
Structural, Interfacial, and Electrochemical Stability of La0.3Ca0.7Fe0.7Cr0.3O3-δ Electrode Material for Application as the Oxygen Electrode in Reversible Solid Oxide Cells

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
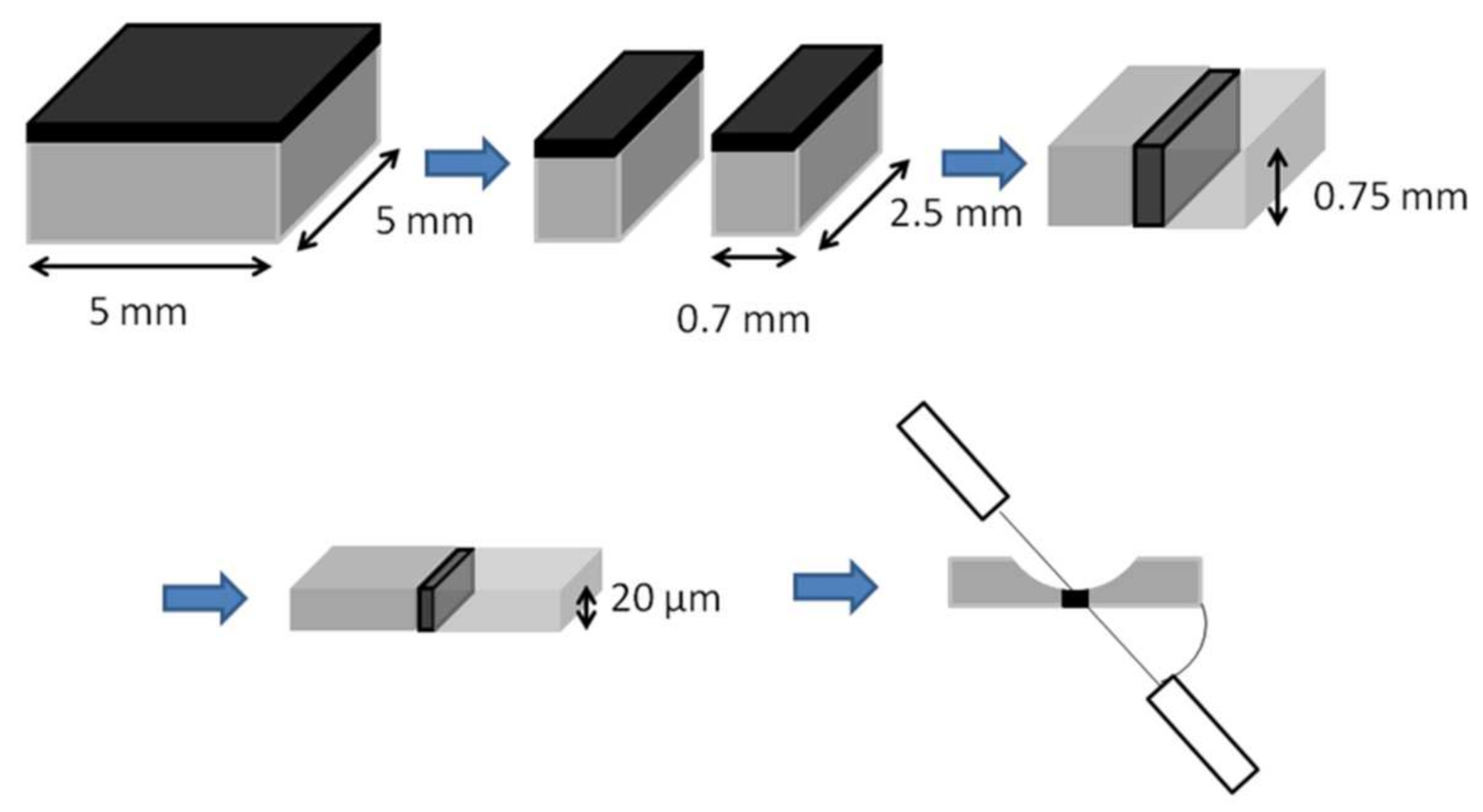
2.1. Cell Fabrication and Testing
2.2. Cell Characterization
2.3. HRTEM Analysis
3. Results
3.1. SEM and 3D X-ray Microscopy (XRM) of As-Prepared Cells
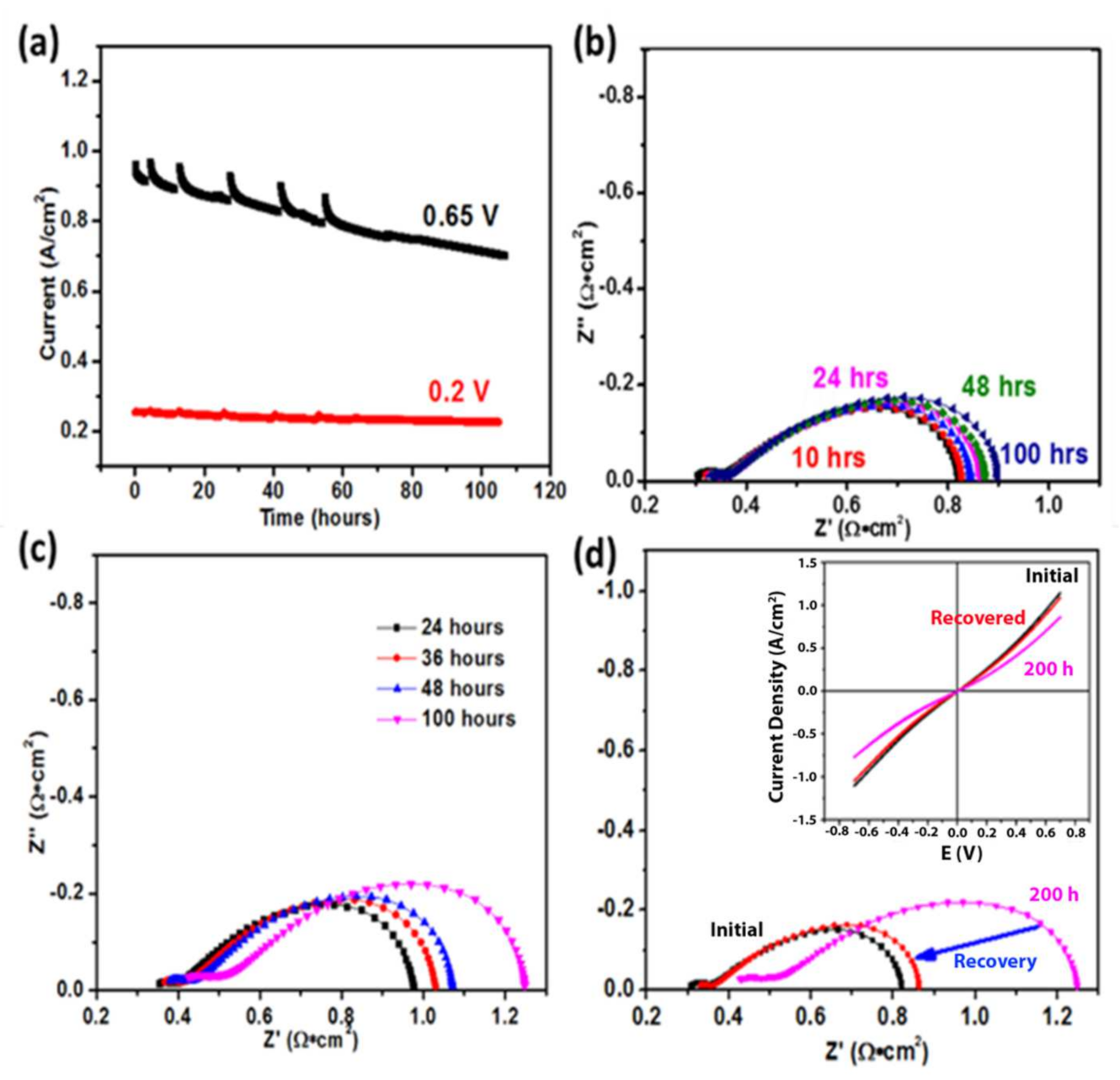
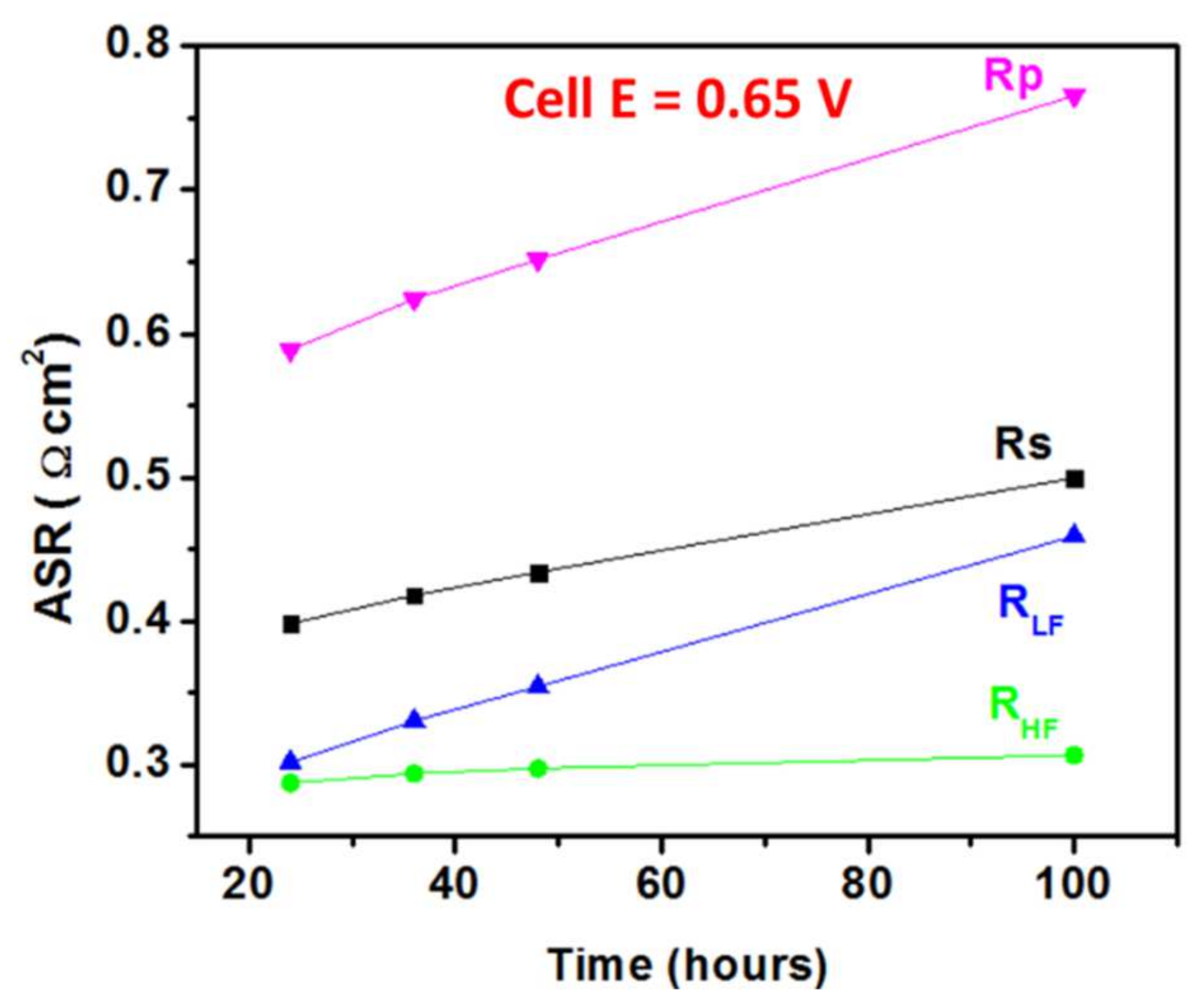
3.2. Electrochemical Testing
3.3. Post Mortem Cell Microstructure Studies
3.3.1. SEM Analysis of La0.3Ca0.7Fe0.7Cr0.3O3-δ (LCFCr)-Based Cells
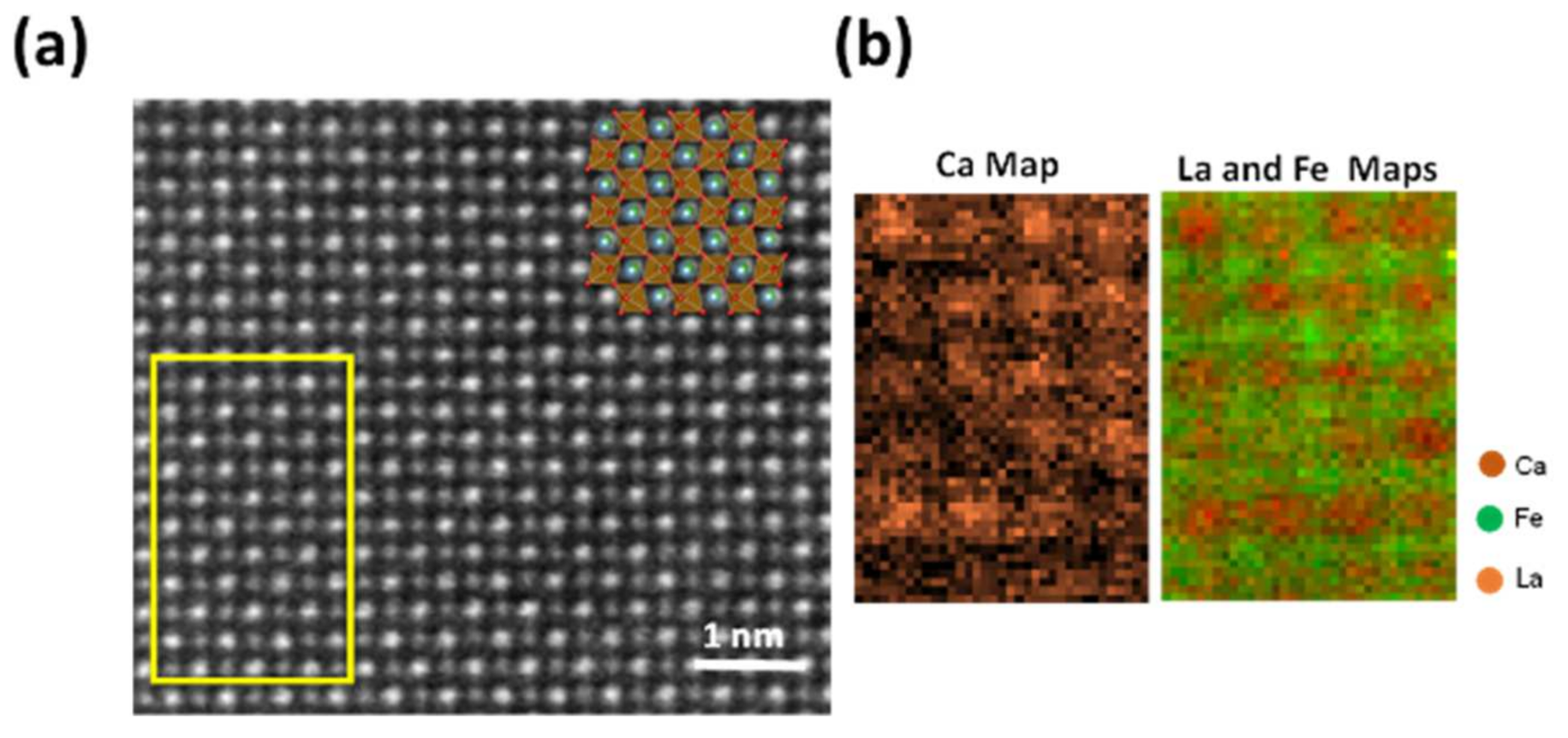
3.3.2. Transmission Electron Microscopy Examination of LCFCr/GDC Interfaces
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hauch, A.; Kungas, R.; Blennow, P.; Hansen, A.B.; Hansen, J.B.; Mathiesen, B.V.; Mogensen, M.B. Recent Advances in Solid Oxide Cell Technology for Electrolysis. Science 2020, 370, eaba6118. [Google Scholar] [CrossRef] [PubMed]
- Ansari, H.M.; Addo, P.K.; Mulmi, S.; Yuan, H.; Botton, G.A.; Thangadurai, V.; Birss, V.I. Deciphering the Interaction of Single-Phase La0.3Sr0.7Fe0.7Cr0.3O3-δ with CO2/CO Environments for Application in Reversible Solid Oxide Cells. ACS Appl. Mater. Interfaces 2022, 14, 13388–13399. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Liu, Q.; Yan, Z.; Jiao, Z.; Bi, L.; Chan, S.H.; Zhong, Z. On the Delamination of Air Electrodes of Solid Oxide Electrolysis Cells: A Mini-Review. Electrochem. Commun. 2022, 137, 107267. [Google Scholar] [CrossRef]
- Shen, M.; Ai, F.; Ma, H.; Xu, H.; Zhang, Y. Progress and Prospects of Reversible Solid Oxide Fuel Cell Materials. iScience 2021, 24, 103464. [Google Scholar] [CrossRef] [PubMed]
- Virkar, A.V. Mechanism of Oxygen Electrode Delamination in Solid Oxide Electrolyzer Cells. Int. J. Hydrogen Energy 2010, 35, 9527–9543. [Google Scholar] [CrossRef]
- Jin, X.; Cook, K.; Huang, K. Interrelationship between Extended Transport Pathways of Mixed Conducting Electrode and Delamination in Solid Oxide Electrolyzer Cells. ECS Meet. Abstr. 2021, MA2021-01, 1143. [Google Scholar] [CrossRef]
- Keane, M.; Mahapatra, M.K.; Verma, A.; Singh, P. LSM-YSZ Interactions and Anode Delamination in Solid Oxide Electrolysis Cells. Int. J. Hydrogen Energy 2012, 37, 16776–16785. [Google Scholar] [CrossRef]
- Chen, K.; Jiang, S.P. Failure Mechanism of (La,Sr)MnO3 Oxygen Electrodes of Solid Oxide Electrolysis Cells. Int. J. Hydrogen Energy 2011, 36, 10541–10549. [Google Scholar] [CrossRef]
- Mocooteguy, P.; Brisse, A. A Review and Comprehensive Analysis of Degradation Mechanisms of Solid Oxide Electrolysis Cells. Int. J. Hydrogen Energy 2013, 38, 15887–15902. [Google Scholar] [CrossRef]
- Pan, Z.; Liu, Q.; Ni, M.; Lyu, R.; Li, P.; Chan, S.H. Activation and Failure Mechanism of La0.6Sr0.4Co0.2Fe0.8O3-δ Air Electrode in Solid Oxide Electrolyzer Cells under High-Current Electrolysis. Int. J. Hydrogen Energy 2018, 43, 5437–5450. [Google Scholar] [CrossRef]
- Shao, Z.; Haile, S.M. A High-Performance Cathode for the Next Generation of Solid-Oxide Fuel Cells. Nature 2004, 431, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; Wang, H.; Lu, M.Y.; Zhang, A.-P.; Mogni, L.V.; Liu, Q.; Li, C.-X.; Li, C.-J.; Barnett, S.A. Cobalt-Substituted SrTi0.3Fe0.7O3-δ: A Stable High-Performance Oxygen Electrode Material for Intermediate-Temperature Solid Oxide Electrochemical Cells. Energy Environ. Sci. 2018, 11, 1870–1879. [Google Scholar] [CrossRef]
- Chen, Y.; Yoo, S.; Choi, Y.; Kim, J.H.; Ding, Y.; Pei, K.; Murphy, R.; Zhang, Y.; Zhao, B.; Zhang, W.; et al. A Highly Active, CO2-Tolerant Electrode for the Oxygen Reduction Reaction. Energy Environ. Sci. 2018, 11, 2458–2466. [Google Scholar] [CrossRef]
- Wu, J.; Pramana, S.S.; Skinner, S.J.; Kilner, J.A.; Horsfield, A.P. Why Ni Is Absent from the Surface of La2NiO4+δ? J. Mater. Chem. A 2015, 3, 23760–23767. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Zhou, F.; Yang, S.; Zhong, S.; Wei, M.; Liu, Y. Performance and Stability of Ruddlesden-Popper La2NiO4+δ Oxygen Electrodes under Solid Oxide Electrolysis Cell Operation Conditions. Ceram. Int. 2017, 43, 10927–10933. [Google Scholar] [CrossRef]
- Molero-Sanchez, B.; Prado-Gonjal, J.; Avila-Brande, D.; Chen, M.; Moran, E.; Birss, V. High Performance La0.3Ca0.7Cr0.3Fe0.7O3-δ Air Electrode for Reversible Solid Oxide Fuel Cell Applications. Int. J. Hydrogen Energy 2015, 40, 1902–1910. [Google Scholar] [CrossRef]
- Molero-Sanchez, B.; Addo, P.; Buyukaksoy, A.; Paulson, S.; Birss, V. Electrochemistry of La0.3Sr0.7Fe0.7Cr0.3O3-δ as an Oxygen and Fuel Electrode for RSOFCs. Faraday Discuss. 2015, 182, 159–175. [Google Scholar] [CrossRef]
- Chen, M.; Paulson, S.; Kan, W.H.; Thangadurai, V.; Birss, V. Surface and Bulk Study of Strontium-Rich Chromium Ferrite Oxide as a Robust Solid Oxide Fuel Cell Cathode. J. Mater. Chem. A 2015, 3, 22614–22626. [Google Scholar] [CrossRef]
- Addo, P.K.; Molero-Sanchez, B.; Chen, M.; Paulson, S.; Birss, V. Co/Co2 Study of High Performance La0.3Sr0.7Fe0.7Cr0.3O3-δ Reversible SOFC Electrodes. Fuel Cells 2015, 15, 689–696. [Google Scholar] [CrossRef]
- Addo, P.K.; Molero-Sanchez, B.; Buyukaksoy, A.; Paulson, S.; Birss, V. Sulfur Tolerance of La0.3M0.7Fe0.7Cr0.3O3-δ (M = Sr, Ca) Solid Oxide Fuel Cell Anodes. ECS Trans. 2015, 66, 219–228. [Google Scholar] [CrossRef]
- Molero-Sanchez, B.; Addo, P.; Buyukaksoy, A.; Birss, V. Performance Enhancement of La0.3Ca0.7Fe0.7Cr0.3O3-δ Air Electrodes by Infiltration Methods. J. Electrochem. Soc. 2017, 164, F3123–F3130. [Google Scholar] [CrossRef]
- Ansari, H.M.; Bass, A.S.; Ahmad, N.; Birss, V.I. Unraveling the Evolution of Exsolved Fe-Ni Alloy Nanoparticles in Ni-Doped La0.3Ca0.7Fe0.7Cr0.3O3-δ and Their Role in Enhancing CO2-CO Electrocatalysis. J. Mater. Chem. A 2022, 10, 2280–2294. [Google Scholar] [CrossRef]
- Molero-Sanchez, B.; Prado-Gonjal, J.; Avila-Brande, D.; Birss, V.; Moran, E. Microwave-Assisted Synthesis and Characterization of New Cathodic Material for Solid Oxide Fuel Cells: La0.3Ca0.7Fe0.7Cr0.3O3-δ. Ceram. Int. 2015, 41, 8411–8416. [Google Scholar] [CrossRef]
- Chen, M.; Paulson, S.; Thangadurai, V.; Birss, V. Sr-Rich Chromium Ferrites as Symmetrical Solid Oxide Fuel Cell Electrodes. J. Power Sources 2013, 236, 68–79. [Google Scholar] [CrossRef]
- Xiao, J.; Xu, Q.; Huang, D.-P.; Chen, M.; Zhao, K.; Kim, B.-H. Evaluation of La0.3Ca0.7Fe1-yCryO3-δ (y = 0.1 − 0.3) Cathodes for Intermediate Temperature Solid Oxide Fuel Cells. Mater. Res. Bull. 2017, 90, 104–110. [Google Scholar] [CrossRef]
- Tkachuk, A.; Duewer, F.; Cui, H.; Feser, M.; Wang, S.; Yun, W. X-Ray Computed Tomography in Zernike Phase Contrast Mode at 8 keV with 50-nm Resolution Using Cu Rotating Anode X-Ray Source. Z. Fur Krist.-Cryst. Mater. 2007, 222, 650–655. [Google Scholar] [CrossRef]
- Merkle, A.P.; Gelb, J.D. The Ascent of 3D X-Ray Microscopy in the Laboratory. Microsc. Today 2013, 21, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Zhu, J.; Lin, Z. Effects of Electrode Composition and Thickness on the Mechanical Performance of a Solid Oxide Fuel Cell. Energies 2018, 11, 1735. [Google Scholar] [CrossRef] [Green Version]
- Molero-Sanchez, B. Development of Oxygen Electrodes for Reversible Solid Oxide Fuel Cells. Ph.D. Thesis, University of Calgary, Calgary, AB, Canada, 2017. [Google Scholar]
- Sharma, V.I.; Yildiz, B. Degradation Mechanism in La0.8Sr0.2CoO3 as Contact Layer on the Solid Oxide Electrolysis Cell Anode. J. Electrochem. Soc. 2010, 157, B441. [Google Scholar] [CrossRef]
- Mawdsley, J.R.; David Carter, J.; Jeremy Kropf, A.; Yildiz, B.; Maroni, V.A. Post-Test Evaluation of Oxygen Electrodes from Solid Oxide Electrolysis Stacks. Int. J. Hydrogen Energy 2009, 34, 4198–4207. [Google Scholar] [CrossRef]
- Addo, P.K. Development of Fuel Electrodes for Reversible Solid Oxide Fuel Cell Applications. Ph.D. Thesis, University of Calgary, Calgary, AB, Canada, 2017. [Google Scholar]
- Hjalmarsson, P.; Sogaard, M.; Mogensen, M. Electrochemical Behaviour of (La1-xSrx)Co1-yNiyO3 as Porous SOFC Cathodes. Solid State Ion. 2009, 180, 1395–1405. [Google Scholar] [CrossRef]
- Liu, S.-S.; Jiao, Z.; Shikazono, N.; Matsumura, S.; Koyama, M. Observation of the Ni/YSZ Interface in a Conventional SOFC. J. Electrochem. Soc. 2015, 162, F750–F754. [Google Scholar] [CrossRef]
- Baque, L.C.; Jorgensen, P.S.; Zhang, W.; Hansen, K.V.; Sogaard, M. Effect of Aging on the Electrochemical Performance of LSM-YSZ Cathodes. J. Electrochem. Soc. 2015, 162, F971–F981. [Google Scholar] [CrossRef] [Green Version]
- Soldati, A.L.; Baque, L.; Troiani, H.; Cotaro, C.; Schreiber, A.; Caneiro, A.; Serquis, A. High Resolution FIB-TEM and FIB-SEM Characterization of Electrode/Electrolyte Interfaces in Solid Oxide Fuel Cells Materials. Int. J. Hydrogen Energy 2011, 36, 9180–9188. [Google Scholar] [CrossRef]
- Baque, L.; Padmasree, K.P.; Ceniceros Reyes, M.A.; Troiani, H.; Serquis, A.; Soldati, A. Improved Sr0.6La0.4Co0.8Fe0.2O3-δ/Ce0.8Y0.2O2-δ Interface for IT-SOFC Applications. Int. J. Hydrogen Energy 2016, 41, 1958–1965. [Google Scholar] [CrossRef]
- Kindermann, L.; Das, D.; Nickel, H.; Hilpert, K. Chemical Compatibility of the Lafeo3 Base Perovskites (La0.6Sr0.4)zFe0.8M0.2O3-δ (z = 1, 0.9; M = Cr, Mn, Co, Ni) with Yttria Stabilized Zirconia. Solid State Ion. 1996, 89, 215–220. [Google Scholar] [CrossRef]
- Kindermann, L.; Das, D.; Bahadur, D.; Wei, R.; Nickel, H.; Hilpert, K. Chemical Interactions between La-Sr-Mn-Fe-O-Based Perovskites and Yttria-Stabilized Zirconia. J. Amer. Ceram. Soc. 1997, 80, 909–914. [Google Scholar] [CrossRef]
- Kinderann, L.; Das, D.; Nickel, H.; Hilpert, K.; Appel, C.C.; Poulson, F.W. Chemical Compatibility of (La0.6Ca0.4)x Fe0.8Mn0.2O3 with Yttria-Stabilized Zirconia. J. Electrochem. Soc. 1997, 144, 717–720. [Google Scholar] [CrossRef]
- Lee, W.; Han, J.W.; Chen, Y.; Cai, Z.; Yildiz, B. Cation Size Mismatch and Charge Interactions Drive Dopant Segregation at the Surfaces of Manganite Perovskites. J. Amer. Chem. Soc. 2013, 135, 7909–7925. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LCFCr (%) | Connected Porosity (%) | Isolated Porosity (%) | Interfacial Contact Area (µm2) | Surface Area (µm2) | Average Pore Size (µm) |
|---|---|---|---|---|---|
| 51.21 | 48.66 | 0.13 | 170 | 3809.93 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, H.M.; Avila-Brande, D.; Kelly, S.; Addo, P.K.; Molero-Sánchez, B. Structural, Interfacial, and Electrochemical Stability of La0.3Ca0.7Fe0.7Cr0.3O3-δ Electrode Material for Application as the Oxygen Electrode in Reversible Solid Oxide Cells. Crystals 2022, 12, 847. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12060847
Ansari HM, Avila-Brande D, Kelly S, Addo PK, Molero-Sánchez B. Structural, Interfacial, and Electrochemical Stability of La0.3Ca0.7Fe0.7Cr0.3O3-δ Electrode Material for Application as the Oxygen Electrode in Reversible Solid Oxide Cells. Crystals. 2022; 12(6):847. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12060847
Chicago/Turabian StyleAnsari, Haris Masood, David Avila-Brande, Steve Kelly, Paul Kwesi Addo, and Beatriz Molero-Sánchez. 2022. "Structural, Interfacial, and Electrochemical Stability of La0.3Ca0.7Fe0.7Cr0.3O3-δ Electrode Material for Application as the Oxygen Electrode in Reversible Solid Oxide Cells" Crystals 12, no. 6: 847. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12060847