
Transcriptomic Mapping of Neural Diversity, Differentiation and Functional Trajectory in iPSC-Derived 3D Brain Organoid Models

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Curation
2.2. ScRNA-Seq Data Preprocessing
2.3. Normalization
2.4. Data Integration
2.5. Dimensionality Reduction and Clustering
2.6. Trajectory Calculation with PAGA Initialization and Pseudotime Analysis
2.7. Geneset Enrichment Analysis (GSEA) and Differential Expression
3. Results and Discussion
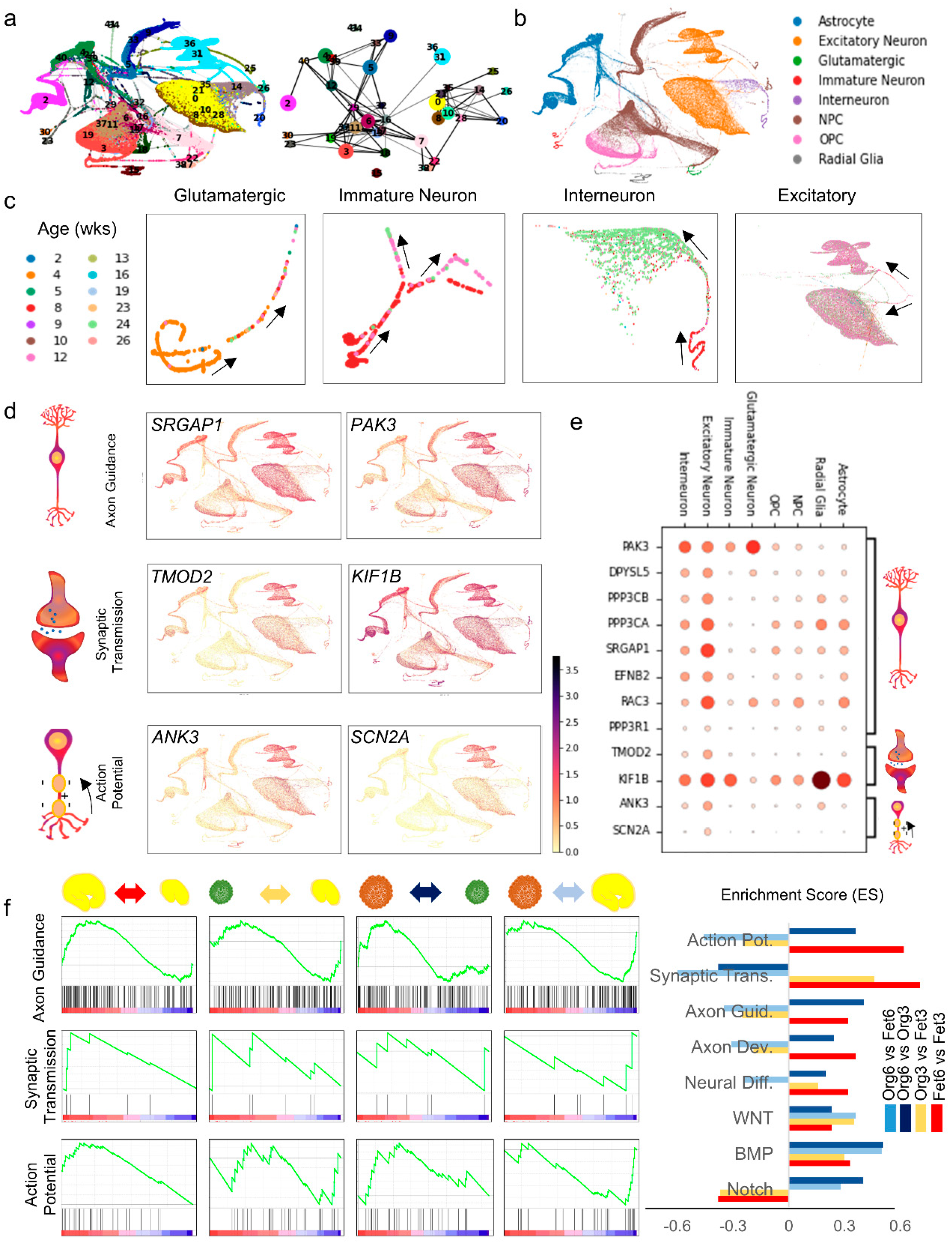
3.1. Integrated scRNA-Seq Dataset Reveals Cellular Diversity and Distribution Landscape across Organoid Generation Protocols and Faithful Reproduction of Fetal Brain Development Program
3.2. Quantifying Cell Maturity and Cellular Signaling in Organoids at Different Timepoints
3.3. Cortical Functional Trajectories during Neural Maturation in Late-Stage Organoids with PAGA Initialization and Pseudotime Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Storch, A.; Paulb, G.; Csetecd, M.; Boehm, B.O.; Carvey, P.M.; Kupschb, A.; Schwarzad, J. Long-Term Proliferation and Dopaminergic Differentiation of Human Mesencephalic Neural Precursor Cells. Exp. Neurol. 2001, 170, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-C.; Wernig, M.; Duncan, I.D.; Brüstle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef]
- Koblar, S.; Turnley, A.; Classon, B.J.; Reid, K.L.; Ware, C.B.; Cheema, S.S.; Murphy, M.; Bartlett, P.F. Neural precursor differentiation into astrocytes requires signaling through the leukemia inhibitory factor receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 3178–3181. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camp, J.G.; Treutlein, B. Advances in mini-brain technology. Nature 2017, 545, 39–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Zhang, X.-J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef]
- Tanaka, Y.; Cakir, B.; Xiang, Y.; Sullivan, G.; Park, I.-H. Synthetic Analyses of Single-Cell Transcriptomes from Multiple Brain Organoids and Fetal Brain. Cell Rep. 2020, 30, 1682–1689.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.; Anderson, A.G.; Merullo, D.P.; Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019, 58, 129–136. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.; Yang, S.M.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Yoon, S.-J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.-J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.-Y.; Lombroso, A.P.; Hwang, S.-M.; et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 2017, 21, 383–398.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, E.; Barrett, T. The gene expression omnibus database. In Statistical Genomics; Springer: New York, NY, USA, 2016; pp. 93–110. [Google Scholar]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef] [Green Version]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Lun, A.T.; McCarthy, D.; Marioni, J. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 2016, 5, 2122. [Google Scholar] [CrossRef]
- Borella, M.; Martello, G.; Risso, D.; Romualdi, C. PsiNorm: A scalable normalization for single-cell RNA-seq data. bioRxiv 2021, btab641. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Kotliar, D.; Veres, A.; Nagy, M.A.; Tabrizi, S.; Hodis, E.; Melton, D.A.; Sabeti, P.C. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife 2019, 8, e43803. [Google Scholar] [CrossRef]
- Liu, Z. Visualizing Single-Cell RNA-seq Data with Semisupervised Principal Component Analysis. Int. J. Mol. Sci. 2020, 21, 5797. [Google Scholar] [CrossRef] [PubMed]
- Hinton, G.; Roweis, S.T. Stochastic neighbor embedding. NIPS 2002, 15, 833–840. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J.J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Ko, H.-K.; Jo, J.; Noh, Y.-K.; Seo, J. Uniform Manifold Approximation with Two-phase Optimization. Available online: https://openreview.net/forum?id=gkOYZpeGEK (accessed on 28 September 2020).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Luecken, M.D.; Büttner, M.; Chaichoompu, K.; Danese, A.; Interlandi, M.; Mueller, M.F.; Strobl, D.C.; Zappia, L.; Dugas, M.; Colomé-Tatché, M.; et al. Benchmarking atlas-level data integration in single-cell genomics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Clark, J.; Iulianella, A. MLLT11/AF1q is differentially expressed in maturing neurons during development. Gene Expr. Patterns 2014, 15, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Esparcia, P.; Hernández-Ortega, K.; Koneti, A.; Gil, L.; Delgado-Morales, R.; Castaño, E.; Carmona, M.; Ferrer, I. Altered machinery of protein synthesis is region- and stage-dependent and is associated with α-synuclein oligomers in Parkinson’s disease. Acta Neuropathol. Commun. 2015, 3, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Li, Y.; Zhang, W.; Fang, F.; Sun, J.; Liu, M.; Li, K.; Dong, L. Long Non-coding RNA MALAT1 Inhibits Neuron Apoptosis and Neuroinflammation While Stimulates Neurite Outgrowth and Its Correlation With MiR-125b Mediates PTGS2, CDK5 and FOXQ1 in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Hackland, J.O.; Frith, T.; Thompson, O.; Navarro, A.M.; Garcia-Castro, M.I.; Unger, C.; Andrews, P.W. Top-Down Inhibition of BMP Signaling Enables Robust Induction of hPSCs Into Neural Crest in Fully Defined, Xeno-free Conditions. Stem Cell Rep. 2017, 9, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, V.M.; Salti, A.; Tilleman, H.; Zega, K.; Jukic, M.M.; Zou, H.; Friedel, R.H.; Prakash, N.; Blaess, S.; Edenhofer, F.; et al. BMP/SMAD Pathway Promotes Neurogenesis of Midbrain Dopaminergic Neurons In Vivo and in Human Induced Pluripotent and Neural Stem Cells. J. Neurosci. 2018, 38, 1662–1676. [Google Scholar] [CrossRef] [Green Version]
- Lasky, J.L.; Wu, H. Notch Signaling, Brain Development, and Human Disease. Pediatr. Res. 2005, 57, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Sueda, R.; Kageyama, R. Regulation of active and quiescent somatic stem cells by Notch signaling. Dev. Growth Differ. 2019, 62, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Sun, T.; Kobayashi, K.; Gao, P.; Griffin, J.D. Identification of a Family of Mastermind-Like Transcriptional Coactivators for Mammalian Notch Receptors. Mol. Cell. Biol. 2002, 22, 7688–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekooki-Machida, Y.; Hagiwara, H. Role of tubulin acetylation in cellular functions and diseases. Med Mol. Morphol. 2020, 53, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, C.; Xue, G.; Dong, Q.; Zhao, L.; Zhang, S. Parental warmth interacts with several genes to affect executive function components: A genome-wide environment interaction study. BMC Genet. 2020, 21, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Maday, S. Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res. 2016, 1649, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Song, H.; Ming, G.-L. Brain organoids: Advances, applications and challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [Green Version]
- Tambalo, M.; Lodato, S. Brain organoids: Human 3D models to investigate neuronal circuits assembly, function and dysfunction. Brain Res. 2020, 1746, 147028. [Google Scholar] [CrossRef]
- Papouin, T.; Dunphy, J.; Tolman, M.; Foley, J.C.; Haydon, P.G. Astrocytic control of synaptic function. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, Z.; Sievert, D.; Smith, D.E.C.; Mendes, M.I.; Chen, D.Y.; Stanley, V.; Ghosh, S.; Wang, Y.; Kara, M.; et al. Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat. Commun. 2020, 11, 4038. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, F.; Tang, H.; Wu, H.; Li, L.; Wu, R.; Zhao, J.; Wu, Y.; Liu, Z.; Chen, J. Tranylcypromine Causes Neurotoxicity and Represses BHC110/LSD1 in Human-Induced Pluripotent Stem Cell-Derived Cerebral Organoids Model. Front. Neurol. 2017, 8, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skipor, J.; Thiery, J.-C. The choroid plexus—Cerebrospinal fluid system: Undervaluated pathway of neuroendocrine signaling into the brain. Acta Neurobiol. Exp. 2008, 68, 414–428. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| scRNA-Seq Data | Reference | Identifier |
|---|---|---|
| Human Brain Organoids | Trujillo et al., 2019 | GSE130238 |
| Velasco et al., 2019 | GSE129519 | |
| Giandomenico et al., 2019 | GSE124174 | |
| Fiddes et al., 2018 | GSE106245 | |
| Madhavan et al., 2018 | GSE110006 | |
| Quadrato et al., 2017 | GSE86153 | |
| Birey et al., 2017 | GSE93811, GSE96045 | |
| Xiang et al., 2017 | GSE97882 | |
| Fetal Brain | Zhong et al., 2020 | GSE104276 |
| Software/Tools | ||
| GSEA (v4.1.0) | Shi et al., 2007 | |
| Harmonypy (v0.0.5) | Korsunsky et al., 2019 | |
| Python (v3.8.8) and libraries (matplotlib (v3.3.4), numpy (v1.19.5)) | Ranjani et al., 2019 | |
| Scanpy (v1.8.1), anndata (v0.7.6) | Wolf et al., 2018 | |
| GSEAPY (v0.10.5) | Mubeen et al., 2019 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiaee, K.; Jodat, Y.A.; Bassous, N.J.; Matharu, N.; Shin, S.R. Transcriptomic Mapping of Neural Diversity, Differentiation and Functional Trajectory in iPSC-Derived 3D Brain Organoid Models. Cells 2021, 10, 3422. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123422
Kiaee K, Jodat YA, Bassous NJ, Matharu N, Shin SR. Transcriptomic Mapping of Neural Diversity, Differentiation and Functional Trajectory in iPSC-Derived 3D Brain Organoid Models. Cells. 2021; 10(12):3422. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123422
Chicago/Turabian StyleKiaee, Kiavash, Yasamin A. Jodat, Nicole J. Bassous, Navneet Matharu, and Su Ryon Shin. 2021. "Transcriptomic Mapping of Neural Diversity, Differentiation and Functional Trajectory in iPSC-Derived 3D Brain Organoid Models" Cells 10, no. 12: 3422. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123422