Polyoxotungstates in Molecular Boxes of Purine Bases

Department für Chemie, Institut für Anorganische Chemie, Universität zu Köln, Greinstraße 6, Köln D-50939, Germany
*
Author to whom correspondence should be addressed.
Crystals 2014, 4(1), 64-73; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst4010064
Submission received: 31 December 2013
/
Revised: 17 February 2014
/
Accepted: 20 February 2014
/
Published: 12 March 2014
Abstract
:Three new compounds, (GuaH)4[W10O32](H2O)4 (1), (ThbH)3(H3O)[(W10O32](H2O)7.5 (2) and (ThbH)2[W6O19](H2O)2 (3) (GuaH = guaninium, thbH = theobrominium) were synthesized in acidified acetonitrile solutions. The polyoxotungstates in all of these compounds are surrounded by an organic matrix consisting of protonated purine bases and water molecules. The distinctive structural arrangement of the aromatic organic cations around the polyoxoanions parallel to their faces is reminiscent of nanosized boxes. The results of IR spectroscopy are consistent with previously reported results for polyoxotungstates and neat organic compounds. The polyoxoanions are reduced to tungsten(IV) oxide upon heating over 400 °C in an intramolecular redox reaction.

1. Introduction
Polyoxometalates (POMs) are condensed oxyanions of transition metals usually from the groups 5 and 6 of the periodic chart. They have already been known for two centuries [1,2], and found numerous applications in a multitude of areas ranging from catalysis to pharmacology and remain of continuous interest, especially in the areas of design and synthesis of functional nanomaterials [3,4]. Polyoxotungstates occupy a special position among POMs, due to a multitude of fascinating structures and a wide scope of current and potential applications [5,6,7,8,9].
Purines such as adenine and guanine are nitrogen bases belonging to the class of alkaloids with an extensive importance in life sciences. As major constituents of the nucleic acids and their vital role in energy metabolism and molecular signal transduction they can be considered central biomolecules [10].
The investigation of interactions between POMs and biomolecules in general [11] as well as between POMs and purine bases in particular are interesting for three major reasons: (1) They are interesting from the structural point of view, due to a multitude of possibilities of non-covalent interactions of the purine bases [12]; (2) The resulting hybrid organic-inorganic solids are in focus of current scientific research in order to elucidate the possibilities of a targeted design of functional materials; (3) They might shed some light onto the mechanism of the pharmacological action of polyoxoanions [9].
In spite of these compelling reasons to conduct research in the area, only a limited number of publications were released on the topic of interactions between purine bases and POMs. To the best of our knowledge, only structures of caffeinium decavanadate and of a hybrid of adenosine and the Strandberg anion were described and discussed [13,14,15]. We recently published two further articles within the area, describing materials consisting of theobromine silver(I) complexes and POMs [16,17]. In the present work we discuss three new compounds consisting of protonated purine bases and polyoxotungstates focusing on their unusual and aesthetically appealing crystal structures.
2. Results and Discussion
2.1. Crystal Structures
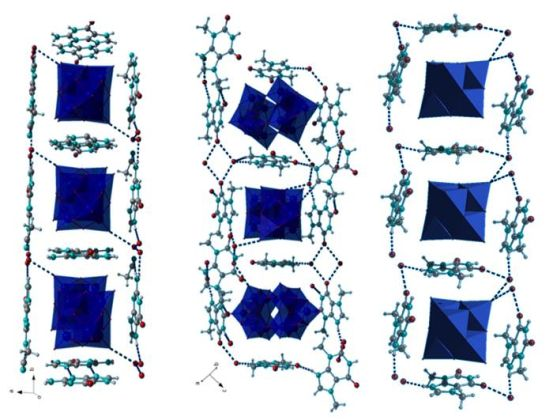
The decatungstates (GuaH)4[W10O32](H2O)4 (1) and (ThbH)3(H3O)[W10O32](H2O)7.5 (2) (GuaH = guaninium, thbH = Theobrominium) both crystallize from acetonitrile solutions acidified with nitric acid. There are considerable differences in the non-covalent frameworks of these compounds which are based on the different functional groups of the two constituent organic molecules (Figure 1). Guanine is capable of conducting a higher amount of H-bonds due to its not methylated nitrogen atoms. Accordingly, there are four guaninium ions per formula unit of 1 with four additional water molecules that are sufficient to sustain the highly polar framework. Theobrominium, on the other hand, displays two methyl substituents on two of its nitrogen atoms rendering the molecule more hydrophobic. Thus, only three theobrominium ions are present per formula unit of 2, with a lot of additional water molecules needed to stabilize the crystal structure. The bulk purity of 2 was confirmed by powder X-ray diffraction (Figure S1).
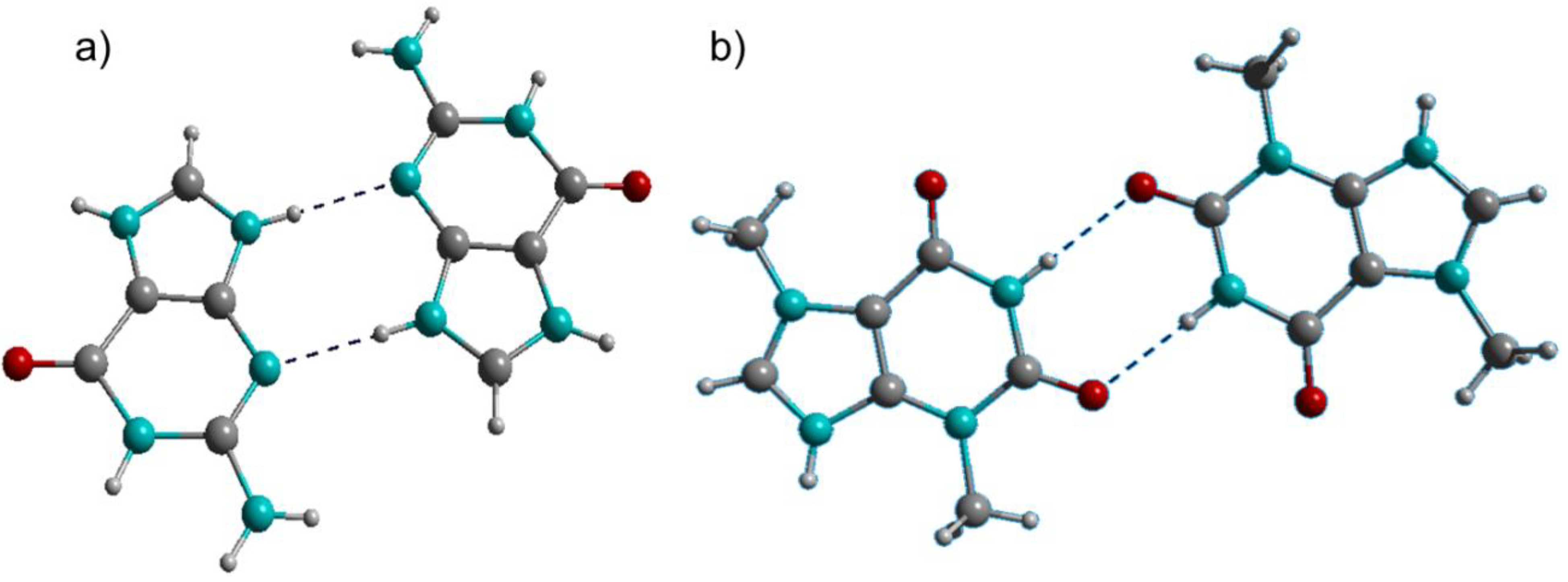
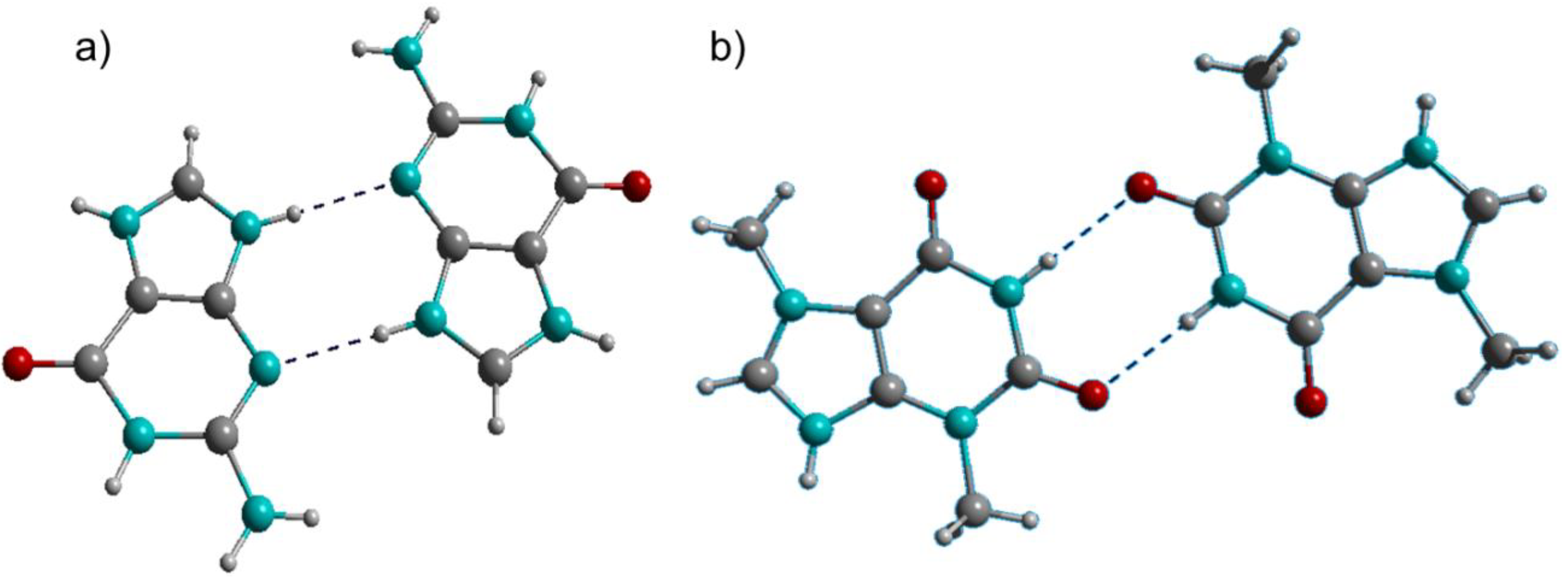
Interactions between the guaninium and theobrominium ions are pronouncedly different (Figure 1). The guaninium dimer is stabilized by interactions between protonated and deprotonated N-atoms, while the theobrominium counterpart is stabilized by amide-to-amide H-bonds reminiscent of the adenine-thymine base pair in the duplex nucleic acids [18]. The H-bond donor-acceptor distances (N···N 2.91(2) Å) and the directionality (N–
H···N = 162.2(8)°) indicate medium strength interactions for the guaninium dimer [19]. The same is true for the theobrominium dimer (N···O 2.88(1) Å; N–
H···N = 169.1(8)°).
Figure 1.
(a) Guaninium dimer; (b) Theobrominium dimer. C: grey, H: white, N: turquoise, O: red. This color scheme is also used in subsequent figures.
Figure 1.
(a) Guaninium dimer; (b) Theobrominium dimer. C: grey, H: white, N: turquoise, O: red. This color scheme is also used in subsequent figures.
The type of non-covalent interactions between purine bases and decatungstate anions important in 1 and 2 are so-called lone pair—π interactions (Figure 2). These are interactions between a lone pair donator and a π-acidic (electron deficient) aromatic system. The decatungstate anions share guaninium dimers in 1 along the crystallographic b-axis (Figure 2a). In 2, on the other hand, theobrominium monomers are shared. They build amide-to-amide dimers interconnecting the structure to a three-dimensional framework. The electron accepting properties of the π-acidic purines, among them especially of adenine and guanine, were thoroughly explored by means of theoretical calculations and structural analysis, due to their importance for the three-dimensional structure of nucleic acids [20].
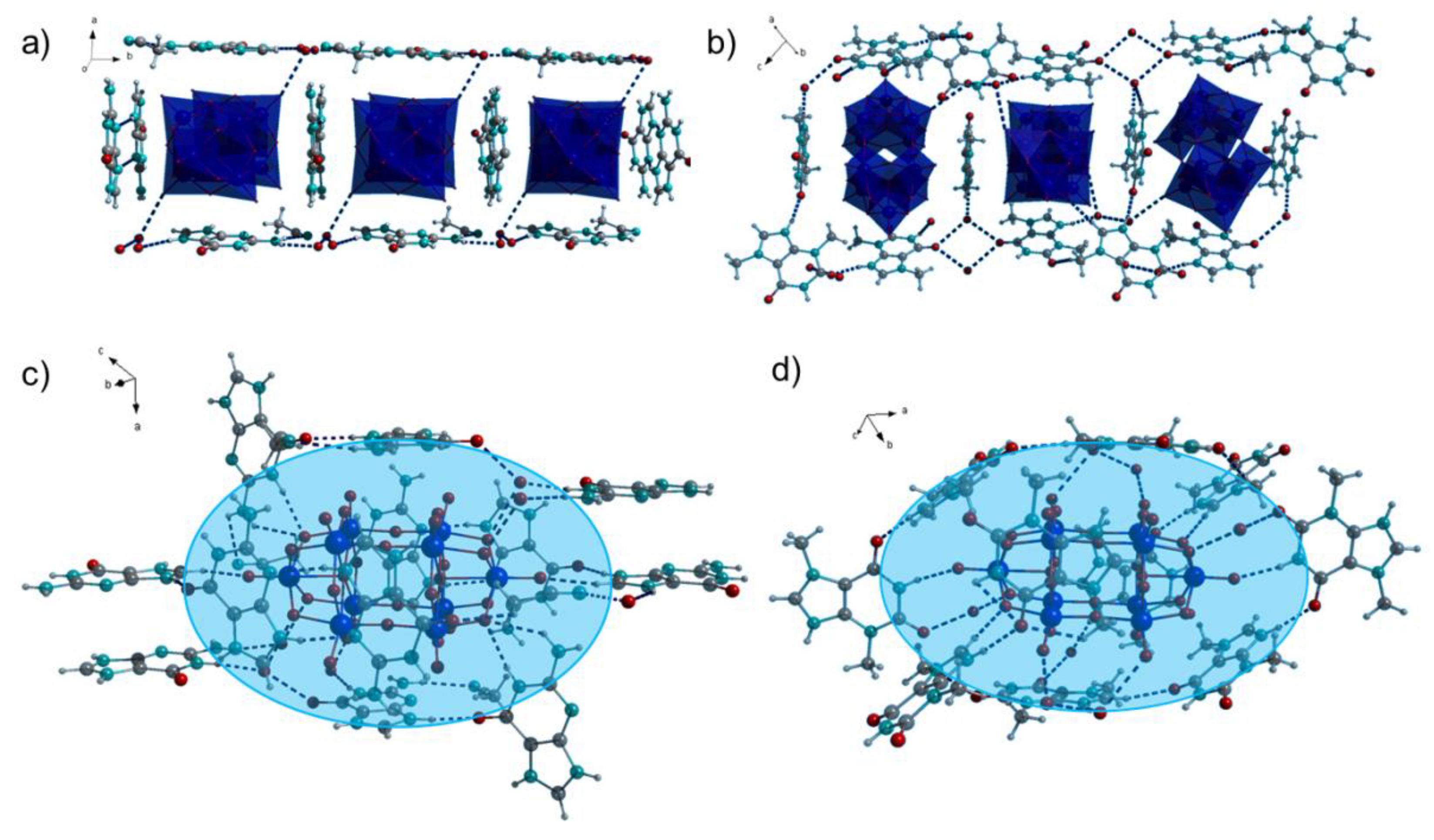
Figure 2.
(a,b) Nanoboxes surrounding decatungstate anions in 1 and 2; (c,d) Decatungstate anions embedded into the organic matrix consisting of protonated purine bases and water molecules. Blue area indicates the organic matrix environment of the POM. W: light blue, color kept in the Figure 3.
Figure 2.
(a,b) Nanoboxes surrounding decatungstate anions in 1 and 2; (c,d) Decatungstate anions embedded into the organic matrix consisting of protonated purine bases and water molecules. Blue area indicates the organic matrix environment of the POM. W: light blue, color kept in the Figure 3.
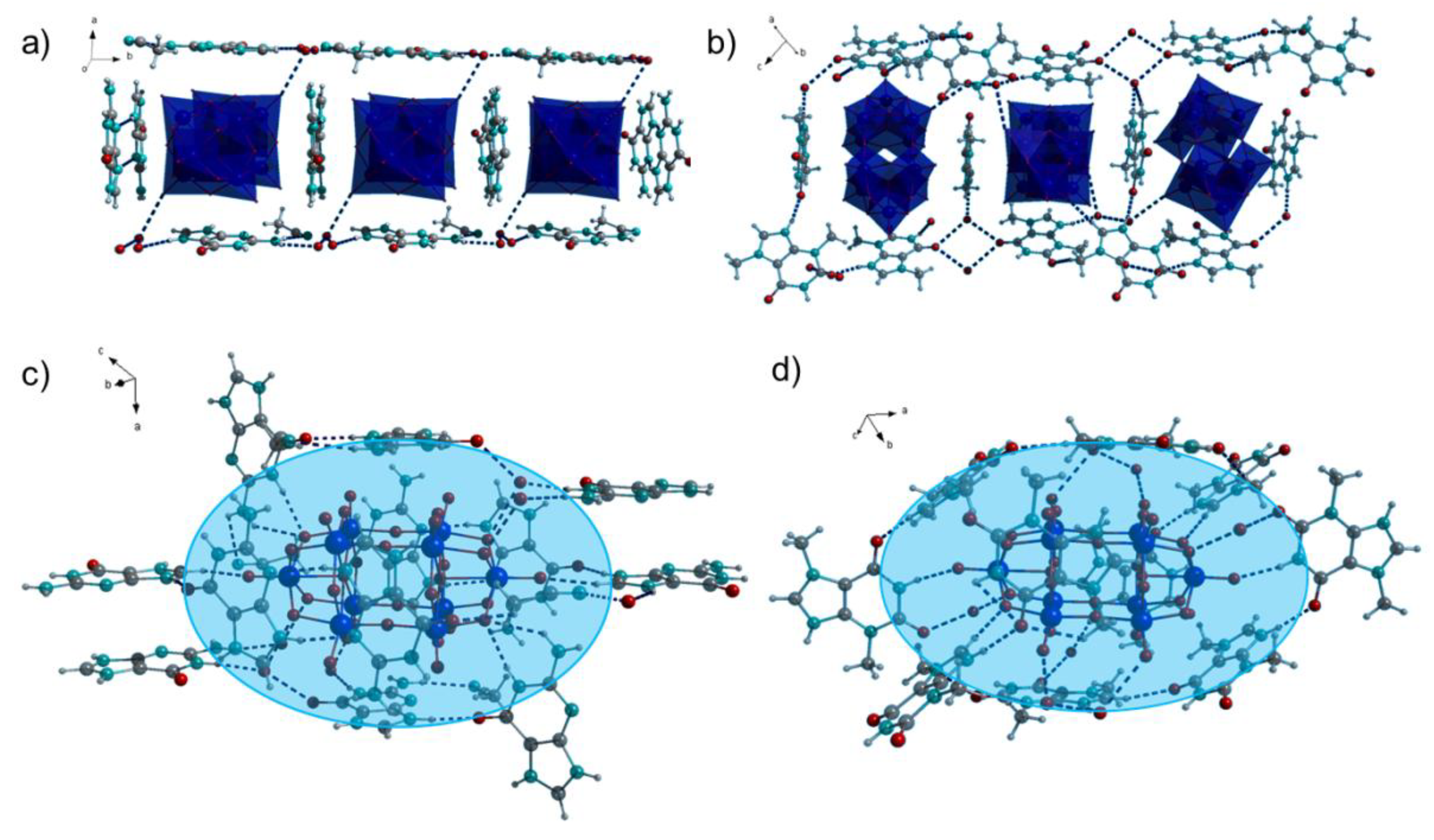
Further H-bonding interactions can be inferred from the crystal structures of 1 and 2 (Figure 2c,d, Tables S1–S3). The H-bonds are observable directly between the aromatic ring protons as well as amino protons of guaninium and terminal oxygen atoms of the decatungstate anion in 1. In some cases H-bonding interactions between the guaninium protons and µ2 bridging oxygen atoms of the decatungstate can be observed as well. Although these H-bond acceptors are more sterically hindered, their higher negative charge renders the interactions energetically favorable. Similar non-covalent interactions are observed in 2.
The organic molecules create a matrix surrounding the decatungstate anions supported by water molecules (indicated by the shaded blue area in the Figure 2c,d). The POMs are situated in the middle of the matrix and the organic cations interact with them by diverse non-covalent interactions.
The decatungstate anion consists of the two symmetry equivalent [W5O18] groups sharing four oxygen atoms with overall D4h symmetry (Figure 2) [21]. The bridging oxygen atoms of the POMs are more nucleophilic than the terminal ones [22]. Theoretical calculations indicated that the µ2 bridging atoms within the [W5O18] groups can be more easily protonated than those shared between them [23]. The positions of the protons on the purinium (theobrominium and guaninium) cations could not be determined from the Fourier difference map but by applying the riding model. Hence, there is a certain possibility that the protons are not attached to the N atoms of the purine rings, but to the oxygen atoms of the POMs.
Bond Valence Sum (BVS) calculations are often performed to assess the consistency of the charge distribution of the crystallographic data [24]. The calculated BVS of the terminal and parts of the bridging oxygen atoms is slightly lower than 2 (Tables S4–S6), due to partial charge transfer to the H-bond donors (Figure 2 and Figure 3). The BVS of the bridging oxygen atoms connecting both halves of the decatungstate is slightly higher than 2, as these do not accept any hydrogen bonds. The BVS of the central oxygen atoms is very low due to the surrounding by five W(VI) atoms in the decatungstate and six W(VI) atoms in the hexatungstate anion. Thus, BVS calculations confirm our model, in which oxygen atoms of the polyoxotungstates function as hydrogen bond acceptors but are not protonated themselves.
Except for these highly directional non-covalent interactions, further electrostatic interactions are inferable from the crystal structures of both compounds. Positively charged protonated purine bases surround the decatungstate anions to maximize the electrostatic stabilization of the crystal structures and gain additional stabilization from packing effects. (Figure 2a,b). The planes of the protonated purine bases are perpendicular to one another, due to the D4h symmetry of the polyoxoanion. The water molecules reinforce the crystal structure by H-bonds. The resulting geometric arrangement, in which the organic cations surround the polyoxoanions from all sides, resembles a nanosized box.
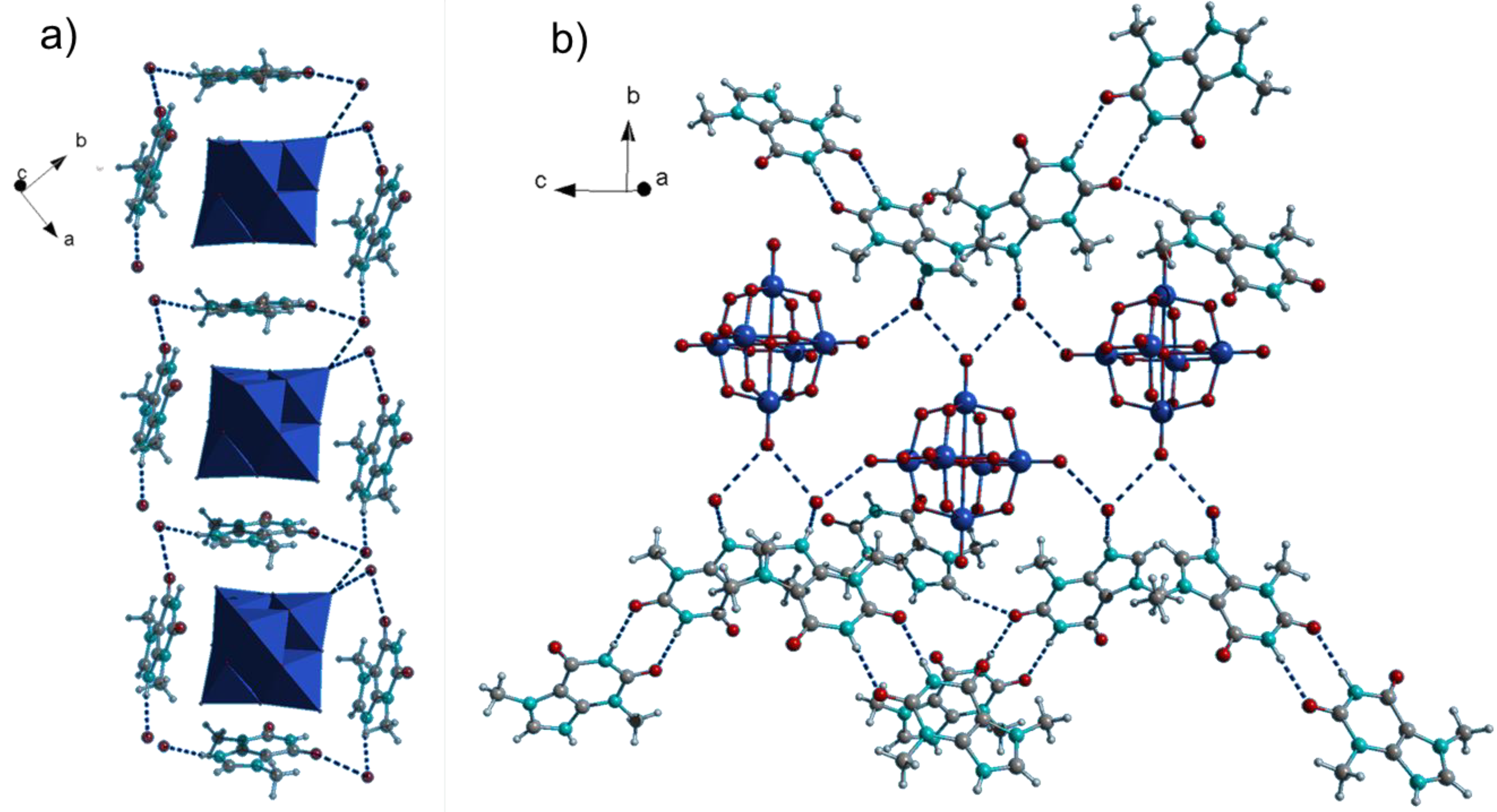
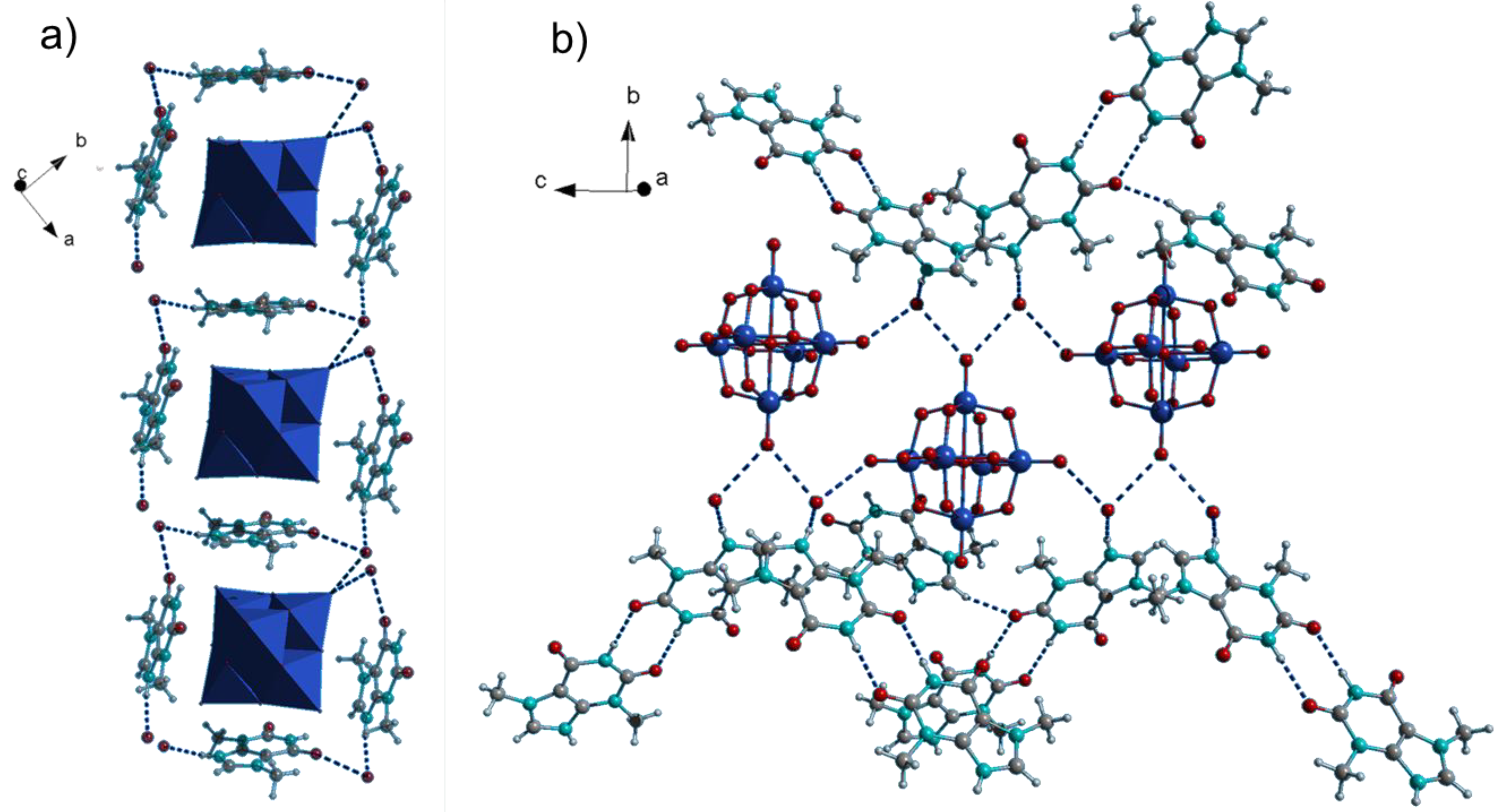
(ThbH)2[W6O19](H2O)2 (3) was obtained under similar synthetic conditions as 1 and 2. Its bulk purity was confirmed by powder X-ray diffraction (Figure S2). Several attempts to synthesize the respective compound with guaninium cations were unsuccessful, most likely due to higher solubility of guanine in acetonitrile. 3 contains the octahedral Lindqvist-type anion [W6O19]2− instead of the decatungstate anion as in 1 and 2 (Figure 3). The general geometrical traits such as the surrounding of the anions by theobrominium cations parallel to the POM’s faces remain the same (Figure 3a). On the other hand, 3 displays a quite distinctive H-bonding framework: The anions are interconnected by H-bonds through water molecules into chains running along the crystallographic c axis (Figure 3b). There is an additional H-bond between the water molecules and the theobrominium cations which build amide-to-amide dimers interconnecting the chains.
Figure 3.
Crystal structure of (ThbH)2[W6O19](H2O)2 (3). (a) Molecular boxes of organic cations connect the Lindqvist anions to chains; (b) H-bond framework of Lindqvist anions, water molecules and theobrominium dimers.
Figure 3.
Crystal structure of (ThbH)2[W6O19](H2O)2 (3). (a) Molecular boxes of organic cations connect the Lindqvist anions to chains; (b) H-bond framework of Lindqvist anions, water molecules and theobrominium dimers.
2.2. IR Spectroscopy
The IR absorption spectra are consistent with the crystal structures. Absorption bands characteristic for theobromine, water and decatungstate as well as hexatungstate are observed in the spectra of 2 and 3, respectively (see Experimental Section for assignments).
The strongest absorption bands between 400 and 1000 cm−1 where assigned to the stretching vibrations of the W–O bonds based on the assignments for polyoxoanions as described in the literature [21,25]. The absorption bands display a slight hypsochromic shift compared to the previously reported tributylammonium decatungstate [21], most likely owing to the stronger polarization of the anion by the protonated purine bases rather than soft trialkylammonium cations. The C=N and C=O double bonds of the theobrominium ions strongly absorb in the usual region between 1750 and 1500 cm−1. All absorption bands in the double bond region of the spectrum display hypsochromic shifts compared with neat theobromine. This fact can be accounted for by the positive charge of the protonated theobromine and accordingly stronger attraction of the carbonyl oxygen atom to the purine ring. The most prominent absorption bands in the fingerprint region (1500–1000 cm−1) are caused by the C–N stretching vibrations [26]. The N–H and C–H bonds of the purine rings absorb slightly above 3000 cm−1, the water molecules display a usual broad absorption in the region between 3300 and 3600 cm−1, with two distinct bands for symmetric and asymmetric H2O vibrations in the spectrum of 3. No distinct absorption bands are observed in this region in the IR spectrum of 2, due to the large amount of water embedded into the crystal structure which leads to a broadening of the absorption bands.
2.3. Thermal Properties
The behavior of 2 and 3 upon heating to higher temperatures was scrutinized in N2 atmosphere by DTA and TG (Figures S3 and S4). Both compounds exhibit exothermic reactions and emission of heat in the temperature ranges 100–350 °C and 150–400 °C, respectively. This thermodynamic behavior can be explained by redox reactions in course of which the organic parts are oxidized to volatile products such as CO2 and H2O, whereas the polyoxotungstates(VI) are reduced to tungsten oxides in lower oxidation states.
Indeed, powder X-ray diffraction experiments show the presence of tungsten(IV) oxide in both thermolysis residues (Figures S5 and S6). The measurements were undertaken in an alumosilicate crucible. The black sample material was stuck firmly at the bottom of the crucible; hence some of the crucible material was scratched off with a spatula together with a sample of compound 2.
3. Experimental Section
The precursors (NBu4)2[W6O19] and (NBu4)4[W10O32] were prepared according to literature procedures [27]. Phase purities of bulk samples of 2 and 3 were verified by X-ray powder diffraction on a STOE STADI transmission powder diffractometer (STOE & Cie GmbH, Darmstadt, Germany, Figures S1 and S2). FT-IR spectra were recorded on a Bruker IFS v/s spectrophotometer (Bruker Corporation, Billerica, MA, USA) using KBr pellets. UV/Vis measurements were accomplished on a Varian Cary 5E spectrophotometer (Varian, brand of Agilent Technologies PLC., Santa Clara, CA, USA) using KBr pellets as well. DTA and TG analyses were performed on a Netzsch STA 409 thermal analyzer (NETZSCH-Gerätebau GmbH, Selb, Germany) with a heating rate of 20 °C/min in a nitrogen atmosphere (Figures S3 and S4), the residues scrutinized with powder diffraction (Figures S5 and S6). Elemental analyses were carried out on a HEKAtech Euro EA 3000 elemental analyzer (HEKAtech GmbH, Wegberg, Germany). The BVS-calculations (Tables S4–S6) were performed with the VaList-software [24].
(GuaH)4[W10O32](H2O)2(MeCN) (1). Guanine (45 mg, 0.3 mmol, Acros Organics, Nidderau, Germany) was dissolved in a mixture of 26 mL of acetonitrile (BASF SE, Ludwigshafen, Germany) and 8 mL of 27% nitric acid (Merck KGaA, Darmstadt, Germany). (NBu4)4[W10O32] (200 mg, 0.06 mmol) was dissolved in 8 mL acetonitrile and added to the reaction mixture. The reaction mixture was filtered after stirring for 1.5 h. Yellow crystals precipitated after 2 weeks. The amount of material so obtained was too low for any further investigations. Attempts to upscale the reaction were not successful.
(ThbH)3(H3O)[W10O32](H2O)7.5 (2). Theobromine (270 mg, 1.50 mmol, Acros Organics, Nidderau, Germany) was dissolved in a mixture of 130 mL acetonitrile and 40 mL of 27% nitric acid. (NBu4)4[W10O32] (996 mg, 0.3 mmol) dissolved in 40 mL acetonitrile was added to the solution. After addition of 20 mL MeOH (BASF SE, Ludwigshafen, Germany) the reaction mixture was kept in a closed vessel for two weeks. The rest of the yellow crystalline product was washed twice with 5 mL portions of acetone and dried in air for a day. Yield: 540 mg, 0.18 mmol, 60% based on decatungstate. Elemental analysis calcd. (%) for C21H45N12O46.5W10: C 8.27, H 1.49, N 5.51; found: C 9.09, H 1.49, N 5.76. Characteristic IR absorption bands (KBr): ῦ (cm−1) = 3450 (br, s, H2O, υs and υ), 3163 (m, NH of thb, υ), 3045 (w, CH of thb, υ), 2826 (w, CH3, thb, υ), 1699 (s, C=N of thb, υ), 1651 (s, C=O of thb, υas), 1580 (m, C=C, thb, υ), 1549 (m, C=O, thb, υs), 1393 (m, C–N of thb, υ), 1279 (m, C–N of thb, υ), 1167 (m, C–N of thb, υ), 1030 (m, C–N of thb, υ), 964 (s, W–Oterminal, υas), 895 (s, W–Ocorner shared–W, υas), 795 (br, s, W–Oedge shared-W, υas), 677 (m, C=C–N of thb, δ), 604 (s, W–Oedge shared–W, υs), 513 (m, C=C–C of thb, δ), 432 (m, C–N–C of thb, δas), 401 (m, C–N–C of thb, δs). UV/Vis (KBr): λ (nm) = 203, 273, 326.
(ThbH)2[W6O19](H2O)2 (3). Theobromine (540 mg, 3.00 mmol) (Acros Organics, Nidderau, Germany) was dissolved in a mixture of 130 mL acetonitrile and 40 mL of 27% nitric acid. (NBu4)2[W6O19] (1.135 mg, 0.6 mmol) dissolved in 40 mL acetonitrile was added to the solution. After addition of 20 mL of MeOH, the reaction mixture was kept in a closed vessel for five days. The product was obtained as a white crystalline powder and dried over CaCl2 in a desiccator under dynamic vacuum (20 mbar) for a day. Yield: 960 mg, 0.51 mmol, 85% based on hexatungstate. Elemental analysis calcd. (%) for C14H22N8O25W6: C 9.31, H 1.23, N 6.21; found: C 8.80, H 1.27, N 5.99. Characteristic IR absorption bands (KBr): ῦ (cm−1) = 3578 (m, H2O, υas), 3524 (m, H2O, υs), 3141 (m, NH of thb, υ), 3053 (w, CH of thb, υ), 2852 (w, CH3, thb, υ), 1728 (s, C=N of thb, υ), 1684 (s, C=O of thb, υas), 1576 (m, C=C, thb, υ), 1545 (m, C=O, thb, υs), 1385 (m, C–N of thb, υ), 1323 (m, C–N of thb, υ), 1275 (m, C–N of thb, υ), 1163 (m, C–N of thb, υ), 1038 (m, C–N of thb, υ), 978 (s, W–Oterminal, υas), 804 (br, s, W–µ2–O–W, υas), 681 (m, C=C–N of thb, δ), 584 (m, W–µ2–O–W, υs), 511 (m, C=C–C of thb, δ), 444 (s, W–µ6–O–W, υas). UV/Vis: λ (nm) = 206, 275.
X-Ray Crystallography: Crystallographic data as well as structure solution and refinement details are summarized in Table 1 and have been deposited in more detail with the Cambridge Crystallographic Data Centre as supplementary publications Nos. CCDC 949335-949337. The alert level A in the checkcif-report of 1 is due to low electron density often observed on the aromatic carbon atoms of the purine rings. The alert level A in the checkcif-report of 3 is due to small distances between methyl-hydrogen atoms. These can be explained by the hydrophobic interactions between the methyl groups. All non-hydrogen atoms except some O, C and N atoms of the incorporated solvent molecules were refined anisotropically. The isotropic refinement of these atoms in the embedded solvent molecules is due to their high disorders, which are usual for incorporated solvent molecules. Copies of the data can be obtained, free of charge, on application to CHGC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223 336033 or E-Mail: [email protected]).
Suitable single crystals were carefully selected under a visible light microscope and mounted in a glass capillary. Intensity data for all compounds were measured at temperatures given in the Table 1 on an image-plate diffractometer STOE IPDS II equipped with a fine focus sealed X-ray tube, λ(MoKα) = 0.71073 Å, graphite monochromator, operating at 50 kV and 40 mA. Structure solutions and refinements were performed using the program package WinGX [28] which includes the programs SHELX [29] and Platon [29,30]. Refinement of F2 against all reflections. The weighted R-factor wR and goodness of fit are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2 (see CIF-Files for all discussed values). The threshold expression of F2 > 4σ (F2) is used only for calculating R-factors, etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on all data will be even larger. Spherical and numerical absorption corrections were accomplished with the X-red and X-shape software (STOE Darmstadt, Darmstadt, Germany) [31,32].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| empirical formula | C22H37N20O41W10 | C21H45N12O46.5W10 | C14H22N8O25W6 |
| M (g/mol) | 3076.2 | 3048.2 | 1805.47 |
| crystal system | monoclinic | triclinic | monoclinic |
| space group | P 21/c | P 1 | C 2/c |
| a (Å) | 14.477 (1) | 13.491 (1) | 16.810 (3) |
| b (Å) | 10.8678 (9) | 15.171 (1) | 13.051 (2) |
| c (Å) | 21.627 (2) | 17.069 (2) | 15.676 (3) |
| α (°) | 90 | 79.594 (8) | 90 |
| β (°) | 122.780 (6) | 89.386 (8) | 109.73 (1) |
| γ (°) | 90 | 70.001 (7) | 90 |
| ρcalc. (g/cm3) | 3.526 | 3.057 | 3.6960 |
| V (Å3) | 2860.8 (4) | 3223.8 (6) | 3237.2 (9) |
| Z | 2 | 2 | 4 |
| μ(MoKα) (mm−1) | 20.131 | 17.86 | 21.35 |
| T (K) | 170 (2) | 293 (2) | 293 (2) |
| reflns measured | 33411 | 41410 | 21383 |
| independent reflns | 6202 | 14242 | 3590 |
| parameters | 443 | 767 | 242 |
| R1 (I > 4σ) | 0.0434 | 0.0347 | 0.0361 |
| R1 (all data) | 0.0612 | 0.0590 | 0.1037 |
| wR2 (all data) | 0.1316 | 0.0950 | 0.0745 |
4. Conclusions
Three compounds containing protonated guanine and theobromine, respectively, as cations and polyoxotungstates as anions were characterized crystallographically. The organic cations lie parallel to the faces of the POMs in all characterized compounds, surrounding them in a box-like arrangement. The crystal structures are stabilized by a non-covalent framework of H-bonding, anion-π and electrostatic interactions. BVS calculations and IR measurements are consistent with the crystallographic data. The POMs are reduced by the surrounding organic cations upon thermolysis.
Acknowledgments
This work was generously supported by the University of Cologne. Vladislav Kulikov is grateful to the Studienstiftung des Deutschen Volkes for a Ph.D. scholarship.
Author Contributions
Vladislav Kulikov designed and performed the syntheses, solved and refined the crystal structures and drafted the manuscript. Gerd Meyer supervised the research, corrected and submitted the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pope, M.; Müller, A. Polyoxometalate Chemistry from Topology via Self-Assembly to Applications, 1st ed.; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Gouzerh, P.; Che, M. From Scheele and Berzelius to Müller: Polyoxometalates (POMs) revisited and the “missing link” between the bottom up and top down approaches. L’Actualité Chim. 2006, 298, 9–21. [Google Scholar]
- Long, D.-L.; Tsunashima, R.; Cronin, L. Polyoxometalates: Building blocks for functional nanoscale systems. Angew. Chem. Int. Ed. 2010, 49, 1736–1758. [Google Scholar] [CrossRef]
- Yamase, T.; Pope, M.T. Polyoxometalate Chemistry for Nano-Composite Design (Nanostructure Science and Technology); Kluwer Academic/Plenum Publishers: New York, NY, USA, 2002. [Google Scholar]
- Symes, M.D.; Cronin, L. Decoupling hydrogen and oxygen evolution during electrolytic water splitting using an electron-coupled-proton buffer. Nat. Chem. 2013, 5, 403–409. [Google Scholar] [CrossRef]
- Long, D.-L.; Abbas, H.; Kögerler, P.; Cronin, L. A high-nuclearity “celtic-ring” isopolyoxotungstate, [H12W36O120]12−, that captures trace potassium ions. J. Am. Chem. Soc. 2004, 126, 13880–13881. [Google Scholar] [CrossRef]
- Miras, H.N.; Yan, J.; Long, D.-L.; Cronin, L. Structural evolution of “s”-shaped [H4W22O74]12− and “§”-shaped [H12W34O116]18− isopolyoxotungstate clusters. Angew. Chem. Int. Ed. 2008, 47, 8420–8423. [Google Scholar] [CrossRef]
- Ito, T.; Fujimoto, N.; Uchida, S.; Iijima, J.; Naruke, H.; Mizuno, N. Polyoxotungstate-surfactant layered crystal toward conductive inorganic-organic hybrid. Crystals 2012, 2, 362–373. [Google Scholar] [CrossRef]
- Rhule, J.T.; Hill, C.L.; Judd, D.A.; Schinazi, R.F. Polyoxometalates in medicine. Chem. Rev. 1998, 98, 327–358. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; John Wiley & Sons, Inc.: San Francisco, CA, USA, 2011. [Google Scholar]
- Kortz, U.; Savelieff, M.G.; Ghali, F.Y.A.; Khalil, L.M.; Maalouf, S.A.; Sinno, D.I. Heteropolymolybdates of AsIII, SbIII, BiIII, SeIV, and TeIV functionalized by amino acids. Angew. Chem. Int. Ed. 2002, 41, 4070–4073. [Google Scholar] [CrossRef]
- Biradha, K.; Samai, S.; Maity, A.C.; Goswami, S. Supramolecular assembly of protonated xanthine alkaloids in their perchlorate salts. Cryst. Growth Des. 2010, 10, 937–942. [Google Scholar] [CrossRef]
- Zhai, H.; Liu, S.; Peng, J.; Hu, N.; Jia, H. Synthesis, crystal structure, and thermal property of a novel supramolecular assembly: (NH4)2(C8H10N4O2)4[H4V10O28]·2H2O, constructed from decavanadate and caffeine. J. Chem. Crystallogr. 2004, 34, 541–548. [Google Scholar] [CrossRef]
- Ng, S.W. Interpretation of Diammonium Decavanadate(V)-Tetra(caffeine) Dihydrate as Tetra(caffeinium) Decavanadate(V) Tetrahydrate. J. Chem. Crystallogr. 2008, 38, 483. [Google Scholar] [CrossRef]
- Inoue, M.; Yamase, T. Crystal structure of the pentamolybdate complex coordinated by adenosine-5'-monophosphoric acid. Bull. Chem. Soc. Jpn. 1996, 69, 2863–2868. [Google Scholar] [CrossRef]
- Kulikov, V.; Meyer, G. A new strategy for the synthetic assembly of inorganic–organic silver(I)-polyoxometalate hybrid structures employing noncovalent interactions between theobromine ligands. Cryst. Growth Des. 2013, 13, 2916–2927. [Google Scholar] [CrossRef]
- Kulikov, V.; Meyer, G. Hexa- to octamolybdate rearrangement leads to the new coordination polymer [Ag(PhCN)(thb)]4[β-Mo8O26](PhCN)2. Z. Anorg. Allg. Chem. 2014, 640, 19–22. [Google Scholar] [CrossRef]
- Hoogsten, K. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Cryst. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Egli, M.; Sarkhel, S. Lone pair-aromatic interactions: To stabilize or not to stabilize. Acc. Chem. Res. 2007, 40, 197–205. [Google Scholar] [CrossRef]
- Fuchs, J.; Hartl, H.; Schiller, W.; Gerlach, U. Die Kristallstruktur des Tributylammoniumdekawolframats [(C4H9)2NH]4W10O23. Acta Cryst. B 1976, 32, 740–749. [Google Scholar] [CrossRef]
- Poblet, J.M.; Lopez, X.; Bo, C. Ab initio and DFT modelling of complex materials: Towards the understanding of electronic and magnetic properties of polyoxometalates. Chem. Soc. Rev. 2003, 32, 297–308. [Google Scholar] [CrossRef]
- Fernández, J.A.; López, X.; Poblet, J.M. A DFT study on the effect of metal, anion charge, heteroatom and structure upon the relative basicities of polyoxoanions. J. Mol. Catal. A Chem. 2007, 262, 236–242. [Google Scholar] [CrossRef]
- Wills, A.S. VaList-Program. Available online: https://dl.dropboxusercontent.com/u/8933134/Website/Site/Software/Software.html (accessed on 15 February 2014).
- Mattes, R.; Bierbüsse, H.; Fuchs, J. Schwingungsspektren und kraftkonstanten von polyanionen mit M6O19-gruppen. Z. Anorg. Allg. Chem. 1971, 385, 230–242. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Sankari, G.; Ponnusamy, S. Vibrational spectral investigation on xanthine and its derivatives—Theophylline, caffeine and theobromine. Spectrochim. Acta Part A 2005, 61, 117–127. [Google Scholar] [CrossRef]
- Fournier, M. Tetrabutylammonium Isopolyoxometallates. In Inorganic Syntheses; Ginsberg, A.P., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; Volume 27, pp. 74–85. [Google Scholar]
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Spek, A. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Stoe; Cie. X-SHAPE; Stoe & Cie GmbH: Darmstadt, Germany, 1999. [Google Scholar]
- Stoe; Cie. X-RED; Stoe & Cie GmbH: Darmstadt, Germany, 2001. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Kulikov, V.; Meyer, G. Polyoxotungstates in Molecular Boxes of Purine Bases. Crystals 2014, 4, 64-73. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst4010064
AMA Style
Kulikov V, Meyer G. Polyoxotungstates in Molecular Boxes of Purine Bases. Crystals. 2014; 4(1):64-73. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst4010064
Chicago/Turabian StyleKulikov, Vladislav, and Gerd Meyer. 2014. "Polyoxotungstates in Molecular Boxes of Purine Bases" Crystals 4, no. 1: 64-73. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst4010064