Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Search
2.2. Study Selection and Data Extraction
2.3. Statistical Analysis
3. Results
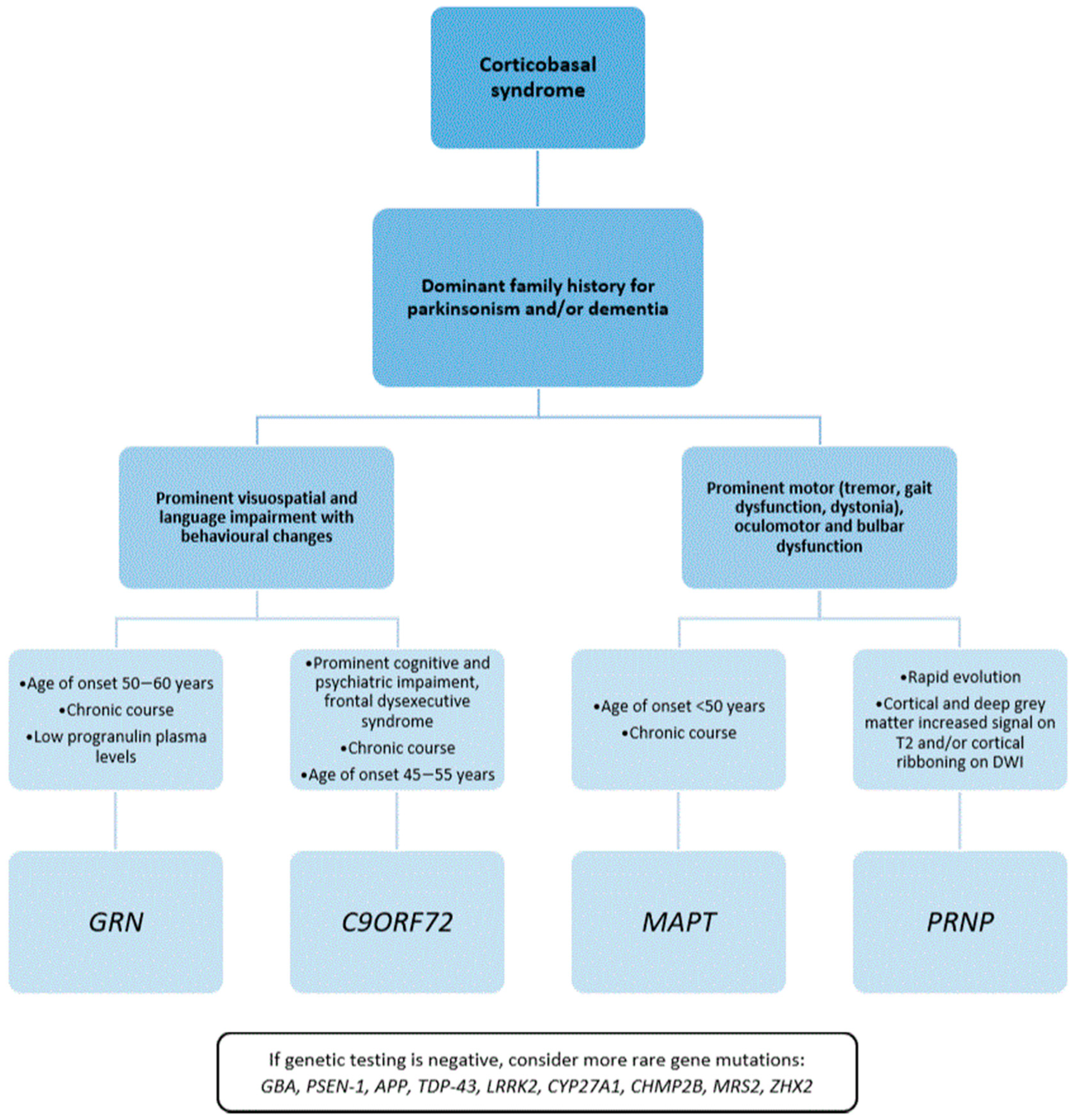
3.1. CBS Caused by a Single-Gene Mutation
3.1.1. GRN Mutations in CBS
3.1.2. MAPT Mutations in CBS
3.1.3. C9ORF72 Expansion in CBS
3.1.4. PRNP Mutations in CBS
3.1.5. Other Genes
3.2. Genetic Risk Factors Associated with CBS and CBD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chahine, L.M.; Rebeiz, T.; Rebeiz, J.J.; Grossman, M.; Gross, R.G. Corticobasal syndrome: Five new things. Neurol. Clin. Pract. 2014, 4, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxer, A.L.; Geschwind, M.D.; Belfor, N.; Gorno-Tempini, M.L.; Schauer, G.F.; Miller, B.L.; Weiner, M.W.; Rosen, H.J. Patterns of Brain Atrophy That Differentiate Corticobasal Degeneration Syndrome From Progressive Supranuclear Palsy. Arch. Neurol. 2006, 63, 81–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togasaki, D.M.; Tanner, C.M. Epidemiologic aspects. Adv. Neurol. 2000, 82, 53–59. [Google Scholar] [PubMed]
- Wenning, G.K.; Litvan, I.; Jankovic, J.; Granata, R.; Mangone, C.A.; McKee, A.; Poewe, W.; Jellinger, K.; Chaudhuri, K.R.; D’Olhaberriague, L.; et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J. Neurol. Neurosurg. Psychiatry 1998, 64, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, R.; Neumann, M.; Forman, M.S.; Farmer, J.; Massimo, L.; Rice, A.; Miller, B.L.; Johnson, J.K.; Clark, C.M.; Hurtig, H.I.; et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007, 68, 1274–1283. [Google Scholar] [CrossRef]
- Lang, A.E.; Riley, D.E.; Bergeron, C. Cortico-Basal Ganglionic Degeneration. In Neurodegenerative Diseases; Calne, D.B., Ed.; WB Saunders: Phila-Delphia, PA, USA, 1994; p. 877.e94. [Google Scholar]
- Boeve, B.F.; Lang, A.E.; Litvan, I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and fronto-temporal dementia. Ann. Neurol. 2003, 54 (Suppl. S5), S15e19. [Google Scholar] [CrossRef]
- Bak, T.H.; Hodges, J.R.; Thomas, H.B. Corticobasal Degeneration: Clinical Aspects. In Handbook of Clinical Neurology; Duyckaerts, C., Litvan, I., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 89, p. 509.e21. [Google Scholar]
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Wadia, P.M.; Lang, A.E. The many faces of corticobasal degeneration. Park. Relat. Disord. 2007, 13, S336–S340. [Google Scholar] [CrossRef]
- Boeve, B.F.; Maraganore, D.M.; Parisi, J.E.; Ahlskog, J.E.; Graff-Radford, N.; Caselli, R.J.; Dickson, D.W.; Kokmen, E.; Petersen, R.C. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999, 53, 795. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F.; Jankovic, J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 2016, 12, 175–185. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Bateman, A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue re-pair and tumorigenesis. J. Mol. Med. 2003, 81, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Chitramuthu, B.P.; Bennett, H.P.J.; Bateman, A. Progranulin: A new avenue towards the understanding and treatment of neurodegenerative disease. Brain 2017, 140, 3081–3104. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ismail, A.; Kriazhev, L.; Sadvakassova, G.; Bateman, A. Progranulin (PC-cell-derived growth factor/acrogranin) regulates invasion and cell survival. Cancer Res. 2002, 62, 5590–5596. [Google Scholar] [PubMed]
- Kumar-Singh, S. Progranulin and TDP-43: Mechanistic Links and Future Directions. J. Mol. Neurosci. 2011, 45, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, R.; He, Z.; Carmichael, K.P.; Halper, J.; Bateman, A. Cellular Localization of Gene Expression for Progranulin. J. Histochem. Cytochem. 2000, 48, 999–1009. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chro-mosome 17q21. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Karch, C.M.; Ezerskiy, L.A.; Redaelli, V.; Giovagnoli, A.R.; Tiraboschi, P.; Pelliccioni, G.; Pelliccioni, P.; Kapetis, D.; D’Amato, I.; Piccoli, E.; et al. Missense mutations in progranulin gene associated with frontotemporal lobar degeneration: Study of pathogenetic features. Neurobiol. Aging 2016, 38, 215.e1–215.e12. [Google Scholar] [CrossRef] [Green Version]
- Finch, N.; Baker, M.; Crook, R.; Swanson, K.; Kuntz, K.; Surtees, R.; Bisceglio, G.; Rovelet-Lecrux, A.; Boeve, B.; Petersen, R.C.; et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 2009, 132, 583–591. [Google Scholar] [CrossRef]
- Galimberti, D.; Bertram, K.; Formica, A.; Fenoglio, C.; Cioffi, S.M.; Arighi, A.; Scarpini, E.; Colosimo, C. Plasma Screening for Progranulin Mutations in Patients with Progressive Supranuclear Palsy and Corticobasal Syndromes. J. Alzheimers Dis. 2016, 53, 445–449. [Google Scholar] [CrossRef]
- Gass, J.; Cannon, A.; MacKenzie, I.R.; Boeve, B.; Baker, M.; Adamson, J.; Crook, R.; Melquist, S.; Kuntz, K.; Petersen, R.; et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006, 15, 2988–3001. [Google Scholar] [CrossRef] [Green Version]
- Van Swieten, J.C.; Heutink, P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological pheno-types of frontotemporal dementia. Lancet Neurol. 2008, 7, 965–974. [Google Scholar] [CrossRef]
- Kelley, B.J.; Haidar, W.; Boeve, B.; Baker, M.; Graff-Radford, N.R.; Krefft, T.; Frank, A.R.; Jack, C.R.; Shiung, M.; Knopman, D.S.; et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol. Aging 2009, 30, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Ber, I.; Camuzat, A.; Hannequin, D.; Pasquier, F.; Guedj, E.; Rovelet-Lecrux, A.; Hahn-Barma, V.; Van Der Zee, J.; Clot, F.; Bakchine, S.; et al. Phenotype variability in progranulin mutation carriers: A clinical, neuropsychological, imaging and genetic study. Brain 2008, 131, 732–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, C.; Rossi, G.; Barbarulo, A.M.; Di Fede, G.; Foglia, C.; Piccoli, E.; Piscosquito, G.; Saracino, D.; Tagliavini, F.; Cotrufo, R. A progranulin mutation associated with cortico-basal syndrome in an Italian family expressing different phenotypes of fronto-temporal lobar degeneration. Neurol. Sci. 2011, 33, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Binetti, G.; Sina, E.; Gigola, L.; Bettecken, T.; Meitinger, T.; Ghidoni, R. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol. Aging 2008, 29, 427–435. [Google Scholar] [CrossRef]
- Canafoglia, L.; Morbin, M.; Scaioli, V.; Pareyson, D.; D’Incerti, L.; Fugnanesi, V.; Tagliavini, F.; Berkovic, S.F.; Franceschetti, S. Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia 2014, 55, e56–e59. [Google Scholar] [CrossRef]
- Kamate, M.; Detroja, M.; Hattiholi, V. Neuronal ceroid lipofuscinosis type-11 in an adolescent. Brain Dev. 2019, 41, 542–545. [Google Scholar] [CrossRef]
- Beck, J.; Rohrer, J.D.; Campbell, T.; Isaacs, A.; Morrison, K.E.; Goodall, E.F.; Warrington, E.K.; Stevens, J.; Revesz, T.; Holton, J.; et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008, 131, 706–720. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.E.; Bird, T.D.; Bekris, L.M.; Montine, T.J.; Leverenz, J.B.; Steinbart, E.; Van Deerlin, V.M. The Spectrum of Mutations in Progranulin. A Collaborative Study Screening 545 Cases of Neurodegeneration. Arch. Neurol. 2010, 67, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Taghdiri, F.; Sato, C.; Ghani, M.; Moreno, D.; Rogaeva, E.; Tartaglia, M.C. Novel GRN Mutations in Patients with Corticobasal Syndrome. Sci. Rep. 2016, 6, 22913. [Google Scholar] [CrossRef] [Green Version]
- Benussi, L.; Ghidoni, R.; Pegoiani, E.; Moretti, D.V.; Zanetti, O.; Binetti, G. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol. Dis. 2009, 33, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Spina, S.; Murrell, J.R.; Huey, E.D.; Wassermann, E.M.; Pietrini, P.; Grafman, J.; Ghetti, B. Corticobasal syndrome associated with the A9D progranulin mutation. J. Neuropathol. Exp. Neurol. 2007, 66, 892–900. [Google Scholar] [PubMed] [Green Version]
- Guerreiro, R.; Santana, I.; Bras, J.; Revesz, T.; Rebelo, O.; Bs, M.H.R.; Santiago, B.; Oliveira, C.R.; Singleton, A.; Hardy, J. Novel progranulin mutation: Screening for PGRN mutations in a Portuguese series of FTD/CBS cases. Mov. Disord. 2008, 23, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Passov, V.; Gavrilova, R.H.; Strand, E.; Cerhan, J.H.; Josephs, K.A. Sporadic Corticobasal Syndrome With Progranulin Mutation Presenting as Progressive Apraxic Agraphia. Arch. Neurol. 2011, 68, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Dopper, E.G.P.; Seelaar, H.; Chiu, W.Z.; De Koning, I.; Van Minkelen, R.; Baker, M.C.; Rozemuller, A.J.M.; Rademakers, R.; Van Swieten, J.C. Symmetrical Corticobasal Syndrome Caused by a Novel c.314dup Progranulin Mutation. J. Mol. Neurosci. 2011, 45, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Whitwell, J.L.; Weigand, S.D.; Boeve, B.F.; Senjem, M.L.; Gunter, J.L.; DeJesus-Hernandez, M.; Rutherford, N.J.; Baker, M.; Knopman, D.S.; Wszolek, Z.K.; et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012, 135, 794–806. [Google Scholar] [CrossRef]
- Whitwell, J.L.; Jack, C.R.; Baker, M.; Rademakers, R.; Adamson, J.; Boeve, B.F.; Knopman, D.S.; Parisi, J.F.; Petersen, R.C.; Dickson, D.W.; et al. Voxel-Based Morphometry in Frontotemporal Lobar Degeneration With Ubiquitin-Positive Inclusions With and Without Progranulin Mutations. Arch. Neurol. 2007, 64, 371–376. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Beck, J.; Warren, J.D.; King, A.; Sarraj, S.A.; Holton, J.L.; Revesz, T.; Collinge, J.; Mead, S. Corticobasal syndrome associated with a novel 1048_1049insG progranulin mutation. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1297–1298. [Google Scholar] [CrossRef]
- Masellis, M.; Momeni, P.; Meschino, W.; Heffner, R.; Elder, J.; Sato, C.; Liang, Y.; George-Hyslop, P.S.; Hardy, J.; Bilbao, J.; et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 2006, 129, 3115–3123. [Google Scholar] [CrossRef] [Green Version]
- Josephs, K.A.; Holton, J.L.; Rossor, M.N.; Godbolt, A.K.; Ozawa, T.; Strand, K.; Khan, N.; Al Sarraj, S.; Revesz, T. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol. Appl. Neurobiol. 2004, 30, 369–373. [Google Scholar]
- Sampathu, D.M.; Neumann, M.; Kwong, L.K.; Chou, T.T.; Micsenyi, M.; Truax, A.; Lee, V.M.Y. Pathological heterogeneity of frontotemporal lobar degeneration with ubiqui-tin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am. J. Pathol. 2006, 169, 1343–1352. [Google Scholar] [PubMed] [Green Version]
- Mackenzie, I.R.A.; Baborie, A.; Pickering-Brown, S.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Mann, D.M. Heterogeneity of ubiquitin pathology in frontotemporal lobar degenera-tion: Classification and relation to clinical phenotype. Acta Neuropathol. 2006, 112, 539–549. [Google Scholar] [PubMed] [Green Version]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Kouri, N.; Whitwell, J.L.; Josephs, K.A.; Rademakers, R.; Dickson, D.W. Corticobasal degeneration: A pathologically distinct 4R tauopathy. Nat. Rev. Neurol. 2011, 7, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Van Swieten, J.C.; Goedert, M. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Neurogenetics 2000, 2, 193–205. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Paviour, D.; Vandrovcova, J.; Hodges, J.; De Silva, R.; Rossor, M. Novel L284R MAPT Mutation in a Family with an Autosomal Dominant Progressive Supranuclear Palsy Syndrome. Neurodegener. Dis. 2011, 8, 149–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casseron, W.; Azulay, J.P.; Guedj, E.; Gastaut, J.L.; Pouget, J. Familial autosomal dominant cortico-basal degeneration with the P301S mutation in the tau gene: An example of phenotype variability. J. Neurol. 2005, 252, 1546–1548. [Google Scholar] [CrossRef]
- Gatto, E.M.; Allegri, R.F.; Da Prat, G.; Mendez, P.C.; Hanna, D.S.; Dorschner, M.O.; Surace, E.I.; Zabetian, C.P.; Mata, I.F. Intrafamili-al variable phenotype including corticobasal syndrome in a family with p.P301L mutation in the MAPT gene: First report in South America. Neurobiol. Aging 2017, 53, 195.e11–195.e17. [Google Scholar]
- Rossi, G.; Marelli, C.; Farina, L.; Laurà, M.; Basile, A.M.; Ciano, C.; Tagliavini, F.; Pareyson, D. The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov. Disord. 2008, 23, 892–895. [Google Scholar] [CrossRef]
- Marshall, C.R.; Guerreiro, R.; Thust, S.; Fletcher, P.; Rohrer, J.D.; Fox, N.C. A Novel MAPT Mutation Causing Corticobasal Syndrome Led by Progressive Apraxia of Speech. J. Alzheimers Dis. 2015, 48, 923–926. [Google Scholar] [CrossRef]
- Bugiani, O.; Murrell, J.R.; Giaccone, G.; Hasegawa, M.; Ghigo, G.; Tabaton, M.; Morbin, M.; Primavera, A.; Carella, F.; Solaro, C.; et al. Frontotemporal Dementia and Corticobasal Degen-eration in a family with a P301S mutation in tau. J. Neuropathol. Exp. Neurol. 1999, 58, 667–677. [Google Scholar] [PubMed] [Green Version]
- Kouri, N.; Carlomagno, Y.; Baker, M.; Liesinger, A.M.; Caselli, R.J.; Wszolek, Z.K.; Petrucelli, L.; Boeve, B.F.; Parisi, J.E.; Josephs, K.A.; et al. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 2014, 127, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Fairen, M.D.; Sabir, M.S.; Pastor, P.; Ding, J.; Ispierto, L.; Butala, A.; Morris, C.M.; Schulte, C.; Gasser, T.; et al. MAPT p.V363I mutation. Neurol. Genet. 2019, 5, e347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, J.D.; Ridgway, G.R.; Modat, M.; Ourselin, S.; Mead, S.; Fox, N.C.; Rossor, M.N.; Warren, J.D. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. NeuroImage 2010, 53, 1070–1076. [Google Scholar] [CrossRef] [Green Version]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endoso-mal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9ORF72 repeat expansion disrupts nucleocytoplasmic transport. Nat. Cell Biol. 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Barker, H.V.; Niblock, M.; Lee, Y.-B.; Shaw, P.; Gallo, J.-M. RNA Misprocessing in C9ORF72-Linked Neurodegeneration. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; MacKenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Boeve, B.; Boylan, K.B.; Graff-Radford, N.R.; DeJesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 135, 765–783. [Google Scholar] [CrossRef]
- Lesage, S.; Le Ber, I.; Condroyer, C.; Brousolle, E.; Gabelle, A.; Thobois, S. The analysis of C9ORF72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013, 136, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Schottlaender, L.V.; Polke, J.M.; Ling, H.; MacDoanld, N.D.; Tucci, A.; Nanji, T.; Pittman, A.; De Silva, R.; Holton, J.L.; Revesz, T.; et al. The analysis of C9ORF72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical parkinsonism. Neurobiol. Aging 2015, 36, 1221.e1–1221.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, S.G.; Duno, M.; Batbayli, M.; Puschmann, A.; Braendgaard, H.; Mardosiene, S.; Svenstrup, K.; Pinborg, L.H.; Vestergaard, K.; Hjermind, L.E.; et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin. Genet. 2013, 83, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Anor, C.J.; Xi, Z.; Zhang, M.; Moreno, D.; Sato, C.; Rogaeva, E.; Tartaglia, C. Mutation analysis of C9ORF72 in patients with corticobasal syndrome. Neurobiol. Aging 2015, 36, 2905.e1–2905.e5. [Google Scholar] [CrossRef] [PubMed]
- Necpál, J.; Stelzer, M.; Koščová, S.; Patarák, M. A Corticobasal Syndrome Variant of Familial Creutzfeldt-Jakob Disease with Stroke-Like Onset. Case Rep. Neurol. Med. 2016, 2016, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.G.; Oh, E.; Park, S.; Kim, Y.-S.; Lee, A.Y. Familial Creutzfeldt–Jakob disease with M232R mutation presented with corticobasal syndrome. Neurol. Sci. 2014, 36, 1291–1293. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Campbell, T.A.; Dickinson, A.; Beck, J.; Holt, M.; Plant, G.; De Pauw, K.W.; Hakin, R.N.; Clarke, C.E.; Howell, S.; et al. Inherited prion disease with an alanine to valine mutation at codon 117 in the prion protein gene. Brain 1999, 122, 1823–1837. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.G.; Seguin, J.; Quadrio, I.; Höftberger, R.; Kapás, I.; Streichenberger, N.; Biacabe, A.G.; Meyronet, D.; Sciot, R.; Vandenberghe, R.; et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: Characterization of a complex proteinopathy. Acta Neuropathol. 2011, 121, 39–57. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.R. GENETICS OF PRIONS. Annu. Rev. Genet. 1997, 31, 139–175. [Google Scholar] [CrossRef]
- Jung, H.H.; Bremer, J.; Streffer, J.R.; Virdee, K.; Spillantini, M.G.; Crowther, R.A.; Brugger, P.; Van Broeckhoven, C.; Aguzzi, A.; Tolnay, M. Phenotypic Variation of Autosomal-Dominant Corticobasal Degeneration. Eur. Neurol. 2012, 67, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Canovas, A.; Katschnig, P.; Tucci, A.; Carecchio, M.; Wood, N.W.; Edwards, M.; Castrillo, J.C.M.; Burke, D.; Heales, S.; Dm, K.P.B. Atypical parkinsonism with apraxia and supranuclear gaze abnormalities in type 1 Gaucher disease. Expanding the spectrum: Case report and literature review. Mov. Disord. 2010, 25, 1506–1509. [Google Scholar] [CrossRef]
- Pilotto, A.; Schulte, C.; Hauser, A.K.; Biskup, S.; Munz, M.; Brockmann, K.; Schaeffer, E.; Synofzik, M.; Maetzler, W.; Suenkel, U.; et al. GBA -associated parkinsonism and dementia: Beyond α-synucleinopathies? Eur. J. Neurol. 2015, 23, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Agusti, I.; Kojovic, M.; Edwards, M.J.; Murphy, E.; Chandrashekar, H.S.; Lachmann, R.H.; Dm, K.P.B. Atypical parkinsonism and cerebrotendinous xanthomatosis: Report of a family with corticobasal syndrome and a literature review. Mov. Disord. 2012, 27, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zee, J.; Urwin, H.; Engelborghs, S.; Bruyland, M.; Vandenberghe, R.; Dermaut, B.; De Pooter, T.; Peeters, K.; Santens, P.; De Deyn, P.P.; et al. CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum. Mol. Genet. 2007, 17, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Yuan, W.; Anderson, C.; Wood, E.M.; Hurtig, H.I.; Clark, C.M.; Miller, B.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Grossman, M.; et al. Corticobasal syndrome and primary progressive aphasia as manifestations of LRRK2 gene mutations. Neurol. 2007, 70, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wszolek, Z.; Vieregge, P.; Uitti, R.; Gasser, T.; Yasuhara, O.; McGeer, P.; Berry, K.; Calne, D.; Vingerhoets, F.; Klein, C.; et al. German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia—Longitudinal observations. Park. Relat. Disord. 1997, 3, 125–139. [Google Scholar] [CrossRef]
- Rajput, A.; Dickson, D.W.; Robinson, C.A.; Ross, O.A.; Dächsel, J.C.; Lincoln, S.J.; Farrer, M.J. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Navarro, E.; De Andrés, C.; Guerrero, C.; Giménez-Roldán, S. Corticobasal Syndrome in a Family with Early-Onset Alzheimer’s Disease Linked to a Presenilin-1 Gene Mutation. Mov. Disord. Clin. Pract. 2015, 2, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Abate, F.; Dati, G.; Ginevrino, M.; Valente, E.M.; Barone, P.; Picillo, M. APP -Related Corticobasal Syndrome: Expanding the List of Corticobasal Degeneration Look Alikes. Mov. Disord. Clin. Pract. 2020, 7, 849–851. [Google Scholar] [CrossRef]
- Huey, E.D.; Ferrari, R.; Moreno, J.H.; Jensen, C.; Morris, C.M.; Potocnik, F.; Kalaria, R.N.; Tierney, M.; Wassermann, E.M.; Hardy, J.; et al. FUS and TDP43 genetic variability in FTD and CBS. Neurobiol. Aging 2012, 33, 1016.e9–1016.e17. [Google Scholar] [CrossRef] [Green Version]
- Fekete, R.; Bainbridge, M.; Baizabal-Carvallo, J.F.; Rivera, A.; Miller, B.; Du, P.; Kholodovych, V.; Powell, S.; Ondo, W.; Kholodovich, V. Exome sequencing in familial corticobasal degeneration. Park. Relat. Disord. 2013, 19, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Höglinger, G.U. Is it Useful to Classify Progressive Supranuclear Palsy and Corticobasal Degeneration as Different Disorders? No. Mov. Disord. Clin. Pract. 2018, 5, 141–144. [Google Scholar] [CrossRef]
- Clark, M.; Geschwind, D.H.; Fogel, B.L. The Neurogenetics of Atypical Parkinsonian Disorders. Semin. Neurol. 2014, 34, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Di Maria, E.; Tabaton, M.; Vigo, T.; Abbruzzese, G.; Bellone, E.; Donati, C.; Frasson, E.; Marchese, R.; Montagna, P.; Munoz, D.G.; et al. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann. Neurol. 2000, 47, 374–377. [Google Scholar] [CrossRef]
- Myers, A.J.; Pittman, A.M.; Zhao, A.S.; Rohrer, K.; Kaleem, M.; Marlowe, L.; Hardy, J. The MAPT H1c risk haplotype is associated with increased expression of tau and es-pecially of 4 repeat containing transcripts. Neurobiol. Dis. 2007, 25, 561–570. [Google Scholar] [CrossRef]
- Litvan, I.; Chism, A.; Litvan, J.; Cambon, A.; Hutton, M. H1/H1 genotype influences symptom severity in corticobasal syndrome. Mov. Disord. 2010, 25, 760–763. [Google Scholar] [CrossRef]
- Houlden, H.; Baker, M.; Morris, H.; Macdonald, N.; Pickering-Brown, S.; Adamson, J.; Lees, A.; Rossor, M.; Quinn, N.; Kertesz, A.; et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 2001, 56, 1702–1706. [Google Scholar] [CrossRef]
- Kouri, N.; Ross, O.A.; Dombroski, B.; Younkin, C.S.; Serie, D.J.; Soto-Ortolaza, A.; Baker, M.; Finch, N.C.A.; Yoon, H.; Kim, J.; et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 2015, 6, 7247. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Höglinger, G.U.; et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017, 133, 825–837. [Google Scholar] [CrossRef]
- Borroni, B.; Del Bo, R.; Goldwurm, S.; Archetti, S.; Bonvicini, C.; Agosti, C.; Bigni, B.; Papetti, A.; Ghezzi, S.; Sacilotto, G.; et al. VEGF Haplotypes are Associated with Increased Risk to Progressive Supranuclear Palsy and Corticobasal Syndrome. J. Alzheimers Dis. 2010, 21, 87–94. [Google Scholar] [CrossRef]
- Moore, K.M.; Nicholas, J.; Grossman, M.; McMillan, C.T.; Irwin, D.J.; Massimo, L.; Van Deerlin, V.M.; Warren, J.D.; Fox, N.C.; Rossor, M.N.; et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: An international retrospective cohort study. Lancet Neurol. 2020, 19, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-W.; Eum, S.-W.; Moon, C.O.; Ma, H.-I.; Kim, Y.J. Corticobasal syndrome associated with antiphospholipid syndrome without cerebral infarction. Neurology 2014, 82, 730–731. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.R.; Baldeiras, I.; Ribeiro, M.H.; Santiago, B.; Machado, C.; Massano, J.; Guimarães, J.; Oliveira, C.R.; Santana, I. Progranulin Peripheral Levels as a Screening Tool for the Identification of Subjects with Progranulin Mutations in a Portuguese Cohort. Neurodegener. Dis. 2013, 13, 214–223. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Number of Cases (% of Total Cases) | Mean Age of Onset * (Years) | Mean Age at Death * (Years) | Male * | Female * | |
|---|---|---|---|---|---|
| GRN | 28 (48%) | 58.08 | 63.57 | 10 | 7 |
| MAPT | 9 (16%) | 48.22 | 56.33 | 4 | 5 |
| C9ORF72 | 6 (10%) | 50.17 | NA | 2 | 4 |
| PRNP | 4 (7%) | 58.75 | 60.5 | 1 | 3 |
| GBA | 3 (5%) | 62.6 | NA | 1 | 2 |
| MRS2/ZHX2 | 2 (3%) | 71 | 74 | 1 | 1 |
| PSEN-1 | 1 (<2%) | 48 | 51 | 1 | 0 |
| APP | 1 (<2%) | 49 | NA | 0 | 1 |
| TDP-43 | 1 (<2%) | 72 | NA | 1 | 0 |
| CHMP2B | 1 (<2%) | 71 | NA | 0 | 1 |
| LRRK2 | 1 (<2%) | 52 | NA | 0 | 1 |
| CYP27A1 | 1 (<2%) | 47 | NA | 1 | 0 |
| Total | 58 | 56.2 (SD 10.45) | 61.31 (SD 9.85) | 22 | 25 |
| GRN | MAPT/C9ORF72 | p Value | |
|---|---|---|---|
| Number of patients | 28 | 15 | − |
| Gender, male (GRN 17; MAPT 9; C9ORF72 6) | 58.8% (10) | 40.0% (6) | 0.288 |
| Age at disease onset, years (GRN 25; MAPT 9; C9ORF72 6) | 58.1 ± 8.1 | 49.0 ± 11.2 | 0.005 |
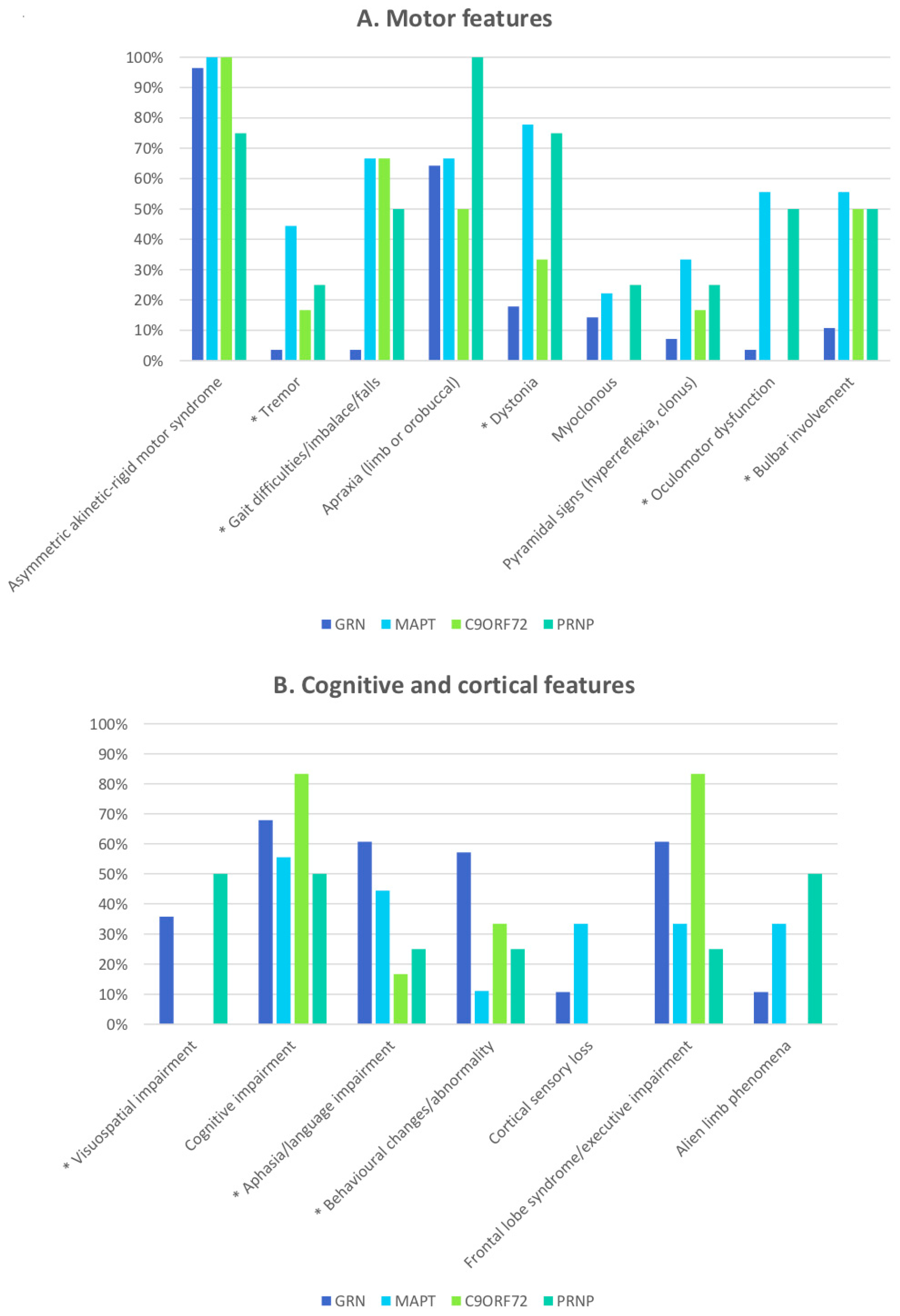
| Asymmetric akinetic–rigid syndrome (GRN 28; MAPT 9; C9ORF72 6) | 96.4% (27) | 100.0% (15) | 0.999 |
| Apraxia (GRN 24; MAPT 9; C9ORF72 6) | 75.0% (18) | 60.0% (9) | 0.323 |
| Gait dysfunction (GRN 28; MAPT 9; C9ORF72 6) | 3.6% (1) | 66.7 % (10) | <0.001 |
| Tremor (GRN 24; MAPT 9; C9ORF72 6) | 4.2% (1) | 33.3% (5) | 0.024 |
| Dystonia (GRN 24; MAPT 9; C9ORF72 6) | 20.8% (5) | 60.0% (9) | 0.013 |
| Myoclonus (GRN 24; MAPT 9; C9ORF72 6) | 16.7% (4) | 13.3% (2) | 0.999 |
| Pyramidal signs (GRN 24; MAPT 9; C9ORF72 6) | 8.3% (2) | 26.7% (4) | 0.180 |
| Oculomotor dysfunction (GRN 23; MAPT 9; C9ORF72 6) | 4.3% (1) | 33.3% (5) | 0.027 |
| Bulbar involvement (GRN 28; MAPT 9; C9ORF72 6) | 10.7% (3) | 53.3% (8) | 0.004 |
| Aphasia/language impairment (GRN 24; MAPT 9; C9ORF72 6) | 70.8% (17) | 33.3% (5) | 0.022 |
| Visuospatial impairment (GRN 20; MAPT 9; C9ORF72 6) | 50.0% (10) | 0% (0) | 0.002 |
| Cognitive impairment (GRN 23; MAPT 9; C9ORF72 6) | 82.6 % (19) | 66.7% (10) | 0.436 |
| Behavioural changes (GRN 24; MAPT 9; C9ORF72 6) | 66.7% (16) | 20.0% (3) | 0.008 |
| Frontal lobe syndrome (GRN 23; MAPT 9; C9ORF72 6) | 73.9% (17) | 53.3% (8) | 0.191 |
| Cortical sensory loss (GRN 24; MAPT 9; C9ORF72 6) | 12.5% (3) | 20.0% (3) | 0.658 |
| Alien limb (GRN 24; MAPT 9; C9ORF72 6) | 12.3% (3) | 20.0% (3) | 0.658 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arienti, F.; Lazzeri, G.; Vizziello, M.; Monfrini, E.; Bresolin, N.; Saetti, M.C.; Picillo, M.; Franco, G.; Di Fonzo, A. Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review. Cells 2021, 10, 171. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010171
Arienti F, Lazzeri G, Vizziello M, Monfrini E, Bresolin N, Saetti MC, Picillo M, Franco G, Di Fonzo A. Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review. Cells. 2021; 10(1):171. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010171
Chicago/Turabian StyleArienti, Federica, Giulia Lazzeri, Maria Vizziello, Edoardo Monfrini, Nereo Bresolin, Maria Cristina Saetti, Marina Picillo, Giulia Franco, and Alessio Di Fonzo. 2021. "Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review" Cells 10, no. 1: 171. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010171