
Application of the Molecular Interaction Volume Model for Calculating Activities of Elements in Ferromanganese Alloys: Mn-C, Mn-Fe, Fe-C, and Mn-Fe-C Systems


Abstract
:1. Introduction
2. Molecular Interaction Volume Model (MIVM)
3. Results and Discussion
3.1. Evaluation of BMnC and BCMn in Mn-C Binary System
3.2. Mn-Fe Binary System: Evaluation of BMnFe and BFeMn
3.3. Verification of MIVM in Liquid Mn-Fe-C Alloys
3.3.1. Evaluation of BFeC and BCFe in Mn-Fe-C Ternary System
3.3.2. Activity Coefficient of Mn, Fe, and C in Liquid Fe-Mn-C
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suh, D.-W.; Kim, S.-J. Medium Mn transformation-induced plasticity steels: Recent progress and challenges. Scr. Mater. 2017, 126, 63–67. [Google Scholar] [CrossRef]
- Djurovic, D.; Hallstedt, B.; Von Appen, J.; Dronskowski, R. Thermodynamic assessment of the Mn–C system. Calphad 2010, 34, 279–285. [Google Scholar] [CrossRef]
- You, B.-D.; Lee, B.-W.; Pak, J.-J. Manganese loss during the oxygen refining of high-carbon ferromanganese melts. Met. Mater. 1999, 5, 497–502. [Google Scholar] [CrossRef]
- Paek, M.-K.; Pak, J.-J.; Kang, Y.-B. Phase equilibria and thermodynamics of Mn–C, Mn–Si, Si–C binary systems and Mn–Si–C ternary system by critical evaluation, combined with experiment and thermodynamic modeling. Calphad 2014, 46, 92–102. [Google Scholar] [CrossRef]
- Coetsee, T.; Reinke, C.; Nell, J.; Pistorius, P.C. Reduction mechanisms in manganese ore reduction. Metall. Mater. Trans. B 2015, 46, 2534–2552. [Google Scholar] [CrossRef]
- Joo, S.-W.; Hong, S.-H.; Lee, G.-H.; You, B.-D. Evaporation behavior of low carbon ferromanganese alloy melt at reduced pressure. Met. Mater. Int. 2013, 19, 585–590. [Google Scholar] [CrossRef]
- Kim, E.-J.; You, B.-D.; Pak, J.-J. Thermodynamics of carbon in liquid manganese and ferromanganese alloys. Mater. Trans. B 2003, 34, 51–59. [Google Scholar] [CrossRef]
- Katsnelson, A.; Tsukihashi, F.; Sano, N. Determination of manganese and carbon activities of Mn-C melts at 1628 K. ISIJ Int. 1993, 33, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Ma, Z.; Wei, S. Thermodynamics of Mn–Fe–C and Mn–Si–C system. Steel Res. 1990, 61, 113–116. [Google Scholar] [CrossRef]
- Li, H.; Morris, A. Evaluation of unified interaction parameter model parameters for calculating activities of ferromanganese alloys: Mn-Fe-C, Mn-Fe-Si, Mn-C-Si, and Mn-Fe-C-Si systems. Metall. Mater. Trans. B 1997, 28, 553–562. [Google Scholar] [CrossRef]
- Djurovic, D.; Hallstedt, B.; Von Appen, J.; Dronskowski, R. Thermodynamic assessment of the Fe–Mn–C system. Calphad 2011, 35, 479–491. [Google Scholar] [CrossRef]
- Lee, Y.E. Thermodynamic assessment of liquid Mn-Fe-C system by unified interaction parameter model. ISIJ Int. 2003, 43, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Wang, S. Thermodynamic properties of Mn-C melts. J. Iron Steel Res. Int. 2008, 15, 1–6. [Google Scholar] [CrossRef]
- Lee, Y.E. Thermodynamic assessment of liquid Fe-Mn-C system. Metall. Mater. Trans. B 1998, 29, 397–403. [Google Scholar] [CrossRef]
- Pelton, A.D.; Bale, C.W. A modified interaction parameter formalism for non-dilute solutions. Metall. Mater. Trans. A 1986, 17, 1211–1215. [Google Scholar] [CrossRef]
- Bale, C.W.; Pelton, A.D. The unified interaction parameter formalism: Thermodynamic consistency and applications. Metall. Trans. A 1990, 21, 1997–2002. [Google Scholar] [CrossRef]
- Shin, J.P.; Lee, Y.E. Assessment of Mn-Fe-Si-C melt in unified interaction parameter formalism. Metall. Mater. Trans. B 2016, 47, 216–227. [Google Scholar] [CrossRef]
- Kim, M.-S.; Kang, Y.-B. Thermodynamic modeling of the Fe-Mn-C and the Fe-Mn-Al systems using the Modified Quasichemical Model for liquid phase. J. Phase Equilib. Diffus. 2015, 36, 453–470. [Google Scholar] [CrossRef]
- Poizeau, S.; Sadoway, D.R. Application of the molecular interaction volume model (MIVM) to calcium-based liquid alloys of systems forming high-melting intermetallics. J. Am. Chem. Soc. 2013, 135, 8260–8265. [Google Scholar] [CrossRef]
- Ping Tao, D. Prediction of activities of three components in the ternary molten slag CaO-FeO-SiO2 by the molecular interaction volume model. Metall. Mater. Trans. B 2006, 37, 1091–1097. [Google Scholar] [CrossRef]
- Newhouse, J.M.; Poizeau, S.; Kim, H.; Spatocco, B.L.; Sadoway, D.R. Thermodynamic properties of calcium–magnesium alloys determined by emf measurements. Electrochim. Acta 2013, 91, 293–301. [Google Scholar] [CrossRef]
- Yang, H.; Yang, B.; Xu, B.; Liu, D.; Tao, D. Application of molecular interaction volume model in vacuum distillation of Pb-based alloys. Vacuum 2012, 86, 1296–1299. [Google Scholar] [CrossRef]
- Liu, K.; Wu, J.; Wei, K.; Ma, W.; Xie, K.; Li, S.; Yang, B.; Dai, Y. Application of molecular interaction volume model on removing impurity aluminum from metallurgical grade silicon by vacuum volatilization. Vacuum 2015, 114, 6–12. [Google Scholar] [CrossRef]
- Tao, D.P. Prediction of the thermodynamic properties of solutes in Pb-based dilute solutions. Thermochim. Acta 2002, 385, 5–10. [Google Scholar] [CrossRef]
- Tao, D.P. Prediction of the thermodynamic properties of solutes in the Bi-based ternary dilute solution. Metall. Mater. Trans. B 2002, 33, 502–506. [Google Scholar] [CrossRef]
- Tao, D.P. Prediction of activities of all components in the lead-free solder systems Bi–In–Sn and Bi–In–Sn–Zn. J. Alloys Compd. 2008, 457, 124–130. [Google Scholar] [CrossRef]
- Yan, W.; Yang, Y.; Chen, W.; Barati, M.; McLean, A. Thermodynamic assessment of Si-P and Si-Fe-P alloys for solar grade silicon refining via vacuum levitation. Vacuum 2017, 135, 101–108. [Google Scholar] [CrossRef]
- Huang, W. Thermodynamic Assessment of the Mn–C System. Scand. J. Metall. 1990, 19, 26–32. [Google Scholar] [CrossRef]
- Witusiewicz, V.T.; Sommer, F.; Mittemeijer, E.J. Enthalpy of formation and heat capacity of Fe-Mn alloys. Metall. Mater. Trans. B 2003, 34, 209–223. [Google Scholar] [CrossRef]
- Huang, W. A thermodynamic assessment of the Fe-Mn-C system. Mater. Trans. A 1990, 21, 2115–2123. [Google Scholar] [CrossRef]
- Ahn, S.H.; Kim, Y.H.; Shin, J.-P.; Lee, Y.E. Thermodynamic assessment of liquid Fe–Si–C system by unified interaction parameter model. ISIJ Int. 2014, 54, 750–755. [Google Scholar] [CrossRef] [Green Version]
- Chipman, J. Activity of interstitial and nonmetallic solutions in dilute metal solutions-lattice ratio as a concentration variable. Aime Met. Soc. Trans. 1967, 239, 1332–1336. [Google Scholar]
- Chipman, J. Thermodynamics of liquid Fe-C solutions. Metall. Trans. 1970, 1, 2163–2168. [Google Scholar] [CrossRef]
- Tanaka, A. Activities of Manganese in Mn–Fe–C, Mn–Si–C and Mn–Fe–Si–C Melts at 1673 K. Trans. Jpn. Inst. Met. 1980, 21, 27–33. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
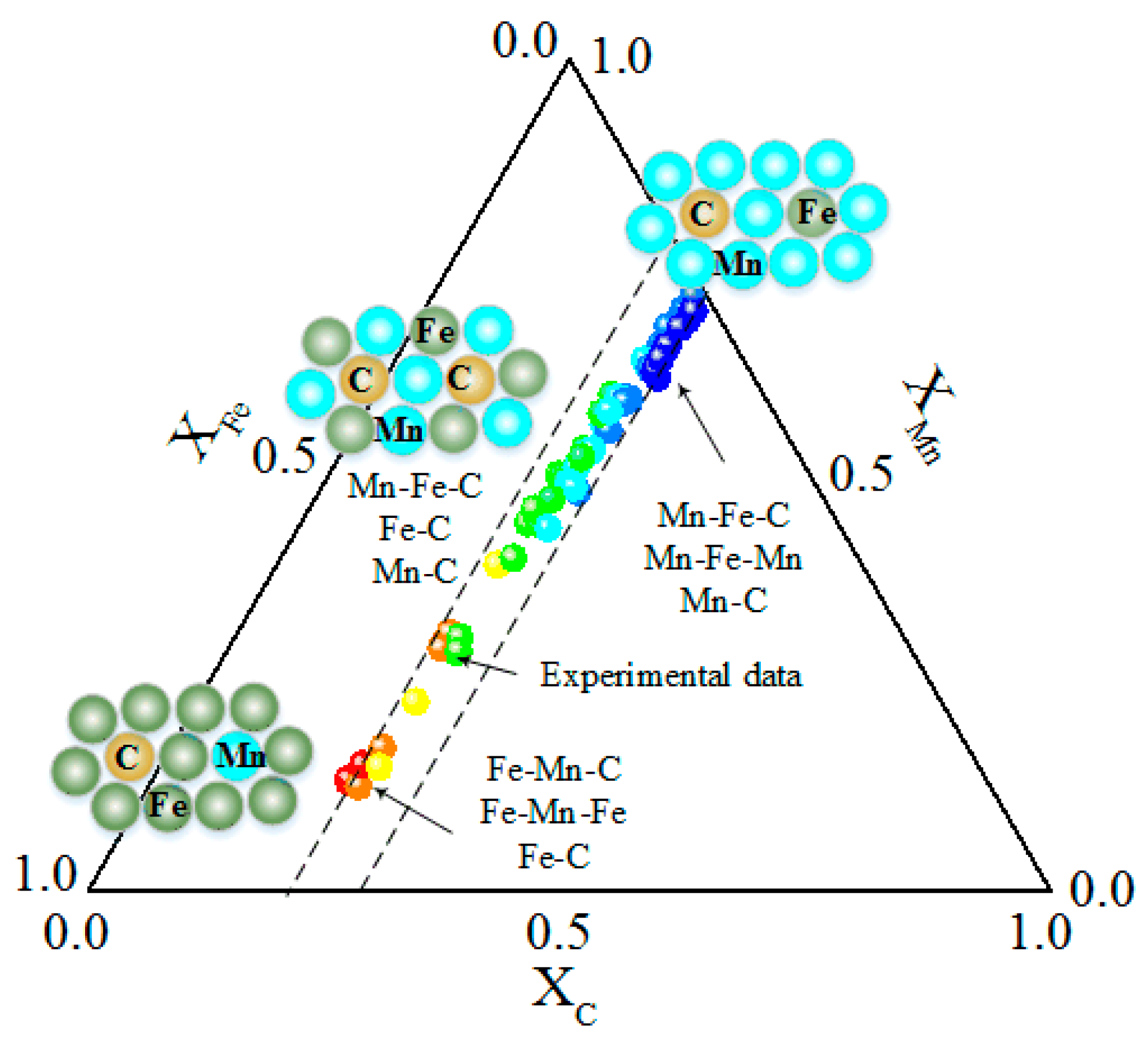{kind=link}
| i | Vmi (cm3·mol−1) | ΔHmi (kJ mol−1) | σi (10−8 cm) | dcovi (10−8 cm) |
|---|---|---|---|---|
| Mn | 9.54·[1 + 1.6 × 10−4·(T − 1517)] | 14.6 | 2.61 | 2.34 |
| Fe | 7.94·[1 + 1.3 × 10−4·(T − 1808)] | 13.77 | 2.55 | 2.34 |
| C | 5.30 | - | 1.82 | 1.54 |
| T(K) | BMnC | BCMn |
|---|---|---|
| 1628 | 2.111 | 0.969 |
| 1673 | 1.809 | 1.201 |
| 1723 | 1.780 | 1.343 |
| 1773 | 1.679 | 1.703 |
| Error | 1628 K | 1673 K | 1723 K | 1773 K |
|---|---|---|---|---|
| MIVM | 5.19 | 4.10 | 3.23 | 5.56 |
| Young et al. | 6.29 | 5.38 | 4.62 | 6.94 |
| Li et al. | 6.35 | 5.46 | 3.64 | 6.83 |
| Chen et al. | 5.18 | 4.21 | 3.25 | 4.52 |
| Katsnelson et al. | 3.46 | 11.24 | - | - |
| Error (kJ/mol) | T/K | MIVM | Li et al. | Young et al. | Dejan et al. | Chen et al. | Kim et al. 1723 K | Katsnelson et al. 1628 K |
|---|---|---|---|---|---|---|---|---|
| Error (aC) | 1673 K | 0.12 | 0.11 | 0.13 | 0.54 | 0.13 | 0.13 | 0.14 |
| 1773 K | 0.14 | 0.61 | 0.18 | 0.42 | 0.22 | |||
| Error (aMn) | 1673 K | 0.59 | 0.71 | 0.64 | 0.78 | 0.60 | 0.56 | 0.61 |
| 1773 K | 0.65 | 1.12 | 1.23 | 0.95 | 0.86 |
| Element | BMnFe | BFeMn | ||
|---|---|---|---|---|
| Mn-Fe | 0.9619 | 0.9853 | 69.8 | 26.3 |
| T/K | 1563 K | 1673 K | 1723 K | 1763 K | 1773 K |
|---|---|---|---|---|---|
| BFeC | 22.38 | 22.84 | 21.60 | 20.17 | 24.94 |
| BCFe | 1.36 | 1.86 | 1.85 | 3.41 | 1.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Gao, L.; Zhan, W.; He, Z.; Zhang, J. Application of the Molecular Interaction Volume Model for Calculating Activities of Elements in Ferromanganese Alloys: Mn-C, Mn-Fe, Fe-C, and Mn-Fe-C Systems. Crystals 2022, 12, 682. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12050682
Chen H, Gao L, Zhan W, He Z, Zhang J. Application of the Molecular Interaction Volume Model for Calculating Activities of Elements in Ferromanganese Alloys: Mn-C, Mn-Fe, Fe-C, and Mn-Fe-C Systems. Crystals. 2022; 12(5):682. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12050682
Chicago/Turabian StyleChen, Haipeng, Lihua Gao, Wenlong Zhan, Zhijun He, and Junhong Zhang. 2022. "Application of the Molecular Interaction Volume Model for Calculating Activities of Elements in Ferromanganese Alloys: Mn-C, Mn-Fe, Fe-C, and Mn-Fe-C Systems" Crystals 12, no. 5: 682. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12050682