The Inflammatory Cytokine IL-3 Hampers Cardioprotection Mediated by Endothelial Cell-Derived Extracellular Vesicles Possibly via Their Protein Cargo

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of Human EC (Human Microvascular Endothelial Cell Line-1, HMEC-1 and Human Umbilical Vein Cells, HUVEC), and Rat Embryonic Cardiac Myoblasts (H9c2)
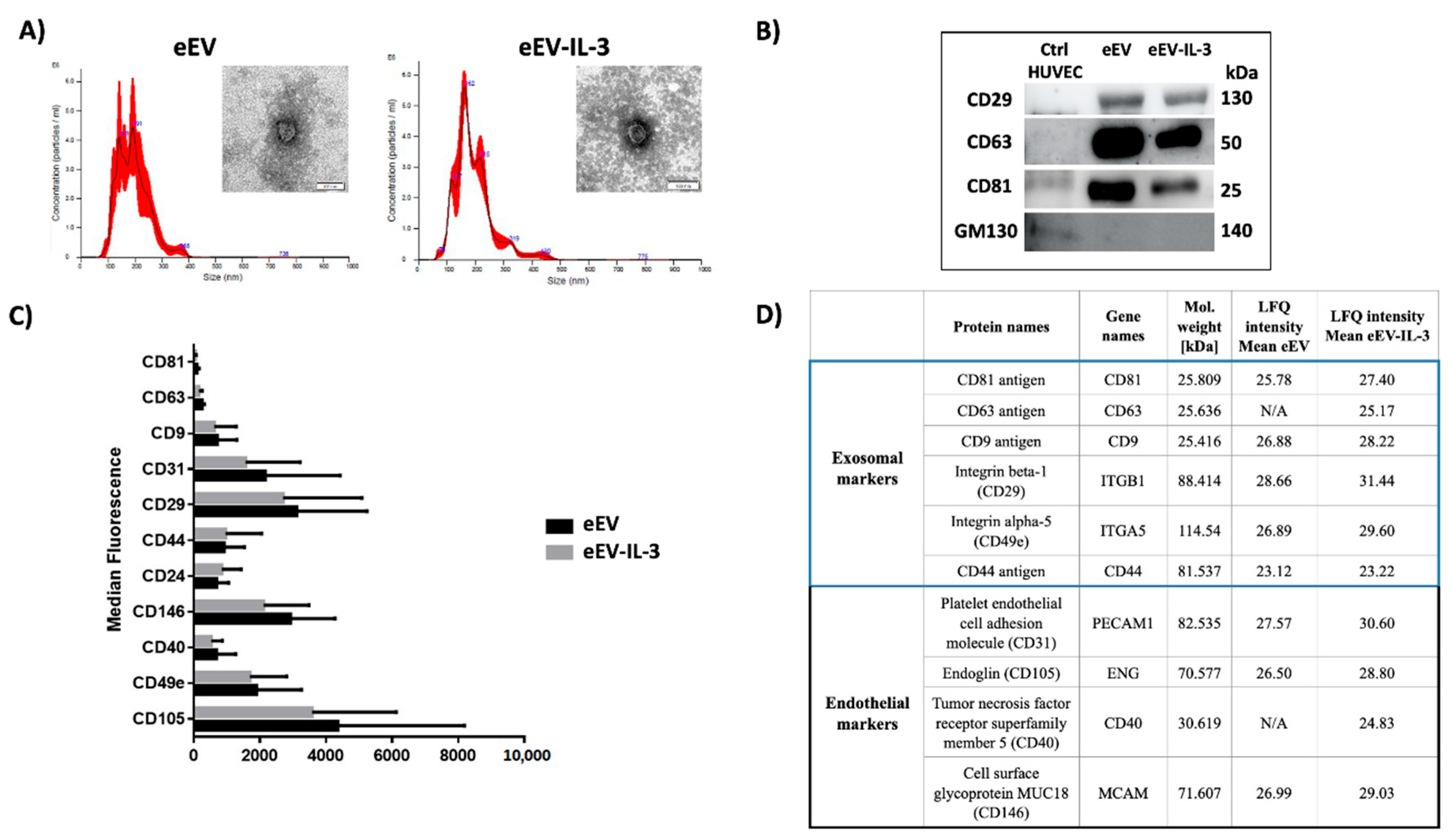
2.2. EC-EV Isolation and Characterization
2.3. Transmission Electron Microscopy
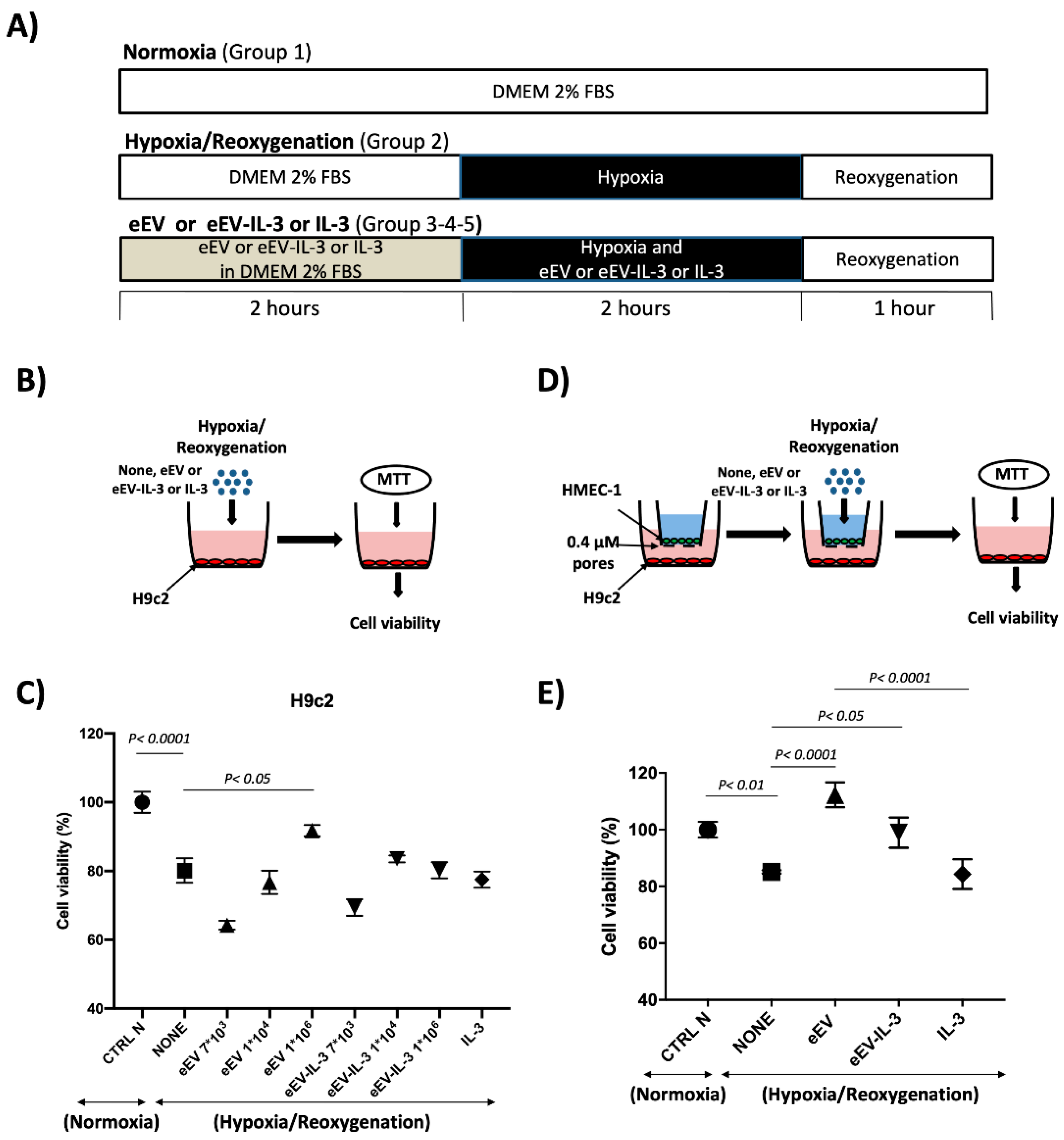
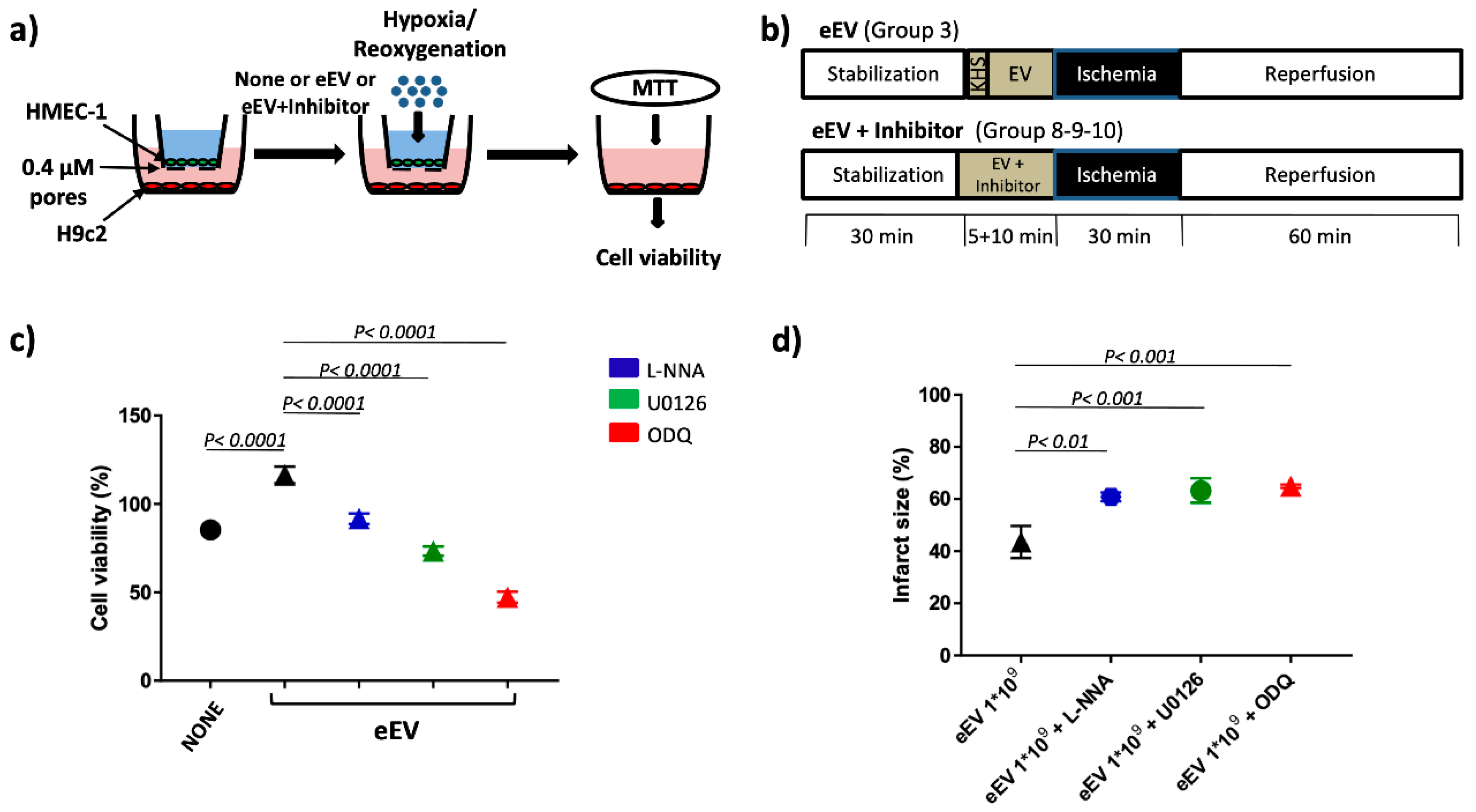
2.4. Hypoxia/Reoxygenation (H/R) Protocol
2.5. MTT Assay
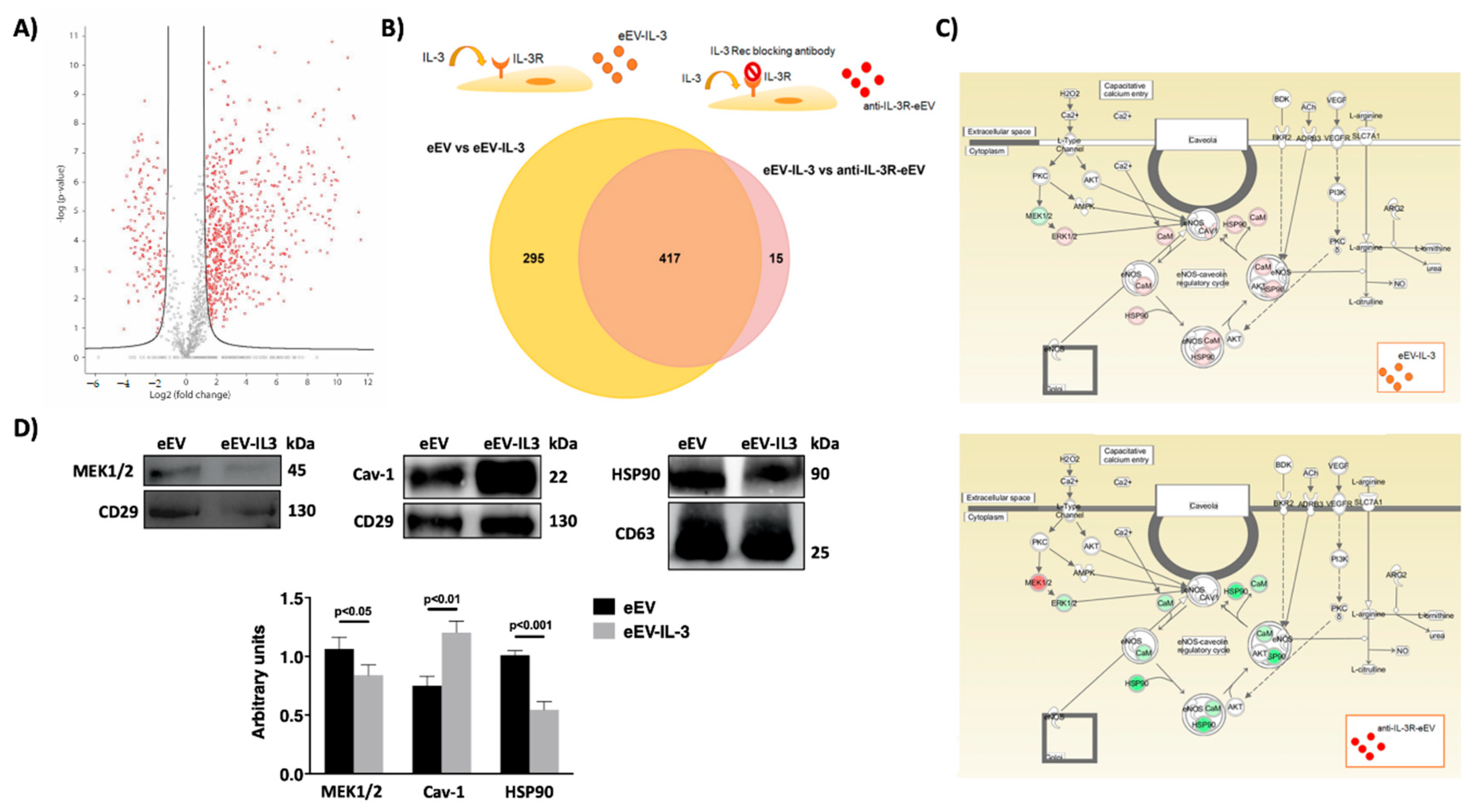
2.6. Label-Free Mass Spectrometry Analysis
2.7. Protein Pathway Analysis
2.8. Animals
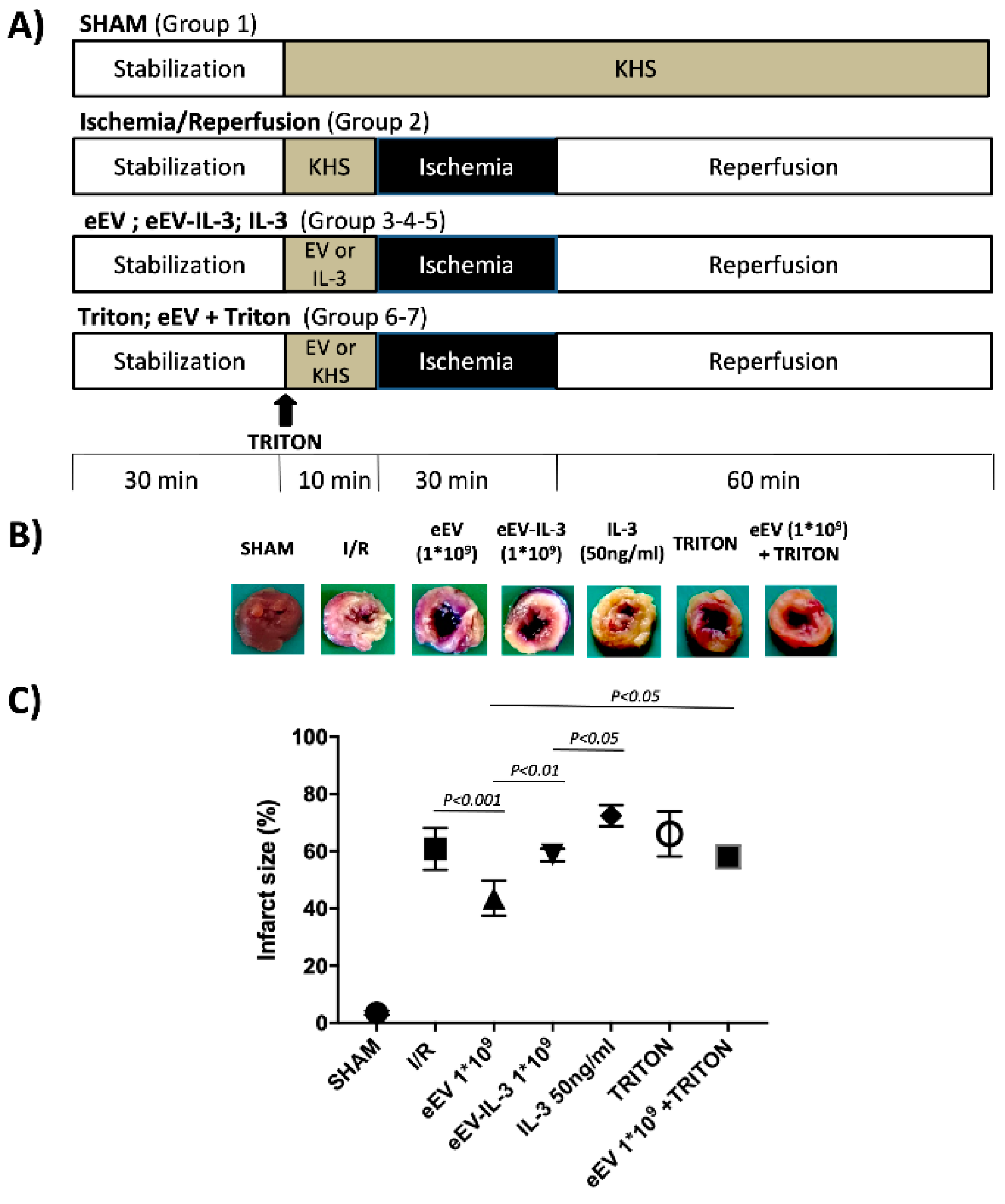
2.9. Ischemia/Reperfusion (I/R) Studies
2.10. Experimental Groups
- (1)
- SHAM (n = 3) only KHS has been infused.
- (2)
- I/R group (n = 8) after stabilization, only I/R protocol was performed [38].
- (3)
- eEV group (n = 5), eEV (1 × 109/mL final concentration) were diluted in KHS and infused into the hearts, through a collateral line for 10 min, then the hearts underwent I/R protocol.
- (4)
- eEV-IL-3 group (n = 5) eEV-IL-3 (1 × 109/mL final concentration) were diluted in KHS and infused into the heart, through a collateral line for 10 min, then hearts underwent I/R protocol.
- (5)
- IL-3 group (n = 5) IL-3 (50 ng/mL) [14] was diluted in KHS and infused into the heart, through a collateral line for 10 min, then the hearts underwent I/R protocol.
- (6)
- (7)
- (8)
- eEV+L-NNA group (n = 3), the eNOS inhibitor N omega-nitro-L-arginine (LNNA, 100 µM) was used to assess the involvement of eNOS enzyme in eEV-induced cardioprotection (1 × 109/mL final concentration) [41].
- (9)
- eEV+U0126 group (n = 3), the MEK1/2 blocker 1,4-Diamino-2,3-dicyano-1,4-bis(2 aminophenylthio) butadiene (U0126: 60 µM) was used to ascertain the involvement of MEK1/2 in eEV-(1 × 109/mL final concentration) induced cardioprotection [38].
- (10)
- eEV+ODQ group (n = 3), the GC blocker 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ;10µM) was used to ascertain the involvement of the GC enzyme in eEV- (1 × 109/mL final concentration) induced cardioprotection [37].
2.11. Infarct Size Assessment
2.12. Western Blot Analysis
2.13. Chemicals
2.14. Statistical Analysis
3. Results
3.1. eEV and eEV-IL-3 Have Similar Size and Surface Markers
3.2. Both eEV and eEV-IL-3 Induce Protection in a Simulated In Situ Condition, While Only eEV Directly Trigger Cardioprotective Signals
3.3. eEV, But Not eEV-IL-3, Exert Endothelial-Dependent Protection against I/R in the Whole Heart
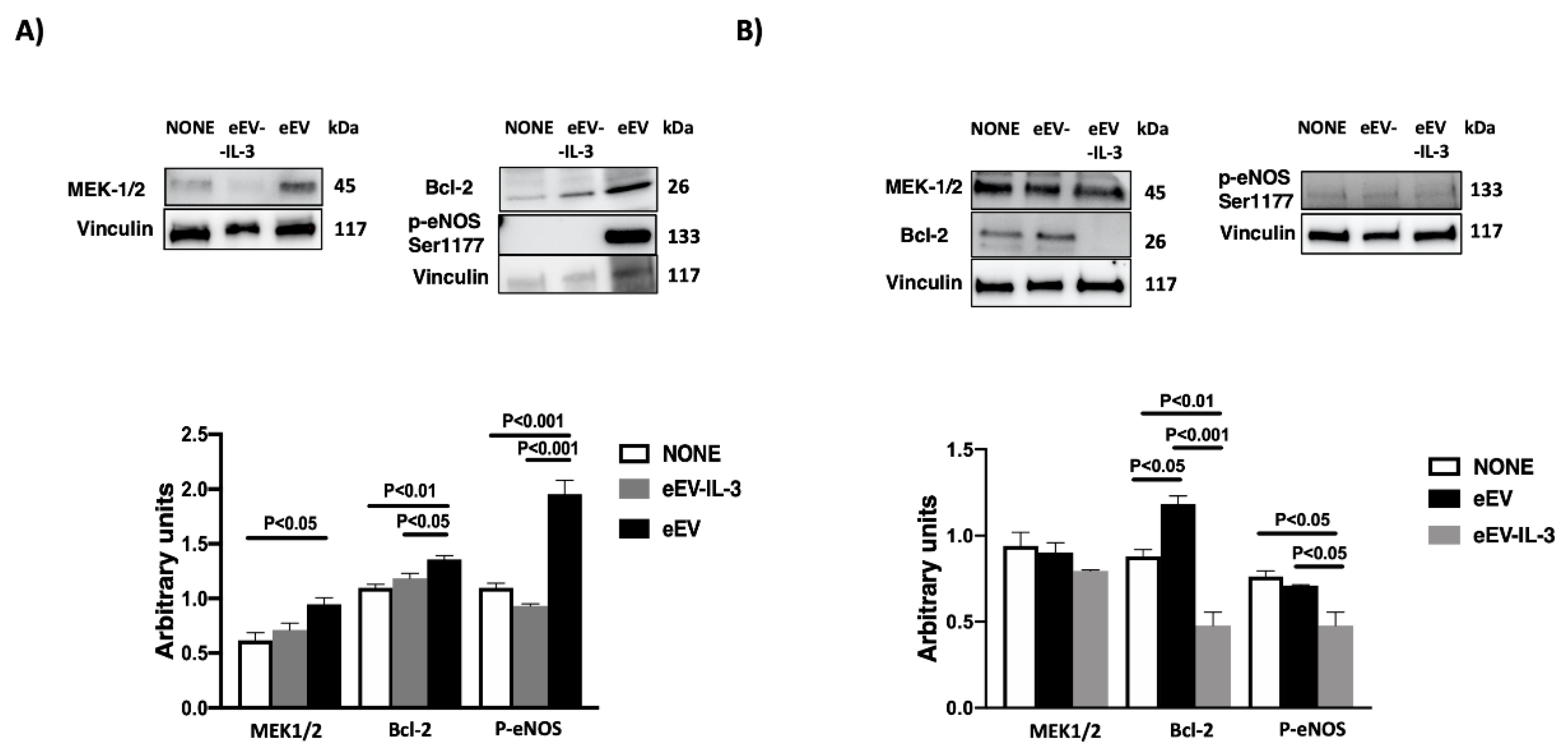
3.4. eEV Are Enriched in MEK1/2 and HPS90 While eEV-IL-3 in the eNOS Antagonist, Caveolin 1
3.5. MEK1/2/eNOS/GC Pathway Is Involved in eEV-Mediated Cardio-Protection
3.6. eEV But Not eEV-IL-3 Treatment Induces the Expression Bcl-2 and the Phosphorylation of eNOS In Vitro
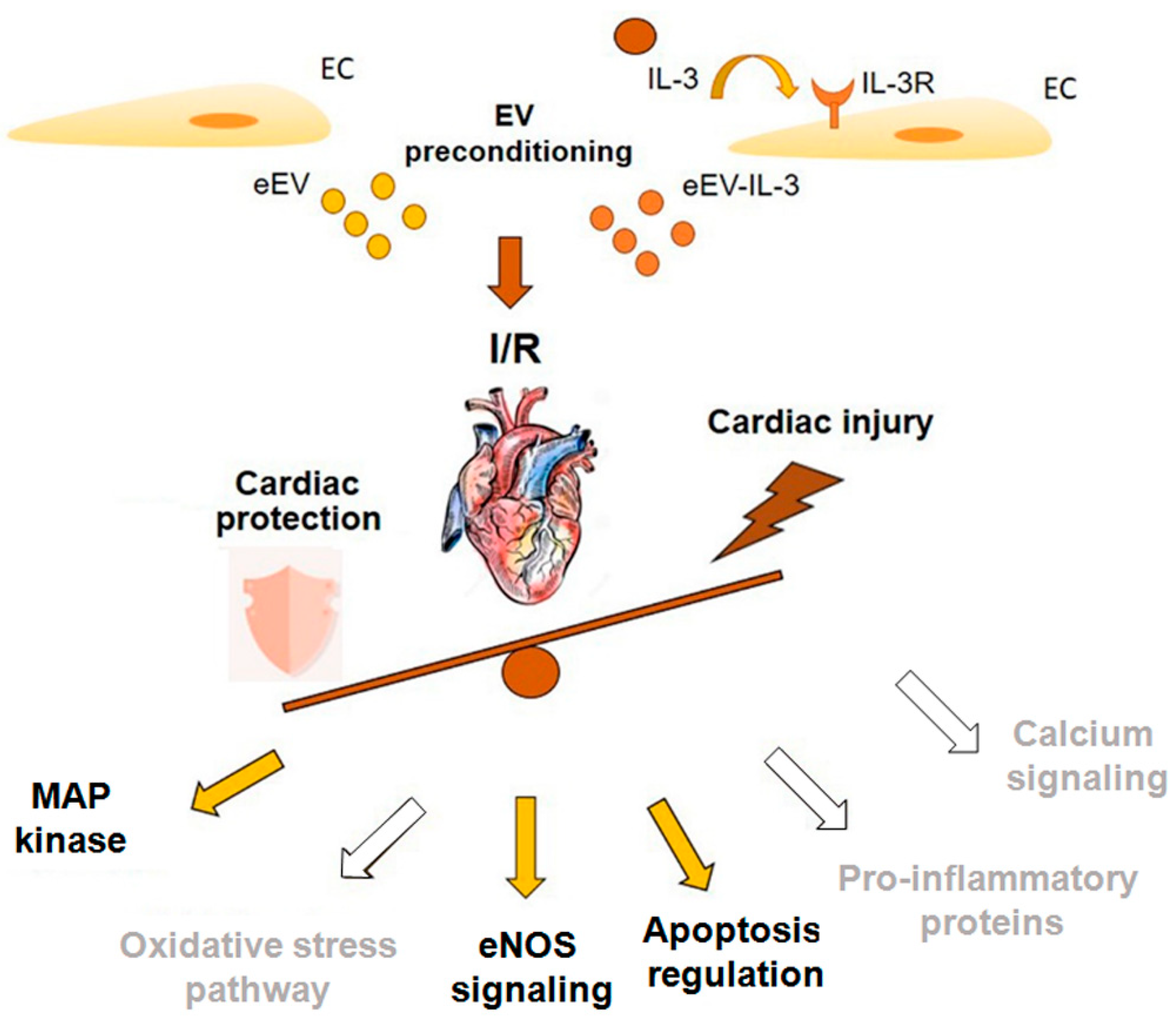
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Author Disclosure Statement
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| Bcl-2 | B-cell-lymphoma-2 |
| CVDs | Cardiovascular Diseases |
| DMSO | Dimethyl Sulfoxide |
| EC | Endothelial Cell |
| eEV | EV released by EC |
| eEV-IL-3 | EV released by EC treated with IL-3 |
| eNOS | Endothelial Nitric Oxide Synthase |
| EV | Extracellular Vesicles |
| FACS | Fluorescence-Activated Cell Sorting |
| FBS | Fetal Bovine Serum |
| FDR | False Discovery Rate |
| GC | Guanylyl Cyclase |
| GO | Gene Ontology |
| H9c2 | Rat Embryonic Cardiac Myoblast |
| HMEC-1 | Human Endothelial Cell-1 |
| HUVEC | Human Umbilical Vein Endothelial Cell |
| I/R | Ischemia/Reperfusion |
| IL-3 | Interleukin-3 |
| IPA | Ingenuity Pathway Analysis |
| KHS | Krebs–Henseleit Buffer Solution |
| L-NNA | N Omega-Nitro-L-Arginine |
| MEK1/2 | Mitogen-Activated Protein Kinase 1/2 |
| MI | Myocardial Infarction |
| NO | Nitric Oxide |
| NTA | Nanoparticle Tracking Analysis |
| ODQ | Oxadiazolo[4,3-a]quinoxalin-1-one |
| PPCI | Primary Percutaneous Coronary Intervention |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| TEM | Transmission Electron Microscopy |
| TFA | Trifluoroacetic Acid |
References
- Shi, A.; Tao, Z.; Wei, P.; Zhao, J. Epidemiological aspects of heart diseases. Exp. Ther. Med. 2016, 12, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Mathur, A.; Rothman, M.T. Recent advances in primary percutaneous intervention for acute myocardial infarction. Heart 2005, 91, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Sezer, M.; Van Royen, N.; Umman, B.; Bugra, Z.; Bulluck, H.; Hausenloy, D.J.; Umman, S. Coronary Microvascular Injury in Reperfused Acute Myocardial Infarction: A View from an Integrative Perspective. J. Am. Hear. Assoc. 2018, 7, 009949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J. Conditioning the heart to prevent myocardial reperfusion injury during PPCI. Eur. Hear. J. Acute Cardiovasc. Care 2012, 1, 13–32. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Granata, R.; Tocchetti, C.G.; Gallo, M.P.; Alloatti, G.; Pagliaro, P. Endogenous Cardioprotective Agents: Role in Pre and Postconditioning. Curr. Drug Targets 2015, 16, 843–867. [Google Scholar] [CrossRef] [Green Version]
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing myocardial infarct size: Challenges and future opportunities. Heart 2015, 102, 341–348. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Zhao, Z.-Q. Oxidative stress-elicited myocardial apoptosis during reperfusion. Curr. Opin. Pharmacol. 2004, 4, 159–165. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Greenberger, J.S.; Eckner, R.J.; Sakakeeny, M.; Marks, P.; Reid, D.; Nabel, G.; Hapel, A.; Ihle, J.N.; Humphries, K.C. Interleukin 3-dependent hematopoietic progenitor cell lines. Fed. Proc. 1983, 42, 2762–2771. [Google Scholar] [PubMed]
- Brizzi, M.F.; Garbarino, G.; Rossi, P.R.; Pagliardi, G.L.; Arduino, C.; Avanzi, G.C.; Pegoraro, L. Interleukin 3 stimulates proliferation and triggers endothelial-leukocyte adhesion molecule 1 gene activation of human endothelial cells. J. Clin. Investig. 1993, 91, 2887–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihle, J.N. Interleukin-3 and Hematopoiesis. Chem. Immunol. Allergy 1992, 51, 65–106. [Google Scholar] [CrossRef]
- Dentelli, P.; Del Sorbo, L.; Rosso, A.; Molinar, A.; Garbarino, G.; Camussi, G.; Pegoraro, L.; Brizzi, M.F. Human IL-3 stimulates endothelial cell motility and promotes in vivo new vessel formation. J. Immunol. 1999, 163, 2151–2159. [Google Scholar]
- Lombardo, G.; Dentelli, P.; Togliatto, G.; Rosso, A.; Gili, M.; Gallo, S.; Deregibus, M.C.; Camussi, G.; Brizzi, M.F. Activated Stat5 trafficking Via Endothelial Cell-derived Extracellular Vesicles Controls IL-3 Pro-angiogenic Paracrine Action. Sci. Rep. 2016, 6, 25689. [Google Scholar] [CrossRef] [Green Version]
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular Vesicles in Angiogenesis. Circ. Res. 2017, 120, 1658–1673. [Google Scholar] [CrossRef] [PubMed]
- Oggero, S.; Austin-Williams, S.; Norling, L.V. The Contrasting Role of Extracellular Vesicles in Vascular Inflammation and Tissue Repair. Front. Pharmacol. 2019, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Dashevsky, O.; Rivo, J.; Gozal, Y.; Varon, D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc. Res. 2005, 67, 30–38. [Google Scholar] [CrossRef]
- Caccioppo, A.; Franchin, L.; Grosso, A.; Angelini, F.; D’Ascenzo, F.; Brizzi, M.F. Ischemia Reperfusion Injury: Mechanisms of Damage/Protection and Novel Strategies for Cardiac Recovery/Regeneration. Int. J. Mol. Sci. 2019, 20, 5024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giricz, Z.; Varga, Z.V.; Baranyai, T.; Bencsik, P.; Pálóczi, K.; Kittel, Á.; Buzás, E.I.; Ferdinandy, P. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J. Mol. Cell. Cardiol. 2014, 68, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, Y.; Zhu, Q.; Zhao, J.; Wang, Y.; Shang, M.; Liu, M.; Wu, Y.; Song, J.; Liu, Y. Protective effects of circulating microvesicles derived from ischemic preconditioning on myocardial ischemia/reperfusion injury in rats by inhibiting endoplasmic reticulum stress. Apoptosis 2018, 23, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shang, M.; Zhang, M.; Wang, Y.; Chen, Y.; Wu, Y.; Liu, M.-L.; Song, J.; Liu, Y. Microvesicles derived from hypoxia/reoxygenation-treated human umbilical vein endothelial cells promote apoptosis and oxidative stress in H9c2 cardiomyocytes. BMC Cell Biol. 2016, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ. Res. 2018, 123, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Nahrendorf, M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc. Res. 2014, 102, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, C.; Cappello, S.; Mancardi, D.; Raimondo, S.; Rastaldo, R.; Gattullo, D.; Losano, G.; Pagliaro, P. Post–conditioning reduces infarct size in the isolated rat heart: Role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res. Cardiol. 2005, 101, 168–179. [Google Scholar] [CrossRef]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Femminò, S.; Penna, C.; Bessone, F.; Caldera, F.; Dhakar, N.K.; Cau, D.; Pagliaro, P.; Cavalli, R.; Trotta, F. α-Cyclodextrin and α-Cyclodextrin Polymers as Oxygen Nanocarriers to Limit Hypoxia/Reoxygenation Injury: Implications from an In Vitro Model. Polymers 2018, 10, 211. [Google Scholar] [CrossRef] [Green Version]
- Wiklander, O.P.B.; Bostancioglu, R.B.; Welsh, J.A.; Zickler, A.M.; Murke, F.; Corso, G.; Felldin, U.; Hagey, D.W.; Evertsson, B.; Liang, X.M.; et al. Systematic Methodological Evaluation of a Multiplex Bead-Based Flow Cytometry Assay for Detection of Extracellular Vesicle Surface Signatures. Front. Immunol. 2018, 9, 1326. [Google Scholar] [CrossRef] [Green Version]
- Giusti, I.; Di Francesco, M.; Cantone, L.; D’Ascenzo, S.; Bollati, V.; Carta, G.; Dolo, V. Time-dependent release of extracellular vesicle subpopulations in tumor CABA I cells. Oncol. Rep. 2015, 34, 2752–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Bi, Y.; Liu, X.; Wei, M.; Zhang, Q. Upregulation of connexin43 by glucose deprivation in H9c2 cells via the extracellular signal-regulated kinase/mitogen-activated protein kinase signaling pathway. Mol. Med. Rep. 2017, 17, 729–734. [Google Scholar] [CrossRef] [Green Version]
- Monastyrskaya, E.A.; Andreeva, L.V.; Duchen, M.R.; Wiegant, F.; Bayda, L.A.; Manukhina, E.B.; Malyshev, I.Y. Adaptation to Heat of Cardiomyoblasts in Culture Protects Them against Heat Shock: Role of Nitric Oxide and Heat Shock Proteins. Biochemistry 2003, 68, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bassino, E.; Fornero, S.; Gallo, M.P.; Gallina, C.; Femminò, S.; Levi, R.; Tota, B.; Alloatti, G. Catestatin Exerts Direct Protective Effects on Rat Cardiomyocytes Undergoing Ischemia/Reperfusion by Stimulating PI3K-Akt-GSK3β Pathway and Preserving Mitochondrial Membrane Potential. PLoS ONE 2015, 10, e0119790. [Google Scholar] [CrossRef] [PubMed]
- Frøyset, A.K.; Edson, A.; Gharbi, N.; Khan, E.A.; Dondorp, D.; Bai, Q.; Tiraboschi, E.; Suster, M.L.; Connolly, J.B.; Burton, E.A.; et al. Astroglial DJ-1 over-expression up-regulates proteins involved in redox regulation and is neuroprotective in vivo. Redox Biol. 2018, 16, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.-S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Tullio, F.; Femminò, S.; Rocca, C.; Angelone, T.; Cerra, M.C.; Gallo, M.P.; Gesmundo, I.; Fanciulli, A.; Brizzi, M.F.; et al. Obestatin regulates cardiovascular function and promotes cardio-protection through the nitric oxide pathway. J. Cell. Mol. Med. 2017, 21, 3670–3678. [Google Scholar] [CrossRef]
- Russo, I.; Femminò, S.; Barale, C.; Tullio, F.; Geuna, S.; Cavalot, F.; Pagliaro, P.; Penna, C. Cardio-protective Properties of Human Platelets Are Lost in Uncontrolled Diabetes Mellitus: A Study in Isolated Rat Hearts. Front. Physiol. 2018, 9, 875. [Google Scholar] [CrossRef]
- Raffaella, R.; Paolocci, N.; Chiribiri, A.; Penna, C.; Gattullo, D.; Pagliaro, P. Cytochrome P-450 metabolite of arachidonic acid mediates bradykinin-induced negative inotropic effect. Am. J. Physiol. Circ. Physiol. 2001, 280, H2823–H2832. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, P.; Penna, C.; Raffaella, R.; Mancardi, D.; Crisafulli, A.; Losano, G.; Gattullo, D. Endothelial cytochrome P450 contributes to the acetylcholine-induced cardiodepression in isolated rat hearts. Acta Physiol. Scand. 2004, 182, 11–20. [Google Scholar] [CrossRef]
- Rastaldo, R.; Cappello, S.; Folino, A.; Berta, G.N.; Sprio, A.E.; Losano, G.; Samaja, M.; Pagliaro, P. Apelin-13 limits infarct size and improves cardiac postischemic mechanical recovery only if given after ischemia. Am. J. Physiol. Circ. Physiol. 2011, 300, H2308–H2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bøtker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.M.; Deshwal, S.; Devaux, Y.; Di Lisa, F.; Di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 1–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.-W.; Li, L.; Zhang, Q.; Zhang, H.; Xiu, R. Effects of tumor necrosis factor-α-induced exosomes on the endothelial cellular behavior, metabolism and bioenergetics. Microcirculation 2018, 26, e12515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Javeed, N.; Mukhopadhyay, D. Exosomes and their role in the micro-/macro-environment: A comprehensive review. J. Biomed. Res. 2017, 31, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Riquelme, J.A.; Zheng, Y.; Vicencio, J.M.; Lavandero, S.; Yellon, D.M. Endothelial cells release cardioprotective exosomes that may contribute to ischaemic preconditioning. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Arnal, J.-F.; Dinh-Xuan, A.-T.; Pueyo, M.; Darblade, B.; Rami, J. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell. Mol. Life Sci. 1999, 55, 1078–1087. [Google Scholar] [CrossRef]
- Balligand, J.-L.; Kobzik, L.; Han, X.; Kaye, D.M.; Belhassen, L.; O’Hara, D.S.; Kelly, R.A.; Smith, T.W.; Michel, T. Nitric Oxide-dependent Parasympathetic Signaling Is Due to Activation of Constitutive Endothelial (Type III) Nitric Oxide Synthase in Cardiac Myocytes. J. Biol. Chem. 1995, 270, 14582–14586. [Google Scholar] [CrossRef] [Green Version]
- García-Cardeña, G.; Martasek, P.; Masters, B.S.; Skidd, P.M.; Couet, J.; Li, S.C.; Lisanti, M.P.; Sessa, W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 1997, 272, 25437–25440. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Sellers, S.; Stefanovic, N.; Leung, C.; Tan, S.M.; Huet, O.; Granville, D.J.; Cooper, M.E.; De Haan, J.B.; Bernatchez, P. Direct Endothelial Nitric Oxide Synthase Activation Provides Atheroprotection in Diabetes-Accelerated Atherosclerosis. Diabetes 2015, 64, 3937–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajmani, P.; Yadav, H.N.; Singh, M.; Sharma, P.L. Possible involvement of caveolin in attenuation of cardioprotective effect of ischemic preconditioning in diabetic rat heart. BMC Cardiovasc. Disord. 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, R.; Collino, M.; Penna, C.; Nigro, D.; Chiazza, F.; Fracasso, V.; Tullio, F.; Alloatti, G.; Pagliaro, P.; Aragno, M. Maladaptive Modulations of NLRP3 Inflammasome and Cardioprotective Pathways Are Involved in Diet-Induced Exacerbation of Myocardial Ischemia/Reperfusion Injury in Mice. Oxidative Med. Cell. Longev. 2015, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, R.; Aragno, M.; Alloatti, G.; Collino, M.; Penna, C.; Pagliaro, P. Metaflammation: Tissue-Specific Alterations of the NLRP3 Inflammasome Platform in Metabolic Syndrome. Curr. Med. Chem. 2018, 25, 1294–1310. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Sugimoto, T.; Aoki, M.; Kida, I.; Tomita, N.; Moriguchi, A.; Maeda, K.; Sawa, Y.; Kaneda, Y.; Higaki, J.; et al. In vivo transfection of cis element ‘decoy’ against nuclear factor-kB binding site prevents myocardial infarction. Nat. Med. 1997, 3, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Minghua, W.; Zhijian, G.; Chahua, H.; Qiang, L.; Minxuan, X.; Luqiao, W.; Weifang, Z.; Peng, L.; Biming, Z.; Lingling, Y.; et al. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingenuity Canonical Pathways | −log(p-Value) |
|---|---|
| Mitochondrial Dysfunction | 12.9 |
| Caveolar-Mediated Endocytosis Signaling | 11.3 |
| Integrin Signaling | 9.69 |
| Remodeling of Epithelial Adherens Junctions | 9.26 |
| Oxidative Phosphorylation | 8.34 |
| Regulation of eIF4 and p70S6K Signaling | 8.02 |
| Actin Cytoskeleton Signaling | 7.97 |
| Sirtuin Signaling Pathway | 7.33 |
| NRF2-mediated Oxidative Stress Response | 6.14 |
| PI3K/AKT Signaling | 5.95 |
| Regulation of Actin-based Motility by Rho | 5.88 |
| VEGF Signaling | 5.05 |
| p70S6K Signaling | 4.82 |
| Leukocyte Extravasation Signaling | 4.41 |
| Apoptosis Signaling | 3.32 |
| mTOR Signaling | 3.16 |
| Hypoxia Signaling in the Cardiovascular System | 3.14 |
| Clathrin-mediated Endocytosis Signaling | 3.11 |
| Endoplasmic Reticulum Stress Pathway | 2.96 |
| Protein Kinase A Signaling | 2.75 |
| Antigen Presentation Pathway | 2.42 |
| ERK/MAPK Signaling | 2.34 |
| Granulocyte Adhesion and Diapedesis | 2.23 |
| Cardiac Hypertrophy Signaling | 2.05 |
| Role of NFAT in Regulation of the Immune Response | 1.98 |
| CXCR4 Signaling | 1.98 |
| Glutathione Redox Reactions I | 1.93 |
| Acute Phase Response Signaling | 1.92 |
| Calcium Signaling | 1.89 |
| Glutathione-mediated Detoxification | 1.74 |
| Arginine Biosynthesis IV | 1.64 |
| Thioredoxin Pathway | 1.51 |
| Aspartate Degradation II | 1.51 |
| Calcium-induced T Lymphocyte Apoptosis | 1.46 |
| IL-1 Signaling | 1.46 |
| FcÎ3 Receptor-mediated Phagocytosis in Macrophages and Monocytes | 1.44 |
| Nitric Oxide Signaling in the Cardiovascular System | 1.44 |
| Superoxide Radicals Degradation | 1.39 |
| CCR3 Signaling in Eosinophils | 1.31 |
| Diseases or Functions Annotation | p-Value | Predicted Activation State | Activation z-Score | # Molecules |
|---|---|---|---|---|
| Cell movement of endothelial cells | 4.09 × 109 | Increased | 2.919 | 41 |
| Migration of endothelial cells | 9.55 × 109 | Increased | 2.429 | 38 |
| Vasculogenesis | 2.45 × 108 | Increased | 2.106 | 46 |
| Interaction of endothelial cells | 7.55 × 108 | Increased | 2.587 | 22 |
| Cell death of endothelial cells | 1.21 × 107 | −1.535 | 21 | |
| Binding of endothelial cells | 2.61 × 107 | Increased | 2.402 | 21 |
| Apoptosis of endothelial cells | 6.80 × 107 | −1.591 | 19 | |
| Binding of vascular endothelial cells | 5.83 × 106 | Increased | 2.825 | 15 |
| Adhesion of endothelial cells | 7.63 × 106 | Increased | 2.188 | 15 |
| Apoptosis of vascular endothelial cells | 8.03 × 106 | −0.969 | 14 | |
| Endothelial cell development | 1.60 × 105 | Increased | 2.57 | 30 |
| Movement of vascular endothelial cells | 2.68 × 105 | 1.976 | 21 | |
| Adhesion of vascular endothelial cells | 6.44 × 105 | Increased | 3.087 | 11 |
| Attachment of vascular endothelial cells | 9.14 × 105 | 4 | ||
| Proliferation of endothelial cells | 1.16 × 104 | Increased | 2.363 | 26 |
| Apoptosis of microvascular endothelial cells | 3.45 × 104 | 0.261 | 6 | |
| Migration of vascular endothelial cells | 4.53 × 104 | 1.604 | 17 | |
| Synthesis of reactive oxygen species | 0.0008 | −0.132 | 13 | |
| Cell spreading of endothelial cells | 0.0014 | Increased | 2.219 | 5 |
| Endothelial barrier function of vascular endothelial cells | 0.0022 | 3 | ||
| Cell viability of endothelial cells | 0.0026 | 1.633 | 7 | |
| Formation of endothelial tube | 0.0035 | 4 | ||
| Generation of reactive oxygen species | 0.0045 | 0.714 | 5 | |
| Production of reactive oxygen species | 0.0050 | −0.566 | 10 | |
| Morphology of endothelial cells | 0.0068 | 3 | ||
| Cell movement of muscle cells | 0.0068 | 4 | ||
| Cell spreading of vascular endothelial cells | 0.0090 | 3 | ||
| Survival of vascular endothelial cells | 0.0095 | 1 | 5 | |
| Transendothelial migration of regulatory T lymphocytes | 0.0097 | 2 | ||
| Cell movement of muscle precursor cells | 0.0097 | 2 | ||
| Occlusion of artery | 0.0138 | 5 | ||
| Tubulation of endothelial cells | 0.0147 | 0.632 | 10 | |
| Angiogenesis of endothelial cells | 0.0147 | 3 | ||
| Differentiation of vascular endothelial cells | 0.0147 | 3 | ||
| Transendothelial migration of T lymphocytes | 0.0147 | 3 | ||
| Coronary artery disease | 0.0157 | 2 | ||
| Adhesion of muscle cells | 0.0157 | 2 | ||
| Sliding of myofilaments | 0.0173 | 5 | ||
| Biosynthesis of hydrogen peroxide | 0.0182 | 3 | ||
| Atherosclerosis | 0.0269 | 4 | ||
| Differentiation of endothelial cells | 0.0285 | 0.447 | 5 | |
| Migration of endothelial progenitor cells | 0.0312 | 2 | ||
| Proliferation of myoblasts | 0.0312 | 2 | ||
| Cell proliferation of vascular endothelial cells | 0.0323 | Increased | 2.735 | 12 |
| Shape change of vascular endothelial cells | 0.0371 | 0.555 | 5 | |
| Permeability of endothelial progenitor cells | 0.0413 | 1 | ||
| Injury of cardiomyocytes | 0.0413 | 1 | ||
| Diastolic heart failure | 0.0413 | 1 | ||
| Perfusion of myocardium | 0.0413 | 1 | ||
| Vasoconstriction of artery | 0.0413 | 1 | ||
| Arrest in mid-G1 phase of microvascular endothelial cells | 0.0413 | 1 | ||
| Anoikis of vascular endothelial cells | 0.0413 | 1 | ||
| Delay in initiation of fusion of myoblasts | 0.0413 | 1 | ||
| Aggregation of myoblasts | 0.0413 | 1 | ||
| Activation of myoblasts | 0.0413 | 1 | ||
| Activation of myotube | 0.0413 | 1 | ||
| Morphology of cardiovascular system | 0.0423 | 6 |
| Ingenuity Canonical Pathways | −log(p-Value) | Gene List |
|---|---|---|
| Agranulocyte Adhesion and Diapedesis | 4.76 | ACTA1, ACTG1, CD99, CDH5, FN1, GLG1, GNAI2, ICAM1, ICAM2, ITGA2, ITGA5, ITGA6, ITGB1, MSN, MYH10, MYH9, MYL6, PECAM1, PODXL, RDX |
| Leukocyte Extravasation Signaling | 4.41 | ACTA1, ACTG1, ACTN1, ACTN4, CD99, CDH5, CTNNA1, CTNNB1, F11R, GNAI2, ICAM1, ITGA2, ITGA5, ITGA6, ITGB1, ITGB3, MAPK1, MSN, MYL6, PECAM1, PXN, RDX |
| NF-kB Activation by Viruses | 2.24 | ITGA2, ITGA5, ITGA6, ITGAV, ITGB1, ITGB3, MAPK1, RALA, RALB, RAP2B |
| Granulocyte Adhesion, and Diapedesis | 2.23 | CD99, CDH5, GLG1, GNAI2, ICAM1, ICAM2, ITGA2, ITGA5, ITGA6, ITGB1, ITGB3, MSN, PECAM1, RDX |
| IL-8 Signaling | 2.01 | ANGPT2, CSTB, GNAI2, GNB1, GNG12, ICAM1, IQGAP1, ITGAV, ITGB3, LASP1, MAP2K1, MAPK1, RALA, RALB, RAP2B, RHOC |
| Role of NFAT in Regulation of the Immune Response | 1.98 | CALM1, CHP1, GNA11, GNAI2, GNAQ, GNB1, GNG12, HLA-A, HLA-B, MAP2K1, MAPK1, RALA, RALB, RAP2B, XPO1 |
| Acute Phase Response Signaling | 1.92 | A2M, CP, FGA, FN1, HMOX2, HNRNPK, HP, MAP2K1, MAPK1, RALA, RALB, RAP2B, RBP4, SERPINE1 |
| Complement System | 1.81 | C1QBP, C6, C8B, CD59, MASP1 |
| IL-1 Signaling | 1.46 | GNA11, GNAI2, GNAQ, GNB1, GNG12, MAPK1, PRKAR1A, PRKAR2A |
| CCR3 Signaling in Eosinophils | 1.31 | CALM1, CFL1, GNAI2, GNB1, GNG12, MAP2K1, MAPK1, RALA, RALB, RAP2B |
| Regulation of IL-2 Expression in Activated and Anergic T Lymphocytes | 1.21 | CALM1, CHP1, MAP2K1, MAPK1, RALA, RALB, RAP2B |
| Role of NFAT in Cardiac Hypertrophy | 1.18 | CALM1, CHP1, GNAI2, GNAQ, GNB1, GNG12, MAP2K1, MAPK1, PDIA3, PRKAR1A, PRKAR2A, RALA, RALB, RAP2B |
| IL-12 Signaling and Production in Macrophages | 0.93 | APOB, APOC2, CLU, MAP2K1, MAPK1, MST1, PCYOX1, PON1, RBP4 |
| IL-3 Signaling | 0.78 | CHP1, MAP2K1, MAPK1, RALA, RALB, RAP2B |
| OX40 Signaling Pathway | 0.76 | B2M, HLA-A, HLA-B, TNFSF4 |
| IL-2 Signaling | 0.75 | MAP2K1, MAPK1, RALA, RALB, RAP2B |
| IL-4 Signaling | 0.73 | HLA-A, HLA-B, HMGA1, RALA, RALB, RAP2B |
| GM-CSF Signaling | 0.61 | MAP2K1, MAPK1, RALA, RALB, RAP2B |
| IL-15 Signaling | 0.58 | MAP2K1, MAPK1, RALA, RALB, RAP2B |
| IL-17 Signaling | 0.47 | MAP2K1, MAPK1, RALA, RALB, RAP2B |
| IL-6 Signaling | 0.31 | A2M, MAP2K1, MAPK1, RALA, RALB, RAP2B |
| NF-kB Signaling | 0 | RALA, RALB, RAP2B, UBE2N, UBE2V1 |
| CD40 Signaling | 0 | ICAM1, MAP2K1, MAPK1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penna, C.; Femminò, S.; Tapparo, M.; Lopatina, T.; Fladmark, K.E.; Ravera, F.; Comità, S.; Alloatti, G.; Giusti, I.; Dolo, V.; et al. The Inflammatory Cytokine IL-3 Hampers Cardioprotection Mediated by Endothelial Cell-Derived Extracellular Vesicles Possibly via Their Protein Cargo. Cells 2021, 10, 13. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010013
Penna C, Femminò S, Tapparo M, Lopatina T, Fladmark KE, Ravera F, Comità S, Alloatti G, Giusti I, Dolo V, et al. The Inflammatory Cytokine IL-3 Hampers Cardioprotection Mediated by Endothelial Cell-Derived Extracellular Vesicles Possibly via Their Protein Cargo. Cells. 2021; 10(1):13. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010013
Chicago/Turabian StylePenna, Claudia, Saveria Femminò, Marta Tapparo, Tatiana Lopatina, Kari Espolin Fladmark, Francesco Ravera, Stefano Comità, Giuseppe Alloatti, Ilaria Giusti, Vincenza Dolo, and et al. 2021. "The Inflammatory Cytokine IL-3 Hampers Cardioprotection Mediated by Endothelial Cell-Derived Extracellular Vesicles Possibly via Their Protein Cargo" Cells 10, no. 1: 13. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010013