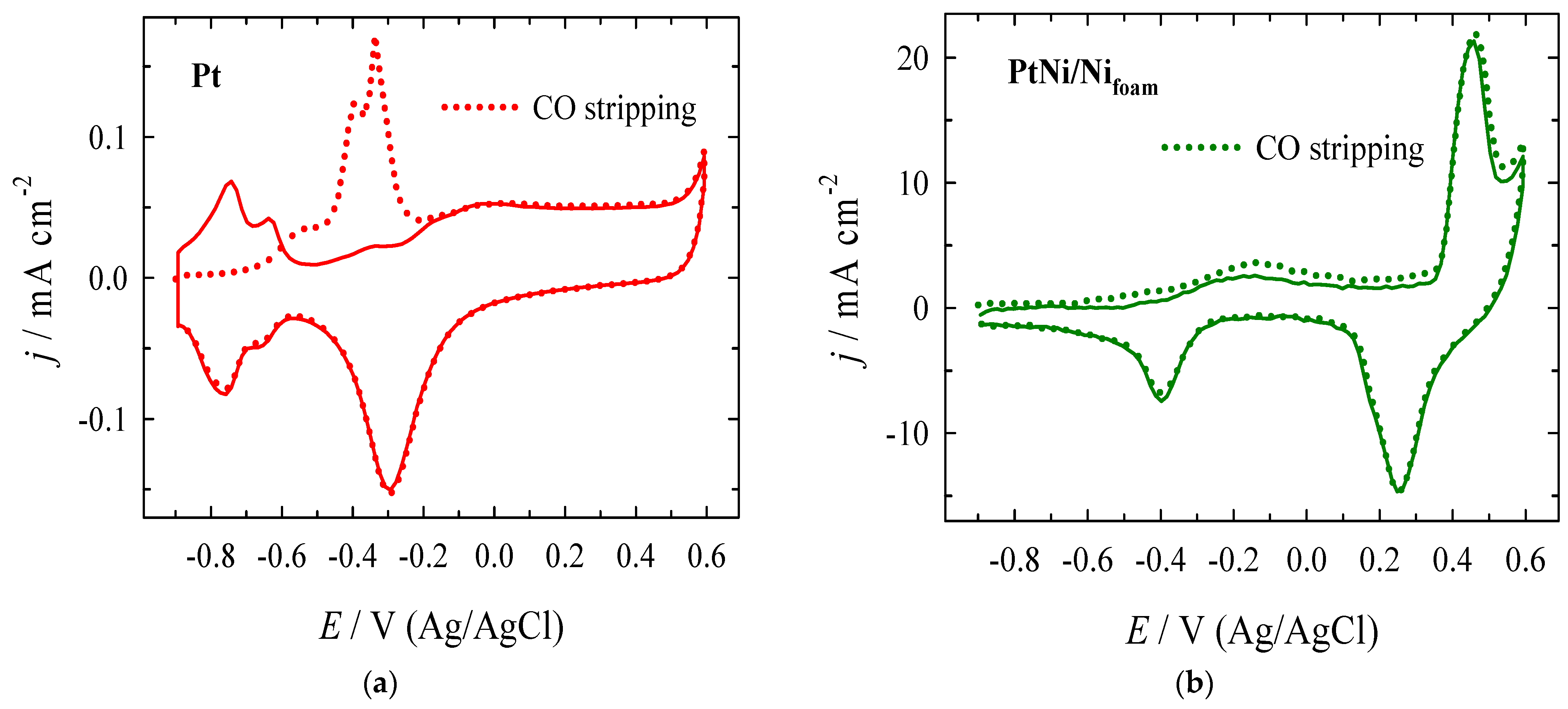
3.2. Electrocatalytic Evaluation for Formate Oxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, Z.; Chen, W. Recent advances in formic acid electro-oxidation: From the fundamental mechanism to electrocatalysts. Nanoscale Adv. 2021, 3, 94–105. [Google Scholar] [CrossRef]
- Folkman, S.J.; González-Cobos, J.; Giancola, S.; Sánchez-Molina, I.; Galán-Mascarós, J.R. Benchmarking Catalysts for Formic Acid/Formate Electrooxidation. Molecules 2021, 26, 4756. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Zhang, J.; Chen, K.; Deng, S.; Wang, D. Recent Progress of Palladium-Based Electrocatalysts for the Formic Acid Oxidation Reaction. Energy Fuels 2020, 34, 9137–9153. [Google Scholar] [CrossRef]
- An, L.; Chen, R. Direct formate fuel cells: A review. J. Power Sources 2016, 320, 127–139. [Google Scholar] [CrossRef]
- Uwitonze, N.; Chen, Y.X. The study of Pt and Pd based anode catalysis for formic acid fuel cell. Chem. Sci. J. 2017, 8, 167. [Google Scholar] [CrossRef]
- Mao, H.; Huang, T.; Yu, A. Electrochemical surface modification on CuPdAu/C with extraordinary behavior toward formic acid/ formate oxidation. Int. J. Hydrogen Energy 2016, 41, 13190–13196. [Google Scholar] [CrossRef]
- Ding, J.; Liu, Z.; Liu, X.; Liu, J.; Deng, Y.; Han, X.; Zhong, C.; Hu, W. Mesoporous decoration of freestanding palladium nanotube arrays boosts the electrocatalysis capabilities toward formic acid and formate oxidation. Adv. Energy Mater. 2019, 9, 1900955. [Google Scholar] [CrossRef]
- Wang, J.; Chen, F.; Jin, Y.; Guo, L.; Gong, X.; Wanga, X.; Johnston, R.L. In situ high-potential-driven surface restructuring of ternary AgPd-Pt dilute aerogels with record-high performance improvement for formate oxidation electrocatalysis. Nanoscale 2019, 11, 14174–14185. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, F.; Tang, Q.; Guo, L.; Gebremarian, T.; Jin, T.; Liu, H.; Kou, B.; Li, Z.; Bian, W. Transition from core-shell to janus segregation pattern in AgPd nanoalloy by Ni doping for the formate oxidation. Appl. Catal. B Environ. 2020, 270, 118861. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, F.; Guo, L.; Jin, T.; Liu, H.; Wang, X.; Gong, X.; Liu, Y. Nanoalloying effects on the catalytic activity of the formate oxidation reaction over AgPd and AgCuPd aerogels. J. Mater. Chem. A 2019, 7, 16122–16135. [Google Scholar] [CrossRef]
- Chen, Y.; Niu, H.; Feng, Y.; Wu, J.; Wang, A.; Huang, H.; Feng, J. Three-dimensional hierarchical urchin-like PdCuPt nanoassembles with zigzag branches: A highly efficient and durable electrocatalyst for formic acid oxidation reaction. Appl. Surf. Sci. 2020, 510, 145480. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, F.; Jin, T.; Guo, L.; Wang, Q.; Liu, H. Alloying in inverse CeO2/Pd nanoparticles to enhance the electrocatalytic activity for the formate oxidation reaction. J. Mater. Chem. A 2019, 7, 22996–23007. [Google Scholar] [CrossRef]
- Hwang, E.; Park, H.; Kim, H.; Ahn, S.H.; Kim, S.K. Electrochemically fabricated Pd–In catalysts for carbon dioxide- formate/formic acid inter-conversion. Bull. Korean Chem. Soc. 2017, 38, 607–613. [Google Scholar] [CrossRef]
- Hong, S.; Chung, S.; Park, J.; Hwang, J.; Lee, C.; Uhm, S.; Bong, S.; Lee, J. Contribution of interstitial boron in a boron-incorporated palladium catalyst toward formate oxidation in an alkaline direct formate fuel cell. ACS Catal. 2021, 11, 4722–4729. [Google Scholar] [CrossRef]
- Yang, L.; Li, G.; Chang, J.; Ge, J.; Liu, C.; Vladimir, F.; Wang, G.; Jin, Z.; Xing, W. Modification of palladium nanocrystals with single atom platinum via an electrochemical self-catalysis strategy for efficient formic acid electrooxidation. Appl. Catal. B 2020, 260, 118200. [Google Scholar] [CrossRef]
- da Silva, S.G.; Silva, J.C.M.; Buzzo, G.S.; Neto, A.O.; Assumpção, M.H.M.T. Use of PtAu/C electrocatalysts toward formate oxidation: Electrochemical and fuel cell considerations. Mater. Renew. Sustain. Energy 2016, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-H.; Liu, H.-M.; Bai, J.; Tian, X.L.; Xia, B.Y.; Zeng, J.-H.; Jiang, J.-X.; Chen, Y. Platinum-silver alloy nanoballoon nanoassemblies with super catalytic activity for the formate electrooxidation. ACS Appl. Energy Mater. 2018, 1, 1252–1258. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Dong, T.; Yuan, X.; Lv, L.; Wei, X.; Wang, J. Formate: A possible replacement for formic acid in fuel cells. Austral. J. Chem. 2017, 70, 757–763. [Google Scholar] [CrossRef]
- John, J.; Wang, H.; Rus, E.D.; Abruna, H.D. Mechanistic studies of formate oxidation on platinum in alkaline medium. J. Phys. Chem. C 2012, 116, 5810–5820. [Google Scholar] [CrossRef]
- Joo, J.; Uchida, T.; Cuesta, A.; Koper, M.T.M.; Osawa, M. Importance of acid-base equilibrium in electrocatalytic oxidation of formic acid on platinum. J. Am. Chem. Soc. 2013, 135, 9991–9994. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Feng, Y.; Sun, X.; He, Y. A sodium-ion-conducting direct formate fuel cell: Generating electricity and producing base. Angew. Chem. 2017, 129, 5828–5831. [Google Scholar] [CrossRef]
- Yu, X.; Manthiram, A. Catalyst-selective, scalable membraneless alkaline direct formate fuel cells. Appl. Catal. B Environ. 2015, 165, 63–67. [Google Scholar] [CrossRef]
- Themsirimongkon, S.; Pongpichayakul, N.; Fang, L.; Jakmunee, J.; Saipanya, S. New catalytic designs of Pt on carbon nanotube-nickel-carbon black for enhancement of methanol and formic acid oxidation. J. Electroanal. Chem. 2020, 876, 114518. [Google Scholar] [CrossRef]
- Kiani, M.; Zhang, J.; Luo, Y.; Chen, Y.; Chen, J.; Fan, J.; Wang, G.; Wang, R. Facile synthesis and enhanced catalytic activity of electrochemically dealloyed platinum-nickel nanoparticles towards formic acid electro-oxidation. J. Energy Chem. 2019, 35, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.-W.; Zhang, Z.-C.; Liao, H.-G.; Gong, Y.; Gu, L.; Qu, X.-M.; You, L.-X.; Liu, S.; Huang, L.; Tian, X.-C.; et al. Tuning Pt-skin to Ni-rich surface of Pt3Ni catalysts supported on porous carbon for enhanced oxygen-reduction reaction and formic electro-oxidation. Nano Energy 2016, 19, 198–209. [Google Scholar] [CrossRef]
- Pei, A.; Ruan, L.; Liu, B.; Chen, W.; Lin, S.; Chen, B.; Liu, Y.; Zhu, L.H.; Chen, B.H. Ultra-low Au decorated PtNi alloy nanoparticles on carbon for high-efficiency electro-oxidation of methanol and formic acid. Int. J. Hydrogen Energy 2020, 45, 22893–22905. [Google Scholar] [CrossRef]
- Lee, J.; Yoo, J.K.; Lee, H.; Kim, S.H.; Sohn, Y.; Rhee, C.K. Formic acid oxidation on Pt deposit model catalysts on Au: Single-layered Pt deposits, plateau-type Pt deposits, and conical Pt deposits. Electrochim. Acta 2019, 310, 38–44. [Google Scholar] [CrossRef]
- Perales-Rondón, J.V.; Ferre-Vilaplana, A.; Feliu, J.M.; Herrero, E. Oxidation mechanism of formic acid on the bismuth adatom-modified Pt(111) surface. J. Amer. Chem. Soc. 2014, 136, 13110–13113. [Google Scholar] [CrossRef]
- Wang, C.Y.; Yu, Z.-Y.; Li, G.; Song, Q.-T.; Li, G.; Luo, C.-X.; Yin, S.-H.; Lu, B.-A.; Xiao, C.; Xu, B.-B.; et al. Intermetallic PtBi nanoplates with high catalytic activity towards electrooxidation of formic acid and glycerol. ChemElectroChem 2020, 7, 239–245. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiao, M.; Huang, Y.; Zou, Y.; Liu, Z.; Tao, L.; Li, Y.; Dong, C.L.; Wang, S. In situ exfoliation and Pt deposition of antimonene for formic acid oxidation via a predominant dehydrogenation pathway. Research 2020, 2020, 5487237. [Google Scholar] [CrossRef] [Green Version]
- Sawy, E.N.E.; Pickup, P.G. Carbon monoxide and formic acid oxidation at Rh@Pt nanoparticles. Electrochim. Acta 2019, 302, 234–240. [Google Scholar] [CrossRef]
- El-Nagar, G.A.; Mohammad, A.M.; El-Deab, M.S.; El-Anadouli, B.E. Electrocatalysis of formic acid electro-oxidation at platinum nanoparticles modified surfaces with nickel and cobalt oxides nanostructures. In Progress in Clean Energy; Dincer, I., Colpan, C., Kizilkan, O., Ezan, M., Eds.; Springer International Publishing: Cham, Switzerland, 2015; Volume 1. [Google Scholar] [CrossRef]
- El-Nagar, G.A.; Mohammad, A.M. Enhanced electrocatalytic activity and stability of platinum, gold, and nickel oxide nanoparticles based ternary catalyst for formic acid electro-oxidation. Int. J. Hydrogen Energy 2014, 39, 11955–11962. [Google Scholar] [CrossRef]
- Mohammad, A.M.; El-Nagar, G.A.; Al-Akraa, I.M.; El-Deab, M.S.; El-Anadouli, B.E. Towards improving the catalytic activity and stability of platinum-based anodes in direct formic acid fuel cells. Int. J. Hydrogen Energy 2015, 40, 7808–7816. [Google Scholar] [CrossRef]
- El-Refaei, S.M.; El-Nagar, G.A.; Mohammad, A.M.; El-Deab, M.S.; El-Anadouli, B.E. Electrocatalytic activity of NiOx nanostructured modified electrodes towards oxidation of small organic molecules. In Springer Proceedings in Energy, Proceedings of the 2nd International Congress on Energy Efficiency and Energy Related Materials (ENEFM2014), 16–19 October 2014; Oral, A., Bahsi Oral, Z., Ozer, M., Eds.; Springer: Cham, Switzerland, 2015. [Google Scholar] [CrossRef]
- El-Nagar, G.A.; Mohammad, A.M.; El-Deab, M.S.; El-Anadouli, B.E. Propitious dendritic Cu2O-Pt nanostructured anodes for direct formic acid, Fuel cells. ACS Appl. Mater. Interf. 2017, 9, 19766–19772. [Google Scholar] [CrossRef]
- Ali Al-Qodami, B.; Farrag, H.H.; Sayed, S.Y.; Allam, N.K.; El-Anadouli, B.E.; Mohammad, A.M. Bifunctional tailoring of platinum surfaces with earth abundant iron oxide nanowires for boosted formic acid electro-oxidation. J. Nanotechnol. 2018, 2018, 4657040. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, A.M.; Al-Akraa, I.M.; El-Deab, M.S. Superior electrocatalysis of formic acid electrooxidation on a platinum, gold and manganese oxide nanoparticle-based ternary catalyst. Int. J. Hydrogen Energy 2018, 43, 139–149. [Google Scholar] [CrossRef]
- Asal, Y.M.; Al-Akraa, I.M.; Mohammad, A.M.; El-Deab, M.S. A competent simultaneously co-electrodeposited Pt-MnOx nanocatalyst for enhanced formic acid electro-oxidation. J. Taiwan Inst. Chem. Eng. 2019, 96, 169–175. [Google Scholar] [CrossRef]
- Santos, A.O.; Silva, J.C.M.; Antoniassi, E.R.M.; Ponzio, A.; Alves, O.C. The formate electrooxidation on Pt/C and PtSnO2/C nanoparticles in alkaline media: The effect of morphology and SnO2 on the platinum catalytic activity. Int. J. Hydrogen Energy 2020, 45, 33895–33905. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Chang, K.-C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni Co, Fe, Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef]
- El-Nagar, G.A.; Mohammad, A.M.; El-Deab, M.S.; El-Anadouli, B.E. Facilitated electrooxidation of formic acid at nickel oxide nanoparticles modified electrodes. J. Electrochem. Soc. 2012, 159, F249–F254. [Google Scholar] [CrossRef]
- El-Nagar, G.A.; Mohammad, A.M.; El-Deab, M.S.; El-Anadouli, B.E. Electrocatalysis by design: Enhanced electrooxidation of formic acid at platinum nanoparticles–nickel oxide nanoparticles binary catalysts. Electrochim. Acta 2013, 94, 62–71. [Google Scholar] [CrossRef]
- Fiameni, S.; Herraiz-Cardona, I.; Musiani, M.; Pérez-Herranz, V.; Vázquez-Gómez, L.; Verlato, E. The HER in alkaline media on Pt-modified three-dimensional Ni cathodes. Int. J. Hydrogen Energy 2012, 37, 10507–10516. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Y.; Shi, J.; Yang, M.; Rui, Y.; An, W.; Men, Y. Well-dispersed Pt nanodots interfaced with Ni(OH)2 on anodized nickel foam for efficient hydrogel evolution reaction. Int. J. Hydrogen Energy 2020, 45, 27067–27077. [Google Scholar] [CrossRef]
- Yuan, S.; Cui, L.; He, X.; Zhang, W.; Asefa, T. Nickel foam-supported Fe, Ni-Polyporphyrin microparticles: Efficient bifunctional catalysts for overall water splitting in alkaline media. Int. J. Hydrogen Energy 2020, 45, 28860–28869. [Google Scholar] [CrossRef]
- Wang, R.; Liu, H.; Zhang, K.; Zhang, G.; Lan, H.; Qu, J. Ni(II)/Ni(III) redox couple endows Ni foam-supported Ni2P with excellent capability for direct ammonia oxidation. Chem. Eng. J. 2021, 404, 126795. [Google Scholar] [CrossRef]
- Yuan, G.; Wang, L.; Zhang, X.; Wang, Q. Self-supported Pt nanoflakes-doped amorphous Ni(OH)2 on Ni foam composite electrode for efficient and stable methanol oxidation. J. Colloid Interf. Sci. 2019, 536, 189–195. [Google Scholar] [CrossRef]
- Sheng, S.; Song, Y.; Sha, L.; Ye, K.; Zhu, K.; Gao, Y.; Yan, J.; Wang, G.; Cao, D. Simultaneous hydrogen evolution and ethanol oxidation in alkaline medium via a self-supported bifunctional electrocatalyst of Ni-Fe phosphide/Ni foam. Appl. Surf. Sci. 2021, 561, 150080. [Google Scholar] [CrossRef]
- Tamašauskaitė-Tamašiūnaitė, L.; Zabielaitė, A.; Balčiūnaitė, A.; Šebeka, B.; Stanionienė, I.; Buzas, V.; Mačiulis, L.; Tumonis, L.; Norkus, E. Deposition of Pt nanoparticles on Ni foam via galvanic displacement. J. Electrochem. Soc. 2017, 164, D53–D56. [Google Scholar] [CrossRef]
- Song, C.; Wang, G.; Li, B.; Miao, C.; Ma, K.; Zhu, K.; Cheng, K.; Ye, K.; Yan, J.; Cao, D.; et al. A novel electrode of ternary CuNiPd nanoneedles decorated Ni foam and its catalytic activity toward NaBH4 electrooxidation. Electrochim. Acta 2019, 299, 395–404. [Google Scholar] [CrossRef]
- Lei, Y.; Liu, Y.; Fan, B.; Mao, L.; Yu, D.; Huang, Y.; Guo, F. Facile fabrication of hierarchically porous Ni foam@Ag-Ni catalyst for efficient hydrazine oxidation in alkaline medium. J. Taiwan Inst. Chem. Eng. 2019, 105, 75–84. [Google Scholar] [CrossRef]
- Chen, N.; Wang, Y.; Du, X.; Zhang, X. Facile synthesis of Ni doped CoWO4 nanoarrays grown on nickel foam substrates for efficient urea oxidation. Int. J. Hydrogen Energy 2021, 46, 25114–25120. [Google Scholar] [CrossRef]
- NIST X-ray Photoelectron Spectroscopy Database. NIST Standard Reference Database Number 20; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2000; p. 20899. [Google Scholar] [CrossRef]
- Igarashi, H.; Fujino, T.; Zhu, Y.; Uchida, H.; Watanabe, M. CO tolerance of Pt alloy electrocatalysts for polymer electrolyte fuel cells and the detoxification mechanism. Phys. Chem. Chem. Phys. 2001, 3, 306–314. [Google Scholar] [CrossRef]
- Zhao, Y.; Yifeng, E.; Fan, L.; Qiu, Y.; Yang, S. A new route for the electrodeposition of platinum-nickel alloy nanoparticles on multi-walled carbon nanotubes. Electrochim. Acta 2007, 52, 5873–5878. [Google Scholar] [CrossRef]
- Kim, K.S.; Winograd, N. X-ray photoelectron spectroscopic studies of nickel oxygen surfaces using oxygen and argon ion-bombardment. Surf. Sci. 1974, 43, 625–643. [Google Scholar] [CrossRef]
- Wittstok, G.; Strübing, A.; Szargan, R.; Werner, G. Glucose oxidation at bismuth-modified platinum electrodes. J. Electroanal. Chem. 1998, 444, 61–73. [Google Scholar] [CrossRef]
- Blasini, D.R.; Rochefort, D.; Fachini, E.; Aldena, L.R.; DiSalvo, J.F.; Cabrera, C.R.; Abruna, H.D. Surface composition of ordered intermetallic compounds PtBi and PtPb. Surf. Sci. 2006, 600, 2670–2680. [Google Scholar] [CrossRef]
- Eiler, K.; Krawiec, H.; Kozina, I.; Sort, J.; Pellicer, E. Electrochemical characterisation of multifunctional electrocatalytic mesoporous Ni-Pt thin films in alkaline and acidic media. Electrochim. Acta 2020, 359, 136952. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O.A. Real surface area measurements in electrochemistry. Pure Appl. Chem. 1991, 63, 711–734. [Google Scholar] [CrossRef]
- Daubinger, P.; Kieninger, J.; Unmussig, T.; Urban, G.A. Electrochemical characteristics of nanostructured platinum electrodes A cyclic voltammetry study. Phys. Chem. Chem. Phys. 2014, 16, 8392–8399. [Google Scholar] [CrossRef]
- Dražić, D.M.; Tripković, A.V.; Popović, K.D.; Lović, D.J. Kinetic and mechanistic study of hydroxyl ion electrosorption at the Pt(111) surface in alkaline media. J. Electroanal. Chem. 1999, 466, 155–164. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Ross, P.N.; Marković, N.M. Temperature-dependent surface electrochemistry on Pt single crystals in alkaline electrolyte: Part 1: CO oxidation. J. Phys. Chem. B 2001, 105, 12082–12086. [Google Scholar] [CrossRef]
- Tilak, B.V.; Conway, B.E.; Angerstain-Kozlowska, H. The real condition of oxidized Pt electrodes: Part III. Kinetic theory of formation and reduction of surface oxides. J. Electroanal. Chem. 1973, 48, 1–23. [Google Scholar] [CrossRef]
- Marković, N.M.; Schmidt, T.J.; Grgur, B.N.; Gasteiger, H.A.; Ross, P.N.; Behm, R.J., Jr. Effect of temperature on surface processes at the Pt(111)-liquid interface: Hydrogen adsorption, oxide formation, and CO oxidation. J. Phys. Chem. B. 1999, 103, 8568–8577. [Google Scholar] [CrossRef]
- Tripković, A.V.; Popović, J.K.D.; Lović, D.J. The influence of the oxygen-containing species on the electrooxidation of the C1–C4 alcohols at some platinum single crystal surfaces in alkaline solution. Electrochim. Acta 2001, 46, 3163–3173. [Google Scholar] [CrossRef]
- Marković, N.M.; Gasteiger, H.A.; Ross, P.N. Oxygen reduction on platinum low-index single-crystal surfaces in alkaline solution: Rotating ring diskPt(hkl) studies. J. Phys. Chem. B 1996, 100, 6715–6721. [Google Scholar] [CrossRef]
- Marković, N.M.; Sarraf, S.T.; Gasteiger, H.A.; Ross, P.N. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J. Chem. Soc., Faraday Trans. 1996, 92, 3719–3725. [Google Scholar] [CrossRef]
- Jiang, J.; Scott, J.; Wieckowski, A. Direct evidence of triple-path mechanism of formate electrooxidation on Pt black in alkaline media at varying temperature. Part I: The electrochemical studies. Electrochim. Acta 2013, 104, 124–133. [Google Scholar] [CrossRef]
- Yan, L.; Yao, S.; Chang, J.; Liu, C.; Xing, W. Pd oxides/hydrous oxides as highly efficient catalyst for formic acid electrooxidation. J. Power Sources 2014, 250, 128–133. [Google Scholar] [CrossRef]
- Okamoto, H.; Kon, W.; Mukouyama, Y. Five current peaks in voltammograms for oxidations of formic acid, formaldehyde, and methanol on platinum. J. Phys. Chem. B 2005, 109, 15659–15666. [Google Scholar] [CrossRef]
- Šljukić, B.; Milikić, J.; Santos, D.M.F.; Sequeira, C.A.C.; Macciò, D.; Saccone, A. Electrocatalytic performance of Pt–Dy alloys for direct borohydride fuel cells. J. Power Sources 2014, 272, 335–343. [Google Scholar] [CrossRef]
- Han, L.; Gonzįlez-Cobos, J.; Sįnchez-Molina, I.; Giancola, S.; Folkman, S.; Tang, P.; Heggen, M.; Dunin-Borkowski, R.; Arbiol, J.; Giménez, S.; et al. Cobalt hexacyanoferrate as a selective and high current density formate oxidation electrocatalyst. ACS Appl. Energy Mater. 2020, 3, 9198–9207. [Google Scholar] [CrossRef]
- Galvan, V.; Glass, D.E.; Baxter, A.F.; Surya Prakash, G.K. Reduced graphene oxide supported palladium nanoparticles for enhanced electrocatalytic activity toward formate electrooxidation in an alkaline medium. ACS Appl. Energy Mater. 2019, 2, 7104–7111. [Google Scholar] [CrossRef]
- Song, S.; Hwang, H.; Kim, J.W.; Lee, J. The effect of synthesis temperature on Pd-H catalyst structure for alkaline direct formate fuel cells. ECS Trans. 2018, 85, 149–158. [Google Scholar] [CrossRef]
- Sankar, S.; Anilkumar, G.M.; Tamaki, T.; Yamaguchi, T. Binary Pd–Ni nanoalloy particles over carbon support with superior alkaline formate fuel electrooxidation performance. ChemCatChem 2019, 11, 4731–4737. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Era, V | Epc, V | jpc, mA cm−2 | Epc, V | jpc, mA cm−2 | jpc PtNi/Nifoam/jpc Pt |
|---|---|---|---|---|---|
| Pt | PtNi/Nifoam | ||||
| +0.1 | −0.205 | −0.069 | −0.235 | −2.151 | 30.77 |
| +0.2 | −0.221 | −0.079 | −0.256 | −2.508 | 31.39 |
| +0.3 | −0.252 | −0.090 | −0.288 | −2.988 | 33.13 |
| +0.4 | −0.281 | −0.105 | −0.321 | −3.711 | 35.32 |
| +0.5 | −0.303 | −0.125 | −0.356 | −5.004 | 39.78 |
| +0.6 | −0.332 | −0.139 | −0.389 | −6.489 | 46.49 |
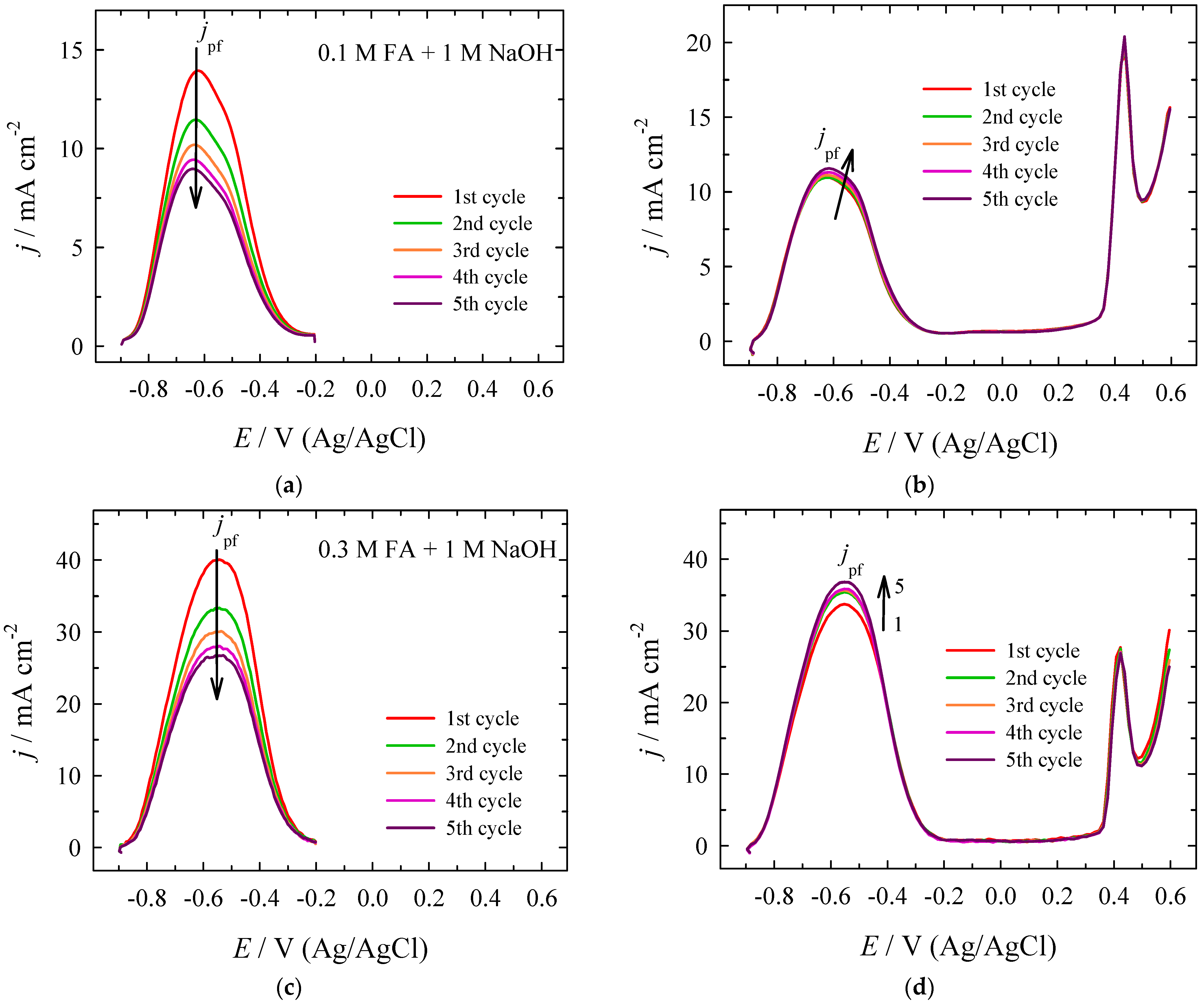
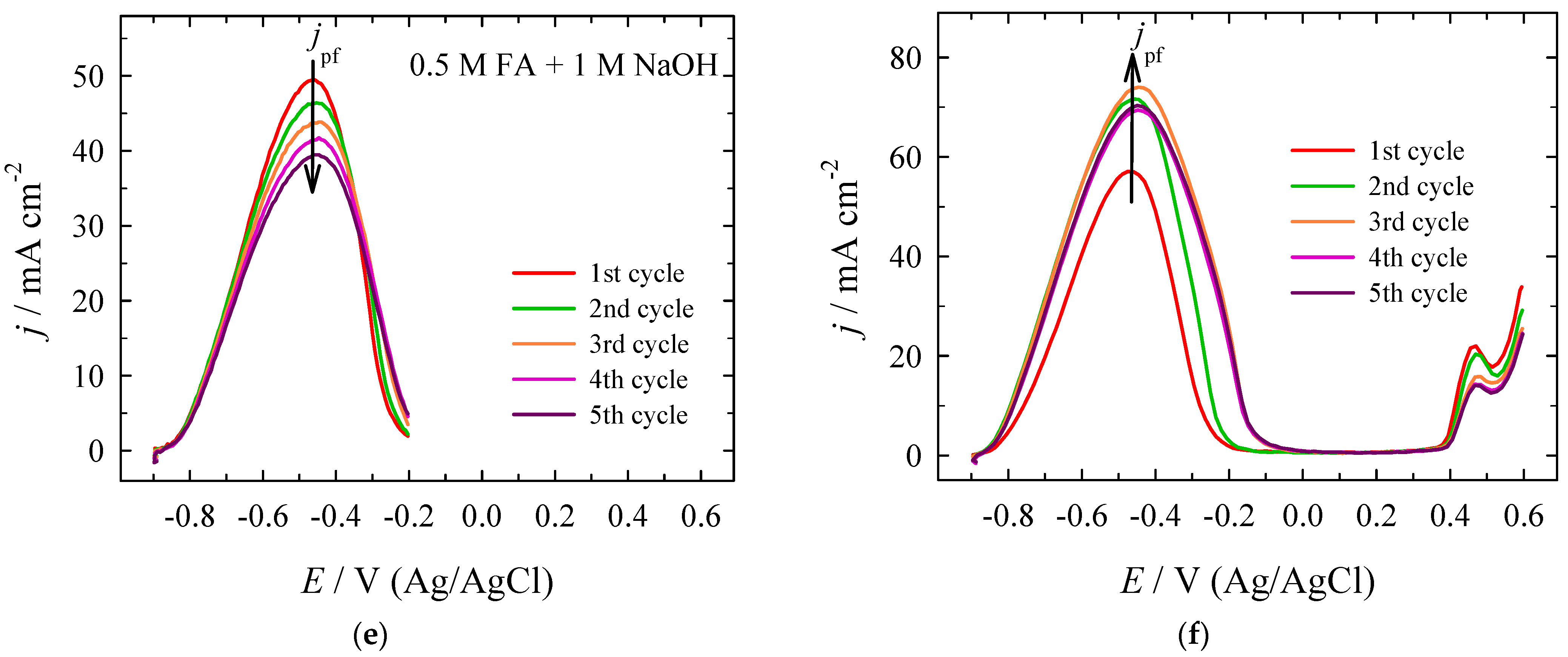
| Era, V | Epf, V | jpf, mA cm−2 | Epb, V | jpb, mA cm−2 | jpf/jpb | jpf at X V/jpf at +0.0 V |
|---|---|---|---|---|---|---|
| +0.0 | −0.493 | 0.1185 | −0.497 | 0.0913 | 1.30 | 1.00 |
| +0.1 | −0.490 | 0.1322 | −0.500 | 0.0976 | 1.35 | 1.12 |
| +0.2 | −0.500 | 0.1423 | −0.507 | 0.0978 | 1.46 | 1.20 |
| +0.3 | −0.495 | 0.1501 | −0.501 | 0.0943 | 1.59 | 1.27 |
| +0.4 | −0.499 | 0.1537 | −0.507 | 0.0875 | 1.76 | 1.30 |
| +0.5 | −0.493 | 0.1549 | −0.509 | 0.0785 | 1.97 | 1.31 |
| +0.6 | −0.507 | 0.1661 | −0.512 | 0.0989 | 1.68 | 1.40 |
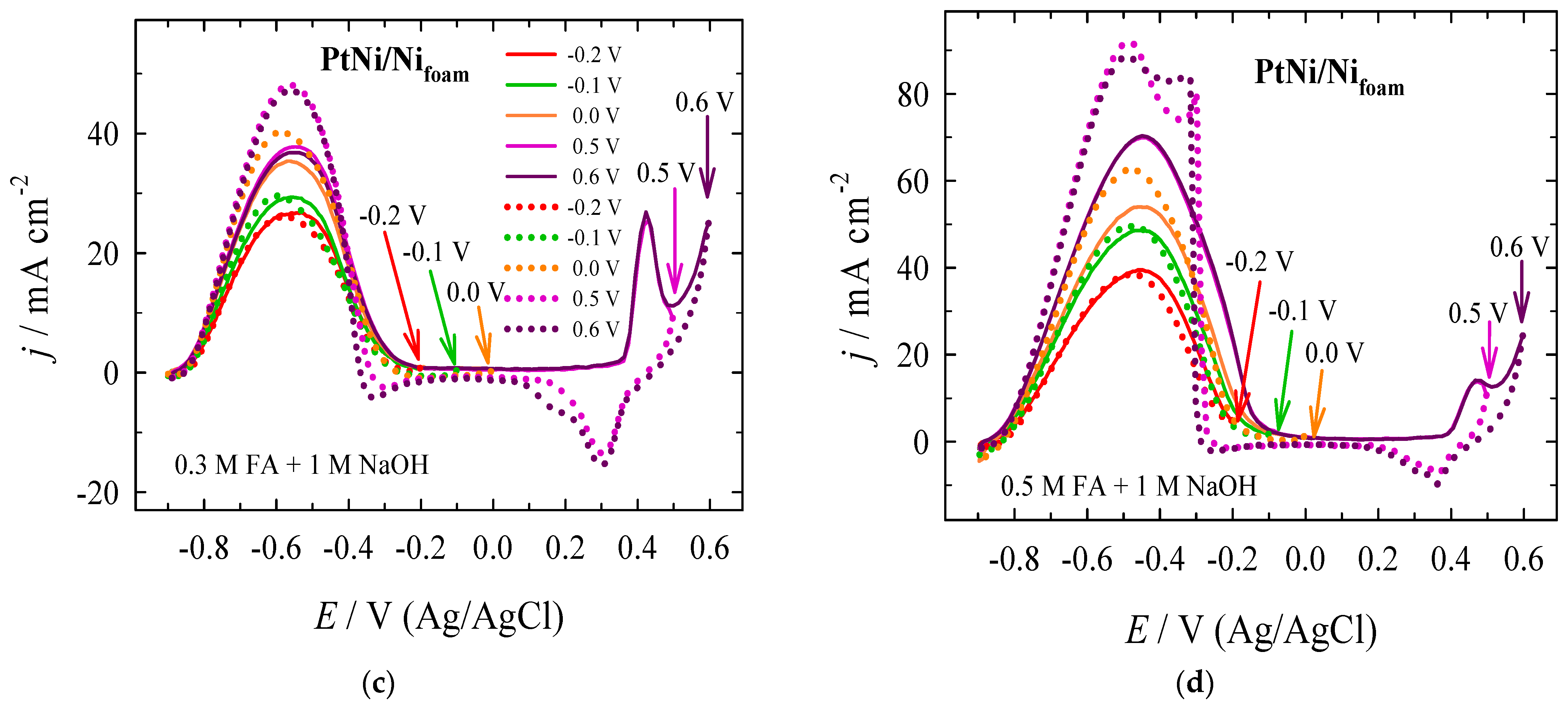
| Era, V | Epf, V | jpf, mA cm−2 | Epb, V | jpb, mA cm−2 | jpf/jpb | jpf at X V/jpf at −0.2 V |
|---|---|---|---|---|---|---|
| −0.2 | −0.530 | 26.75 | −0.591 | 25.99 | 1.03 | 1.00 |
| −0.1 | −0.563 | 29.30 | −0.597 | 29.57 | 0.99 | 1.10 |
| +0.0 | −0.570 | 35.38 | −0.582 | 40.10 | 0.88 | 1.32 |
| +0.5 | −0.549 | 37.77 | −0.565 | 48.04 | 0.79 | 1.41 |
| +0.6 | −0.552 | 36.80 | −0.557 | 47.22 | 0.78 | 1.38 |
| Era, V | Epf, V | jpf, mA cm−2 | Epb, V | jpb, mA cm−2 | jpf/jpb | jpf at X V/jpf at −0.2 V |
|---|---|---|---|---|---|---|
| −0.2 | −0.461 | 39.48 | −0.478 | 38.40 | 1.03 | 1.00 |
| −0.1 | −0.446 | 48.73 | −0.490 | 49.55 | 0.98 | 1.23 |
| +0.0 | −0.454 | 54.00 | −0.483 | 62.53 | 0.86 | 1.37 |
| +0.5 | −0.451 | 69.91 | −0.481 | 91.95 | 0.76 | 1.77 |
| +0.6 | −0.449 | 70.32 | −0.496 | 88.61 | 0.79 | 1.78 |
| Catalyst | Conditions of Experiment | Eonset (V vs. RHE) | Imax (mA cm−2) at Epeak (V vs. RHE) | Ref. |
|---|---|---|---|---|
| Pt disk | 0.2 M HCOONa + 1 M NaOH (pH ≈ 14.0) 20 mV s−1 | +0.3 | 0.1 mA cm−2 (+0.5 V vs. RHE) | [19] |
| Pt disk | 0.4 M HCOO- + 1 M KNO3 (pH 13) 5 mV s−1 | +0.5 | 3.1 mA cm−2 (+1.1 V vs. RHE) | [74] |
| Pt nanoparticles (Pt black) | 0.5 M HCOOK + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | +0.2 | 0.2 mA cm−2 (+0.5 V vs. RHE) | [70] |
| Pt (40%)/ Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | +0.4 | 14.6 mA cm−2 (+0.6 V vs. RHE) | [22] |
| Pt (50%)/C | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | +0.8 | 33.9 mA cm−2 (+1.2 V vs. RHE) | [11] |
| PtAg alloy nanoballoon nanoassembly | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.2 | 32.6 mA cm−2 (+0.7 V vs. RHE) | [17] |
| Pd black | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | +0.8 | 27.3 mA cm−2 (+1.2 V vs. RHE) | [11] |
| Pd/C | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.1 | 108.8 mA cm−2 (+0.7 V vs. RHE) | [4] |
| Pd (20%)-H/Vulcan carbon | 0.5 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | +0.2 | 71.0 mA c cm−2 (+0.8 V vs. RHE) | [76] |
| Pd (20%)/C | 1 M HCOONa + 1 M NaOH (pH ≈ 14.0) 20 mV s−1 | +0.2 | 40.0 mA cm−2 (+0.8 V vs. RHE) | [75] |
| Pd (25%)/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.2 | 4.6 mA cm−2 (+0.7 V vs. RHE) | [8] |
| Pd (40%)/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | +0.2 | 102.0 mA cm−2 (+1.0 V vs. RHE) | [22] |
| Pd/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.4 | 23.0 mA cm−2 (+0.8 V vs. RHE) | [77] |
| Pd (20%)/Reduced graphene oxide | 1 M HCOONa + 1 M NaOH (pH ≈ 14.0) 20 mV s−1 | +0.2 | 57.0 mA cm−2 (+0.8 V vs. RHE) | [75] |
| Pd54Ag46 (core-shell) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.2 | 31.0 mA cm−2 (+0.7 V vs. RHE) | [8] |
| Pd70Cu30/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.2 | 4.3 mA cm−2 (+0.6 V vs. RHE) | [12] |
| Pd72Ce28/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.1 | 19.4 mA cm−2 (+0.6 V vs. RHE) | |
| Pd2.3Co/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | +0.3 | 38.0 mA cm−2 (+0.8 V vs. RHE) | |
| PdNi/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 74.0 mA cm−2 (+0.8 V vs. RHE) | |
| PdNi/Ketjen carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 117.0 mA cm−2 (+0.8 V vs. RHE) | [77] |
| Pd60Ag20Ni20 (alloyed) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 99.6 mA cm−2 (+0.8 V vs. RHE | [9] |
| Pd2Ag1 aerogel | 0.5 M HCOOK + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.2 | 27.5 mA cm−2 (+0.8 V vs. RHE) | [8] |
| Pd2Ag1Pt0.25 aerogel | 0.5 M HCOOK + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.1 | 60.0 mA cm−2 (+0.7 V vs. RHE) | |
| Pd (interstitial B) | 0.5 M HCOOK + 1 M KOH (pH ≈ 14.0) 100 mv s−1 | 0.2 | 90 mA cm−2 (+0.8 V vs. RHE) | [14] |
| PdCuPt (hierarchical zigzag-branched urchin-like superstructure | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.5 | 102.4 mA cm−2 (+1.2 V vs. RHE) | [11] |
| Pt | 0.5 M HCOOH + 1 M NaOH (pH ≈ 14.0) 50 mV s−1 | 0.34 | 0.17 mA cm−2 (+0.53 V vs. RHE) | Our work |
| PtNi/Nifoam | 0.5 M HCOOH + 1 M NaOH (pH ≈ 14.0) 50 mV s−1 | 0.14 | 70.32 mA cm−2 (+0.59 V vs. RHE | Our work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nacys, A.; Šimkūnaitė, D.; Balčiūnaitė, A.; Zabielaitė, A.; Upskuvienė, D.; Šebeka, B.; Jasulaitienė, V.; Kovalevskij, V.; Norkus, E.; Tamašauskaitė-Tamašiūnaitė, L. An Enhanced Oxidation of Formate on PtNi/Ni Foam Catalyst in an Alkaline Medium. Crystals 2022, 12, 362. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030362
Nacys A, Šimkūnaitė D, Balčiūnaitė A, Zabielaitė A, Upskuvienė D, Šebeka B, Jasulaitienė V, Kovalevskij V, Norkus E, Tamašauskaitė-Tamašiūnaitė L. An Enhanced Oxidation of Formate on PtNi/Ni Foam Catalyst in an Alkaline Medium. Crystals. 2022; 12(3):362. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030362
Chicago/Turabian StyleNacys, Antanas, Dijana Šimkūnaitė, Aldona Balčiūnaitė, Aušrinė Zabielaitė, Daina Upskuvienė, Benjaminas Šebeka, Vitalija Jasulaitienė, Vitalij Kovalevskij, Eugenijus Norkus, and Loreta Tamašauskaitė-Tamašiūnaitė. 2022. "An Enhanced Oxidation of Formate on PtNi/Ni Foam Catalyst in an Alkaline Medium" Crystals 12, no. 3: 362. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030362