
The Complex Regulatory Role of Cytomegalovirus Nuclear Egress Protein pUL50 in the Production of Infectious Virus

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Infection
2.2. Purification of ΔUL50 Particles
2.3. Quantitative Real-Time PCR (qPCR)
2.4. Immunogold Labeling and Transmission Electron Microscopy (Immuno-EM)
2.5. Western Blot (Wb) Analysis
2.6. Mass Spectrometry (MS)-Based Proteomic Analyses
2.7. Indirect Immunofluorescence (IF) Analysis and Confocal Laser-Scanning Microscopy
3. Results and Discussion
3.1. The Propagation and Particle Purification of ORF-UL50-Deleted HCMV by the Use of Complementing HFF-UL50 Cells
3.2. Determination of the ΔUL50 Virus Replication Characteristics under Conditions of pUL50 Complementation versus Non-Complementation
3.3. Immunogold EM Analysis of Viral Capsids, Egress Regulator pUL53 and Proteins of the Nuclear Lamina in ΔUL50-Infected Fibroblasts
3.4. Initial Qualitative and Semi-Quantitative Assessment of ΔUL50 Particle Preparations by the Use of Wb Analysis
3.5. Detailed Qualitative and Quantitative Assessment of ΔUL50 Particle Preparations by the Use of MS-Based Proteomics
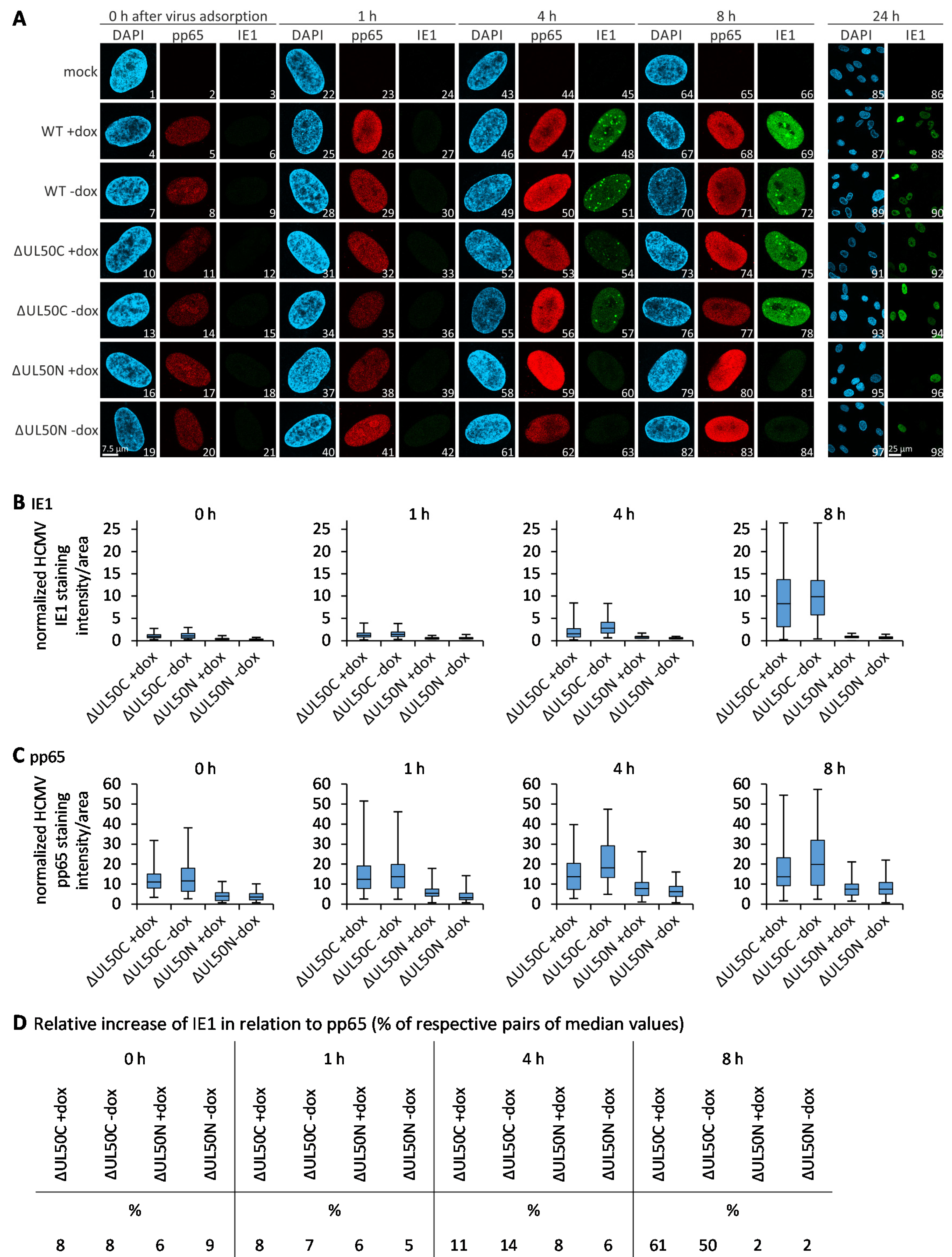
3.6. Confocal Imaging-Based Measurement of the Kinetics of ΔUL50 Viral Onset of Immediate Early Gene Expression
3.7. Identification of Reduced Levels of Genomic DNA Packaging in ΔUL50N Particles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, T.; Passvogel, L.; Schulz, K.S.; Klupp, B.G.; Mettenleiter, T.C. Nuclear egress of herpesviruses: The prototypic vesicular nucleocytoplasmic transport. Adv. Virus Res. 2016, 94, 81–140. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeev-Ben-Mordehai, T.; Weberruss, M.; Lorenz, M.; Cheleski, J.; Hellberg, T.; Whittle, C.; El Omari, K.; Vasishtan, D.; Dent, K.C.; Harlos, K.; et al. Crystal Structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep. 2015, 13, 2645–2652. [Google Scholar] [CrossRef] [Green Version]
- Mettenleiter, T.C.; Muller, F.; Granzow, H.; Klupp, B.G. The way out: What we know and do not know about herpesvirus nuclear egress. Cell Microbiol. 2013, 15, 170–178. [Google Scholar] [CrossRef]
- Rose, A.; Schlieker, C. Alternative nuclear transport for cellular protein quality control. Trends Cell Biol. 2012, 22, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Speese, S.D.; Ashley, J.; Jokhi, V.; Nunnari, J.; Barria, R.; Li, Y.; Ataman, B.; Koon, A.; Chang, Y.T.; Li, Q.; et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 2012, 149, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Bender, B.J.; Kamil, J.P.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.M.; Coen, D.M. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol. 2015, 89, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Hage, S.; Kraut, A.; Hesse, A.M.; Amin, B.; Sonnewald, U.; Coute, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The Prolyl isomerase pin1 promotes the herpesvirus-induced phosphorylation-dependent disassembly of the nuclear lamina required for nucleocytoplasmic egress. PLoS Pathog. 2016, 12, e1005825. [Google Scholar] [CrossRef] [Green Version]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27, e1934. [Google Scholar] [CrossRef]
- Milbradt, J.; Kraut, A.; Hutterer, C.; Sonntag, E.; Schmeiser, C.; Ferro, M.; Wagner, S.; Lenac, T.; Claus, C.; Pinkert, S.; et al. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell Proteom. 2014, 13, 2132–2146. [Google Scholar] [CrossRef] [Green Version]
- Häge, S.; Sonntag, E.; Svrlanska, A.; Borst, E.M.; Stilp, A.C.; Horsch, D.; Müller, R.; Kropff, B.; Milbradt, J.; Stamminger, T.; et al. Phenotypical Characterization of the nuclear egress of recombinant cytomegaloviruses reveals defective replication upon orf-ul50 deletion but not pul50 phosphosite mutation. Viruses 2021, 13, 165. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the human cytomegalovirus pUL50-pUL53 Core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [Green Version]
- Muller, Y.A.; Hage, S.; Alkhashrom, S.; Hollriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical beta- and gamma-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020, 295, 3189–3201. [Google Scholar] [CrossRef]
- Schweininger, J.; Kriegel, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Lösing, J.; Weiler, S.; Tillmanns, J.; Egerer-Sieber, C.; Decker, A.; et al. The crystal structure of the varicella zoster Orf24-Orf27 nuclear egress complex spotlights multiple determinants of herpesvirus subfamily specificity. J. Biol. Chem. 2021. under revision. [Google Scholar]
- Marschall, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Lösing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear Egress complexes of HCMV and Other herpesviruses: Solving the puzzle of sequence coevolution, conserved structures and subfamily-spanning binding properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.E.; Van Sant, C.; Krug, P.W.; Sears, A.E.; Roizman, B. The null mutant of the U(L)31 gene of herpes simplex virus 1: Construction and phenotype in infected cells. J. Virol. 1997, 71, 8307–8315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Tanaka, M.; Kawaguchi, Y.; Baines, J.D. Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. J. Virol. 2004, 329, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.J.; Roizman, B. The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proc. Natl. Acad. Sci. USA 2000, 97, 11002–11007. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 2000, 74, 10063–10073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 2011, 85, 8285–8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, K.S.; Klupp, B.G.; Granzow, H.; Müller, F.M.; Fuchs, W.; Mettenleiter, T.C. Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J. Virol. 2012, 86, 6512–6521. [Google Scholar] [CrossRef] [Green Version]
- Farina, A.; Feederle, R.; Raffa, S.; Gonnella, R.; Santarelli, R.; Frati, L.; Angeloni, A.; Torrisi, M.R.; Faggioni, A.; Delecluse, H.J. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J. Virol. 2005, 79, 3703–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, M.; Feederle, R.; Farina, A.; Gonnella, R.; Santarelli, R.; Hub, B.; Faggioni, A.; Delecluse, H.J. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 2008, 82, 4042–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubeck, A.; Wagner, M.; Ruzsics, Z.; Lötzerich, M.; Iglesias, M.; Singh, I.R.; Koszinowski, U.H. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J. Virol. 2004, 78, 8026–8035. [Google Scholar] [CrossRef] [Green Version]
- Lotzerich, M.; Ruzsics, Z.; Koszinowski, U.H. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J. Virol. 2006, 80, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Couté, Y.; Kraut, A.; Zimmermann, C.; Büscher, N.; Hesse, A.M.; Bruley, C.; De Andrea, M.; Wangen, C.; Hahn, F.; Marschall, M.; et al. Mass spectrometry-based characterization of the virion proteome, phosphoproteome, and associated kinase activity of human cytomegalovirus. Microorganisms 2020, 8, 820. [Google Scholar] [CrossRef] [PubMed]
- Häge, S.; Horsch, D.; Stilp, A.C.; Kicuntod, J.; Müller, R.; Hamilton, S.T.; Egilmezer, E.; Rawlinson, W.D.; Stamminger, T.; Sonntag, E.; et al. A quantitative nuclear egress assay to investigate the nucleocytoplasmic capsid release of human cytomegalovirus. J. Virol. Methods 2020, 283, 113909. [Google Scholar] [CrossRef] [PubMed]
- Lorz, K.; Hofmann, H.; Berndt, A.; Tavalai, N.; Mueller, R.; Schlotzer-Schrehardt, U.; Stamminger, T. Deletion of open reading frame UL26 from the human cytomegalovirus genome results in reduced viral growth, which involves impaired stability of viral particles. J. Virol. 2006, 80, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Webel, R.; Milbradt, J.; Auerochs, S.; Schregel, V.; Held, C.; Nöbauer, K.; Razzazi-Fazeli, E.; Jardin, C.; Wittenberg, T.; Sticht, H.; et al. Two isoforms of the protein kinase pUL97 of human cytomegalovirus are differentially regulated in their nuclear translocation. J. Gen. Virol. 2011, 92, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Casabona, M.G.; Vandenbrouck, Y.; Attree, I.; Couté, Y. Proteomic characterization of Pseudomonas aeruginosa PAO1 inner membrane. Proteomics 2013, 13, 2419–2423. [Google Scholar] [CrossRef]
- Bouyssié, D.; Hesse, A.M.; Mouton-Barbosa, E.; Rompais, M.; Macron, C.; Carapito, C.; Gonzalez de Peredo, A.; Couté, Y.; Dupierris, V.; Burel, A.; et al. Proline: An efficient and user-friendly software suite for large-scale proteomics. Bioinformatics 2020, 36, 3148–3155. [Google Scholar] [CrossRef] [Green Version]
- Couté, Y.; Bruley, C.; Burger, T. Beyond Target-decoy competition: Stable validation of peptide and protein identifications in mass spectrometry-based discovery proteomics. Anal. Chem. 2020, 92, 14898–14906. [Google Scholar] [CrossRef]
- Wieczorek, S.; Combes, F.; Lazar, C.; Giai Gianetto, Q.; Gatto, L.; Dorffer, A.; Hesse, A.M.; Couté, Y.; Ferro, M.; Bruley, C.; et al. DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33, 135–136. [Google Scholar] [CrossRef] [Green Version]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Lenac Rovis, T.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human cytomegalovirus nuclear capsids associate with the core nuclear egress complex and the viral protein kinase pUL97. Viruses 2018, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [Green Version]
- Reyda, S.; Tenzer, S.; Navarro, P.; Gebauer, W.; Saur, M.; Krauter, S.; Büscher, N.; Plachter, B. The tegument protein pp65 of human cytomegalovirus acts as an optional scaffold protein that optimizes protein uploading into viral particles. J. Virol. 2014, 88, 9633–9646. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, C.; Krämer, N.; Krauter, S.; Strand, D.; Sehn, E.; Wolfrum, U.; Freiwald, A.; Butter, F.; Plachter, B. Autophagy interferes with human cytomegalovirus genome replication, morphogenesis, and progeny release. Autophagy 2021, 17, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Irmiere, A.; Gibson, W. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 1983, 130, 118–133. [Google Scholar] [CrossRef]
- Gibson, W. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 2008, 325, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1960–2014. [Google Scholar]
- Kalejta, R.F. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Tomtishen, J.P., 3rd. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol. J. 2012, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.K.; Kim, Y.J.; Kim, Y.E.; Han, T.H.; Milbradt, J.; Marschall, M.; Ahn, J.H. Transmembrane protein pUL50 of Human cytomegalovirus inhibits ISGylation by downregulating UBE1L. J. Virol. 2018, 92, e00462–e00518. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Hyeon, S.; Ahn, J.H. The human cytomegalovirus transmembrane protein pUL50 induces loss of VCP/p97 and is regulated by a small isoform of pUL50. J. Virol. 2020, 94, e00110–e00120. [Google Scholar] [CrossRef]
- Maeda, F.; Arii, J.; Hirohata, Y.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Herpes simplex virus 1 UL34 protein regulates the global architecture of the endoplasmic reticulum in infected cells. J. Virol. 2017, 91, e00271–e00317. [Google Scholar] [CrossRef] [Green Version]
- Gonnella, R.; Dimarco, M.; Farina, G.A.; Santarelli, R.; Valia, S.; Faggioni, A.; Angeloni, A.; Cirone, M.; Farina, A. BFRF1 protein is involved in EBV-mediated autophagy manipulation. Microbes Infect. 2020, 22, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, Y.; Guo, Y.; Xu, Z.; Li, Y.; Ou, X.; Xie, L.; Lu, M.; Zhong, J.; Li, B.; et al. Epstein-barr virus early protein BFRF1 suppresses IFN-β activity by inhibiting the activation of IRF3. Front. Immunol. 2020, 11, 3383. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.T.; Kung, H.N.; Chen, C.K.; Huang, C.; Wang, Y.L.; Yu, C.P.; Lee, C.P. Improving nuclear envelope dynamics by EBV BFRF1 facilitates intranuclear component clearance through autophagy. FASEB J. 2018, 32, 3968–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütz, M.; Steingruber, M.; Socher, E.; Müller, R.; Wagner, S.; Kögel, M.; Sticht, H.; Marschall, M. Functional relevance of the interaction between human cyclins and the cytomegalovirus-encoded CDK-like protein kinase pUL97. Viruses 2021, 13, 1248. [Google Scholar] [CrossRef]
- Schmeiser, C.; Borst, E.; Sticht, H.; Marschall, M.; Milbradt, J. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J. Gen. Virol. 2013, 94, 2056–2069. [Google Scholar] [CrossRef]
- Webel, R.; Hakki, M.; Prichard, M.N.; Rawlinson, W.D.; Marschall, M.; Chou, S. Differential properties of cytomegalovirus pUL97 kinase isoforms affect viral replication and maribavir susceptibility. J. Virol. 2014, 88, 4776–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marschall, M.; Stamminger, T.; Urban, A.; Wildum, S.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. In vitro evaluation of the activities of the novel anticytomegalovirus compound AIC246 (letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob. Agents Chemother. 2012, 56, 1135–1137. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Uniprot | Protein Name | Normalized iBAQ ΔUL50N | Normalized iBAQ ΔUL50C | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| Capsid | ||||||
| MCP/UL86 | P16729 | Major capsid protein | 1.04 × 107 | 5.96 × 104 | 1.06 × 107 | 1.15 × 105 |
| SCP/UL48A | Q7M6N6 | Smallest capsid protein | 5.43 × 105 | 2.06 × 105 | 4.76 × 105 | 1.67 × 105 |
| TRX1//UL46 | P16783 | Triplex capsid protein 1 | 2.16 × 106 | 5.96 × 104 | 1.96 × 106 | 1.15 × 105 |
| TRX2/UL85 | P16728 | Triplex capsid protein 2 | 6.95 × 106 | 1.10 × 105 | 7.09 × 106 | 8.11 × 105 |
| CVC1/UL93 | P16799 | Capsid vertex component 1 | 3.37 × 104 | 9.64 × 103 | 4.56 × 104 | 1.15 × 104 |
| CVC2/UL77 | P16729 | Capsid vertex component 2 | 4.12 × 105 | 3.66 × 104 | 4.54 × 105 | 8.14 × 104 |
| PORT/UL104 | P16735 | Portal protein | 5.30 × 105 | 1.30 × 105 | 3.81 × 105 | 9.03 × 104 |
| UL80 | P16753 | Capsid scaffolding protein | 1.49 × 106 | 1.92 × 105 | 1.57 × 106 | 2.03 × 105 |
| UL80.5 | B8YEA6 | Capsid scaffold protein | 2.75 × 105 | 1.07 × 105 | 2.83 × 105 | 6.45 × 104 |
| Envelope | ||||||
| gB/UL55 | P06473 | Envelope glycoprotein B | 1.30 × 107 | 2.41 × 106 | 1.06 × 107 | 6.73 × 105 |
| gH/UL75 | P12824 | Envelope glycoprotein H | 5.68 × 106 | 8.15 × 105 | 5.01 × 106 | 3.23 × 105 |
| gL/UL115 | P16832 | Envelope glycoprotein L | 1.51 × 106 | 2.14 × 105 | 1.31 × 106 | 9.78 × 104 |
| gM/UL100 | P16733 | Envelope glycoprotein M | 2.52 × 107 | 7.47 × 106 | 2.15 × 107 | 1.29 × 106 |
| gN/UL73 | P16795 | Envelope glycoprotein N | 3.39 × 106 | 1.21 × 106 | 1.96 × 106 | 1.84 × 105 |
| gO/UL74 | P16750 | Glycoprotein O | 2.45 × 105 | 1.44 × 104 | 1.47 × 105 | 3.66 × 104 |
| UL132 | P69338 | Envelope glycoprotein UL132 | 6.71 × 106 | 9.39 × 105 | 5.89 × 106 | 1.37 × 105 |
| UL41A | O39920 | Protein UL41A | 5.35 × 106 | 8.62 × 105 | 5.09 × 106 | 2.86 × 105 |
| TRL10/RL10 | P16808 | Protein IRL10 | 1.37 × 106 | 2.33 × 105 | 1.19 × 106 | 7.28 × 104 |
| UL119/UL118 | P16739 | Viral Fc-gamma receptor-like protein UL119 | 3.31 × 105 | 9.34 × 104 | 4.05 × 105 | 3.00 × 104 |
| UL116 | P16833 | Uncharacterized protein UL116 | 7.65 × 105 | 1.12 × 105 | 6.60 × 105 | 8.10 × 104 |
| UL128 | P16837 | Uncharacterized protein UL128 | 4.53 × 105 | 1.15 × 105 | 5.46 × 105 | 1.59 × 105 |
| Tegument | ||||||
| pp65/UL83 | P06725 | 65 kDa phosphoprotein | 1.01 × 108 | 2.23 × 107 | 9.41 × 107 | 9.45 × 106 |
| pp85/UL25 | P16761 | Phosphoprotein 85 | 7.31 × 106 | 1.57 × 106 | 6.14 × 106 | 8.48 × 105 |
| pp71/UL82 | P06726 | Protein pp71 | 1.32 × 107 | 2.21 × 106 | 1.10 × 107 | 6.57 × 105 |
| pp150/UL32 | P08318 | Large structural phosphoprotein | 6.34 × 106 | 1.33 × 106 | 5.01 × 106 | 2.08 × 105 |
| UL103 | P16734 | Cytoplasmic envelopment protein 1 | 5.75 × 106 | 7.45 × 105 | 5.53 × 106 | 1.72 × 105 |
| UL94 | P16800 | Cytoplasmic envelopment protein 2 | 7.72 × 106 | 1.28 × 106 | 6.99 × 106 | 4.04 × 105 |
| pp28/UL99 | P13200 | Cytoplasmic envelopment protein 3 | 7.58 × 106 | 1.84 × 106 | 6.79 × 106 | 2.25 × 105 |
| UL47 | P16784 | Inner tegument protein | 4.83 × 106 | 6.54 × 105 | 3.99 × 106 | 1.93 × 105 |
| UL48 | P16785 | Large tegument protein deneddylase | 4.16 × 106 | 8.22 × 105 | 3.04 × 106 | 7.10 × 104 |
| UL71 | P16823 | Tegument protein UL51 homolog | 2.32 × 106 | 1.27 × 105 | 2.24 × 106 | 1.70 × 105 |
| UL26 | P16762 | Tegument protein UL26 | 3.50 × 106 | 7.42 × 105 | 2.89 × 106 | 1.98 × 105 |
| UL43 | P16781 | Tegument protein UL43 | 8.14 × 105 | 7.89 × 104 | 8.34 × 105 | 1.18 × 105 |
| US24 | P09700 | Tegument protein US24 | 3.94 × 104 | 4.62 × 104 | 2.91 × 104 | 2.06 × 104 |
| RIR1/UL45 | P16782 | Ribonucleoside-diphosphate reductase large subunit-like protein | 5.11 × 106 | 1.08 × 106 | 4.71 × 106 | 1.91 × 105 |
| US23 | B8YEI2 | Protein US23 | 3.04 × 104 | 1.73 × 104 | 2.56 × 104 | 1.26 × 104 |
| UL88 | P16731 | Protein UL88 | 2.93 × 106 | 1.93 × 105 | 2.20 × 106 | 1.93 × 105 |
| UL24 | P16760 | Protein UL24 | 2.53 × 106 | 4.18 × 105 | 2.16 × 106 | 2.25 × 105 |
| UL35 | P16766 | Protein UL35 | 1.96 × 106 | 3.52 × 105 | 1.60 × 106 | 2.17 × 105 |
| UL96 | P16787 | Protein UL96 | 1.04 × 105 | 1.29 × 105 | 1.85 × 105 | 4.68 × 104 |
| TRS1 | P09695 | Protein HHLF1 | 9.47 × 105 | 1.25 × 104 | 8.73 × 105 | 9.11 × 104 |
| IRS1 | P09715 | Protein IRS1 | 4.88 × 106 | 3.27 × 105 | 4.61 × 106 | 3.53 × 105 |
| UL97 | P16788 | Serine/threonine protein kinase UL97 | 1.32 × 106 | 2.32 × 105 | 1.20 × 106 | 1.06 × 105 |
| UL69 | P16749 | mRNA export factor ICP27 homolog | 1.02 × 106 | 2.49 × 104 | 8.64 × 105 | 9.03 × 104 |
| US22 | P09722 | Early nuclear protein HWLF1 | 9.38 × 106 | 5.17 × 105 | 7.52 × 106 | 9.03 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Häge, S.; Büscher, N.; Pakulska, V.; Hahn, F.; Adrait, A.; Krauter, S.; Borst, E.M.; Schlötzer-Schrehardt, U.; Couté, Y.; Plachter, B.; et al. The Complex Regulatory Role of Cytomegalovirus Nuclear Egress Protein pUL50 in the Production of Infectious Virus. Cells 2021, 10, 3119. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10113119
Häge S, Büscher N, Pakulska V, Hahn F, Adrait A, Krauter S, Borst EM, Schlötzer-Schrehardt U, Couté Y, Plachter B, et al. The Complex Regulatory Role of Cytomegalovirus Nuclear Egress Protein pUL50 in the Production of Infectious Virus. Cells. 2021; 10(11):3119. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10113119
Chicago/Turabian StyleHäge, Sigrun, Nicole Büscher, Victoria Pakulska, Friedrich Hahn, Annie Adrait, Steffi Krauter, Eva Maria Borst, Ursula Schlötzer-Schrehardt, Yohann Couté, Bodo Plachter, and et al. 2021. "The Complex Regulatory Role of Cytomegalovirus Nuclear Egress Protein pUL50 in the Production of Infectious Virus" Cells 10, no. 11: 3119. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10113119