The Inhibition of CDK8/19 Mediator Kinases Prevents the Development of Resistance to EGFR-Targeting Drugs


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Gefitinib and Erlotinib Selection and Sensitivity Testing
2.3. Cetuximab Selection and Sensitivity Testing
3. Results
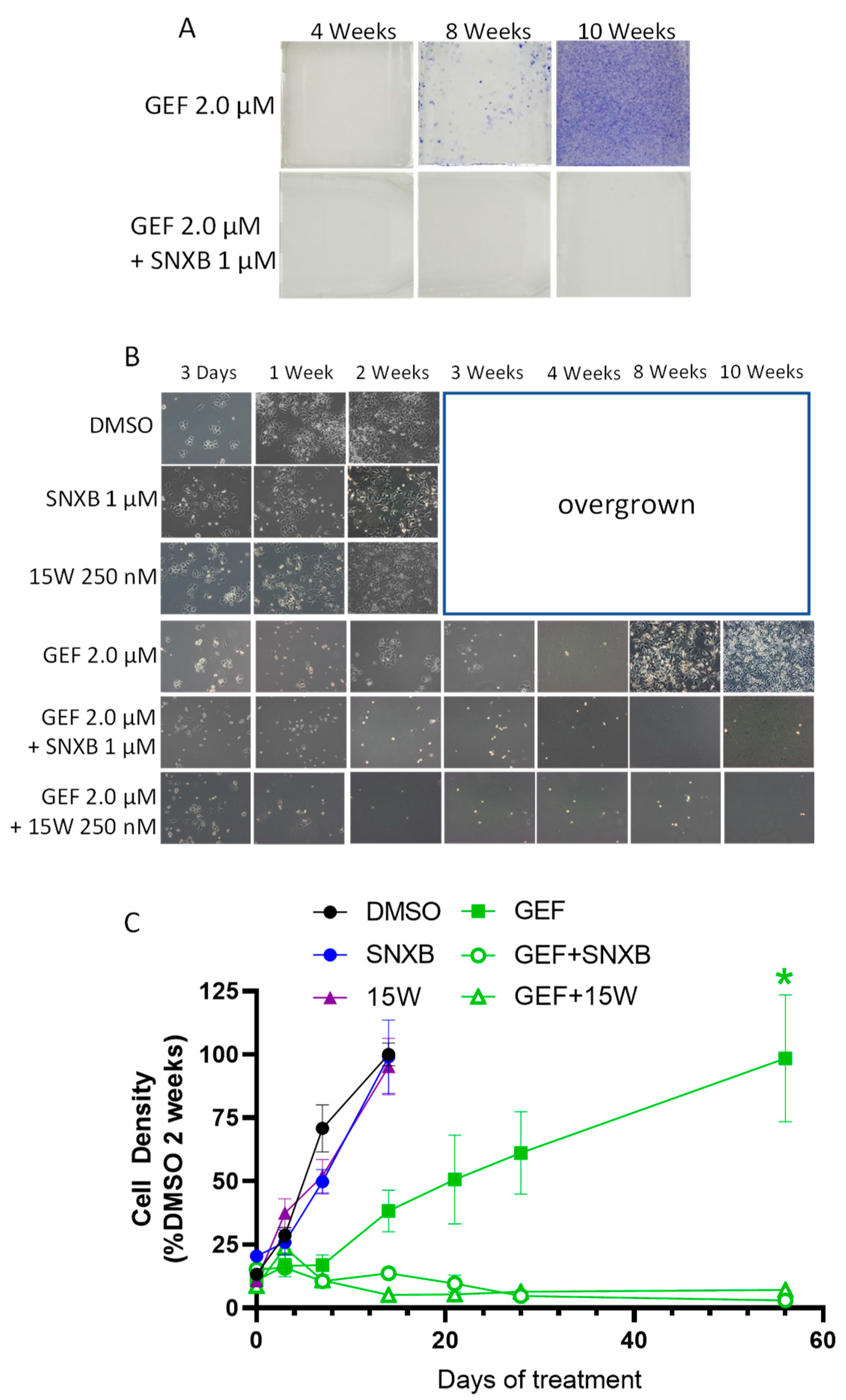
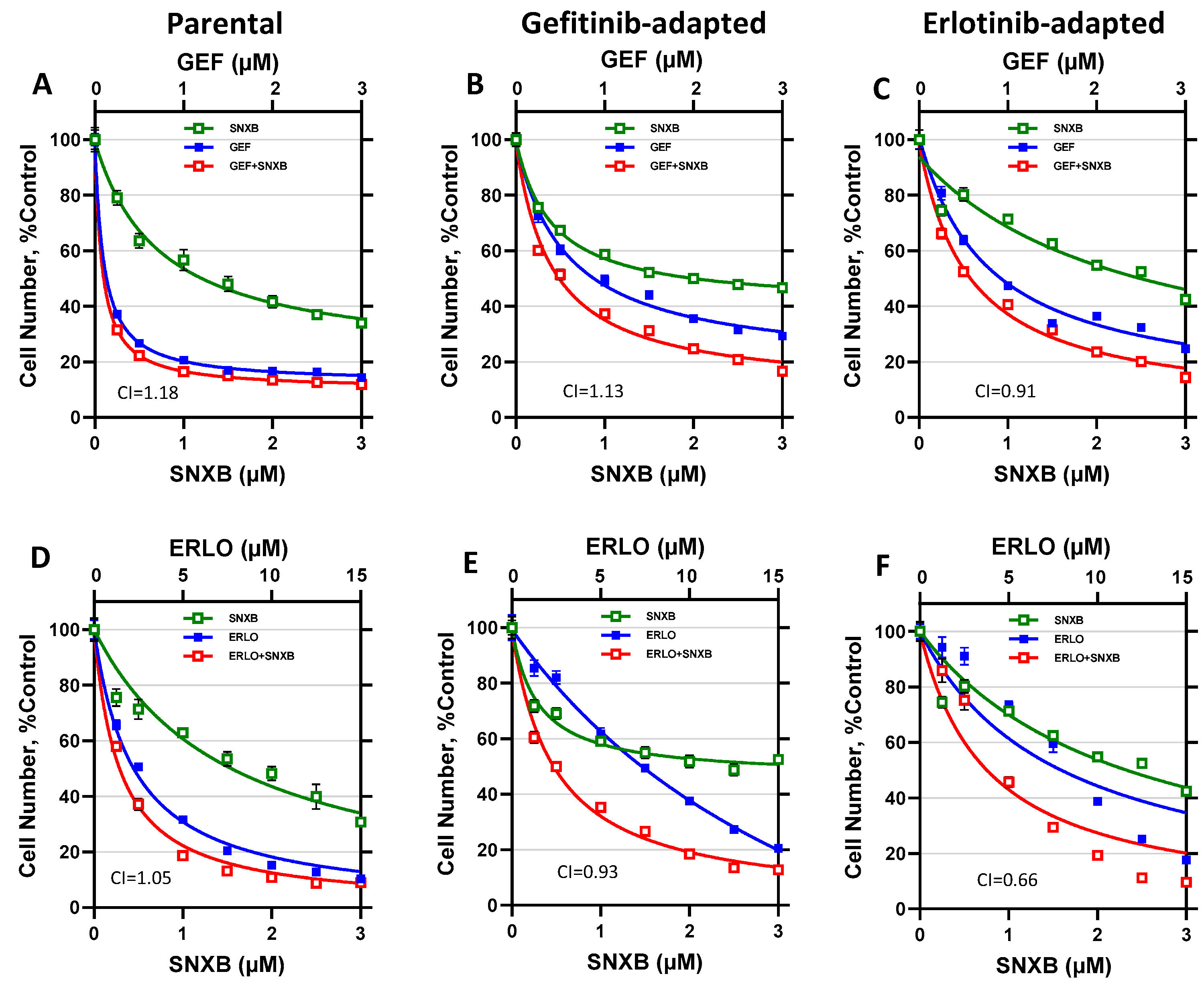
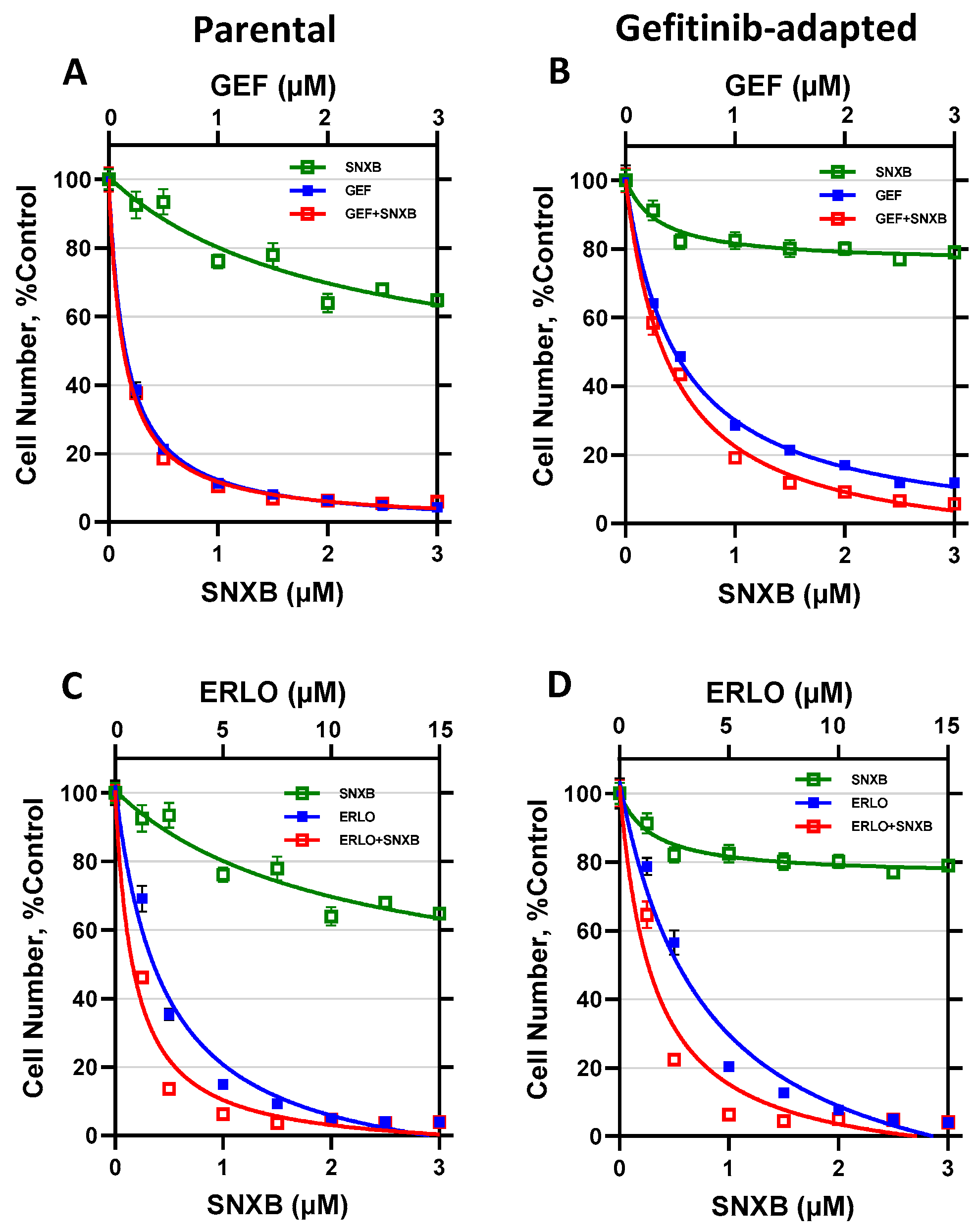
3.1. Effects of CDK8/19 Inhibition on Gefitinib and Erlotinib Resistance
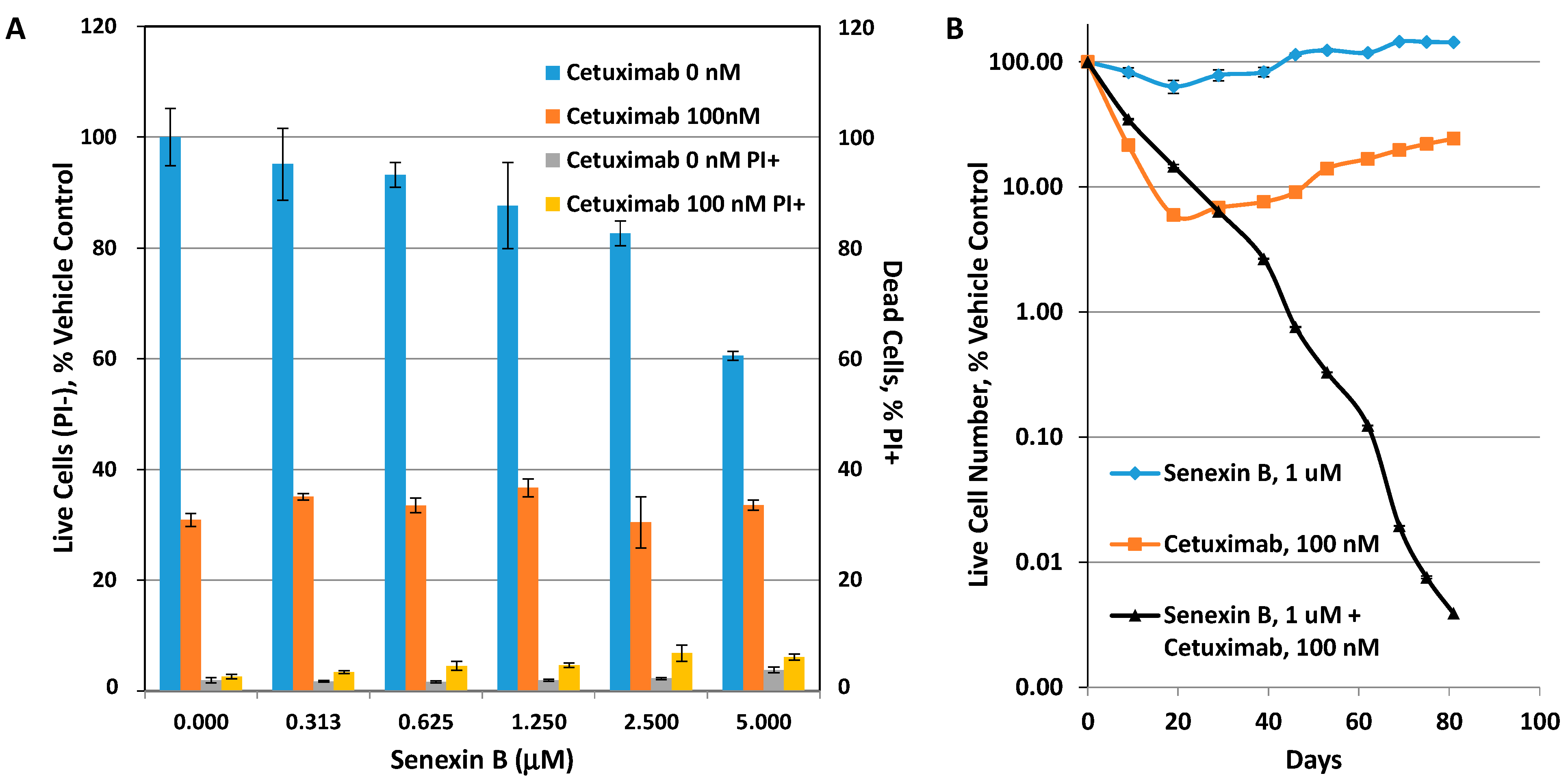
3.2. Effects of CDK8/19 Inhibition on Cetuximab Resistance
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaudhary, P.M.; Roninson, I.B. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J. Natl. Cancer Inst. 1993, 85, 632–639. [Google Scholar] [CrossRef]
- Shtil, A.A. Signal transduction pathways and transcriptional mechanisms as targets for prevention of emergence of multidrug resistance in human cancer cells. Curr. Drug Targets 2001, 2, 57–77. [Google Scholar] [CrossRef]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisco, A.O.; Brock, A.; Zhou, J.; Moor, A.; Mojtahedi, M.; Jackson, D.; Huang, S. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat. Commun. 2013, 4, 2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgia, R.; Kulkarni, P. The Genetic/Non-genetic Duality of Drug ’Resistance’ in Cancer. Trends Cancer 2018, 4, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Kyrochristos, I.D.; Ziogas, D.E.; Roukos, D.H. Drug resistance: Origins, evolution and characterization of genomic clones and the tumor ecosystem to optimize precise individualized therapy. Drug Discov. Today 2019, 24, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Hammerlindl, H.; Schaider, H. Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance. J. Cell Commun. Signal. 2018, 12, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.Y.; Zhong, H. Epigenetic programming contributes to development of drug resistance in hematological malignancies. Front. Biosci. (Landmark Ed.) 2015, 20, 728–742. [Google Scholar]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.R.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Homet Moreno, B.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.L.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, T.D.; Vartanian, S.; Cote, A.; Bellon, S.; Duplessis, M.; Flynn, E.M.; Hewitt, M.; Huang, H.R.; Kiefer, J.R.; Murray, J.; et al. Inhibition of bromodomain-containing protein 9 for the prevention of epigenetically-defined drug resistance. Bioorganic Med. Chem. Lett. 2017, 27, 3534–3541. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.H.; Jia, Y.S.; Shi, Y.H.; Li, Q.Y.; Bao, S.Q.; Lu, W.P.; Tong, Z.S. ACT001 can prevent and reverse tamoxifen resistance in human breast cancer cell lines by inhibiting NF-kappaB activation. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Tsutsui, T.; Fukasawa, R.; Tanaka, A.; Hirose, Y.; Ohkuma, Y. Identification of target genes for the CDK subunits of the Mediator complex. Genes Cells 2011, 16, 1208–1218. [Google Scholar] [CrossRef]
- Fant, C.B.; Taatjes, D.J. Regulatory functions of the Mediator kinases CDK8 and CDK19. Transcription 2019, 10, 76–90. [Google Scholar] [CrossRef]
- Philip, S.; Kumarasiri, M.; Teo, T.; Yu, M.; Wang, S. Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Westerling, T.; Kuuluvainen, E.; Makela, T.P. Cdk8 is essential for preimplantation mouse development. Mol. Cell Biol. 2007, 27, 6177–6182. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.J.; Bernad, R.; Martínez-Val, A.; Shahbazi, M.N.; Nóbrega-Pereira, S.; Calvo, I.; Blanco-Aparicio, C.; Tarantino, C.; Garreta, E.; Richart-Ginés, L.; et al. Global hyperactivation of enhancers stabilizes human and mouse naive pluripotency through inhibition of CDK8/19 Mediator kinases. Nat. Cell Biol. 2020, 22, 1223–1238. [Google Scholar] [CrossRef]
- Postlmayr, A.; Dumeau, C.E.; Wutz, A. Cdk8 is required for establishment of H3K27me3 and gene repression by Xist and mouse development. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; McCleland, M.L.; Truong, T.; Lau, S.; Modrusan, Z.; Soukup, T.M.; Roose-Girma, M.; Blackwood, E.M.; Firestein, R. CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res. 2012, 72, 2129–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, I.B.; Győrffy, B.; Mack, Z.T.; Shtil, A.A.; Shtutman, M.S.; Chen, M.; Broude, E.V. Identifying Cancers Impacted by CDK8/19. Cells 2019, 8, 821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.C.; Farmaki, E.; Altilia, S.; Schools, G.P.; West, D.K.; Chen, M.; Chang, B.D.; Puzyrev, A.T.; Lim, C.U.; Rokow-Kittell, R.; et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. USA 2012, 109, 13799–13804. [Google Scholar] [CrossRef] [Green Version]
- Koehler, M.F.; Bergeron, P.; Blackwood, E.M.; Bowman, K.; Clark, K.R.; Firestein, R.; Kiefer, J.R.; Maskos, K.; McCleland, M.L.; Orren, L.; et al. Development of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. Acs Med. Chem. Lett. 2016, 7, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Chen, M.; Hughes, D.; Chumanevich, A.A.; Altilia, S.; Kaza, V.; Lim, C.U.; Kiaris, H.; Mythreye, K.; Pena, M.M.; et al. CDK8 Selectively Promotes the Growth of Colon Cancer Metastases in the Liver by Regulating Gene Expression of TIMP3 and Matrix Metalloproteinases. Cancer Res. 2018, 78, 6594–6606. [Google Scholar] [CrossRef] [Green Version]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [Green Version]
- Serrao, A.; Jenkins, L.M.; Chumanevich, A.A.; Horst, B.; Liang, J.; Gatza, M.L.; Lee, N.Y.; Roninson, I.B.; Broude, E.V.; Mythreye, K. Mediator kinase CDK8/CDK19 drives YAP1-dependent BMP4-induced EMT in cancer. Oncogene 2018, 37, 4792–4808. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Allen, M.A.; Bensard, C.L.; Wang, X.; Schwinn, M.K.; Qin, B.; Long, H.W.; Daniels, D.L.; Hahn, W.C.; Dowell, R.D.; et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013, 153, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dolken, L.; Strobl, B.; Muller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.S.; Chumanevich, A.A.; Lim, C.U.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H.; et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017, 8, 12558–12575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Liang, J.; Ji, H.; Yang, Z.; Altilia, S.; Hu, B.; Schronce, A.; McDermott, M.S.J.; Schools, G.P.; Lim, C.U.; et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFkappaB. Proc. Natl. Acad. Sci. USA 2017, 114, 10208–10213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Amirhosseini, M.; Bernhardsson, M.; Lang, P.; Andersson, G.; Flygare, J.; Fahlgren, A. Cyclin-dependent kinase 8/19 inhibition suppresses osteoclastogenesis by downregulating RANK and promotes osteoblast mineralization and cancellous bone healing. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Li, J.; Ji, H.; Porter, D.C.; Broude, E.V.; Roninson, I.B.; Chen, M. Characterizing CDK8/19 Inhibitors through a NFκB-Dependent Cell-Based Assay. Cells 2019, 8, 1208. [Google Scholar] [CrossRef] [Green Version]
- Kalykaki, A.; Agelaki, S.; Kallergi, G.; Xyrafas, A.; Mavroudis, D.; Georgoulias, V. Elimination of EGFR-expressing circulating tumor cells in patients with metastatic breast cancer treated with gefitinib. Cancer Chemother. Pharmacol. 2014, 73, 685–693. [Google Scholar] [CrossRef]
- Dickler, M.N.; Cobleigh, M.A.; Miller, K.D.; Klein, P.M.; Winer, E.P. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res. Treat. 2009, 115, 115–121. [Google Scholar] [CrossRef]
- Ferrer-Soler, L.; Vazquez-Martin, A.; Brunet, J.; Menendez, J.A.; De Llorens, R.; Colomer, R. An update of the mechanisms of resistance to EGFR-tyrosine kinase inhibitors in breast cancer: Gefitinib (Iressa) -induced changes in the expression and nucleo-cytoplasmic trafficking of HER-ligands (Review). Int. J. Mol. Med. 2007, 20, 3–10. [Google Scholar] [CrossRef]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. 2011, 11, 777–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Roninson, I.B.; Porter, D.C.; Wentland, M.P. CDK8-CDK19 Selective Inhibitors and Their Use in Anti-Metastatic and Chemopreventative Methods for Cancer. U.S. Patent US 09321737, 26 April 2016. [Google Scholar]
- Chen, M.; Li, J.; Liang, J.; Thompson, Z.S.; Kathrein, K.; Broude, E.V.; Roninson, I.B. Systemic Toxicity Reported for CDK8/19 Inhibitors CCT251921 and MSC2530818 Is Not Due to Target Inhibition. Cells 2019, 8, 1413. [Google Scholar] [CrossRef] [Green Version]
- Acar, A.; Nichol, D.; Fernandez-Mateos, J.; Cresswell, G.D.; Barozzi, I.; Hong, S.P.; Trahearn, N.; Spiteri, I.; Stubbs, M.; Burke, R.; et al. Exploiting evolutionary steering to induce collateral drug sensitivity in cancer. Nat. Commun 2020, 11, 1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.S.J.; Sharko, A.C.; Munie, J.; Kassler, S.; Melendez, T.; Lim, C.U.; Broude, E.V. CDK7 Inhibition is Effective in all the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells 2020, 9, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, S.K.; Pandey, K.; Rengasamy, K.R.R.; Biswal, B.K. Recent updates on the resistance mechanisms to epidermal growth factor receptor tyrosine kinase inhibitors and resistance reversion strategies in lung cancer. Med. Res. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Chan, T.E.; Lombardo, Y.; Corleone, G.; Rotmensz, N.; Bravaccini, S.; Rocca, A.; Pruneri, G.; McEwen, K.R.; Coombes, R.C.; et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat. Commun. 2019, 10, 3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagohara, L.T.; Zamuner, F.; Davis-Marcisak, E.F.; Sharma, G.; Considine, M.; Allen, J.; Yegnasubramanian, S.; Gaykalova, D.A.; Fertig, E.J. Integrated single-cell and bulk gene expression and ATAC-seq reveals heterogeneity and early changes in pathways associated with resistance to cetuximab in HNSCC-sensitive cell lines. Br. J. Cancer 2020, 123, 101–113. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BT474-Par | BT474-GefR | BT474-ErlR | SKBR3-Par | SKBR3-GefR | |
|---|---|---|---|---|---|
| Gefitinib | 0.140 | 0.973 (6.95) | 0.863 (6.16) | 0.619 | 1.530 (2.47) |
| Senexin B | 1.547 | 2.430 (1.57) | 2.561 (1.66) | N/A | N/A |
| Senexin B + Gefitinib (1:1) | 0.087/0.087 | 0.510/0.510 (5.86) | 0.612/0.612 (7.03) | 0.403 | 0.644 (1.60) |
| Erlotinib | 2.489 | 7.329 (2.94) | 8.520 (3.42) | 2.478 | 3.437 (1.39) |
| Senexin B + Erlotinib (1:5) | 0.351/1.753 | 0.473/2.124 (1.35) | 0.915/4.577 (2.61) | 0.258/1.292 | 0.328/1.641 (1.27) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharko, A.C.; Lim, C.-U.; McDermott, M.S.J.; Hennes, C.; Philavong, K.P.; Aiken, T.; Tatarskiy, V.V.; Roninson, I.B.; Broude, E.V. The Inhibition of CDK8/19 Mediator Kinases Prevents the Development of Resistance to EGFR-Targeting Drugs. Cells 2021, 10, 144. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010144
Sharko AC, Lim C-U, McDermott MSJ, Hennes C, Philavong KP, Aiken T, Tatarskiy VV, Roninson IB, Broude EV. The Inhibition of CDK8/19 Mediator Kinases Prevents the Development of Resistance to EGFR-Targeting Drugs. Cells. 2021; 10(1):144. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010144
Chicago/Turabian StyleSharko, Amanda C., Chang-Uk Lim, Martina S. J. McDermott, Chuck Hennes, Kingsavanh P. Philavong, Tiffanie Aiken, Victor V. Tatarskiy, Igor B. Roninson, and Eugenia V. Broude. 2021. "The Inhibition of CDK8/19 Mediator Kinases Prevents the Development of Resistance to EGFR-Targeting Drugs" Cells 10, no. 1: 144. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010144