
Planting Season Impacts Sugarcane Stem Development, Secondary Metabolite Levels, and Natural Antisense Transcription

 , , and
, , and 
Abstract
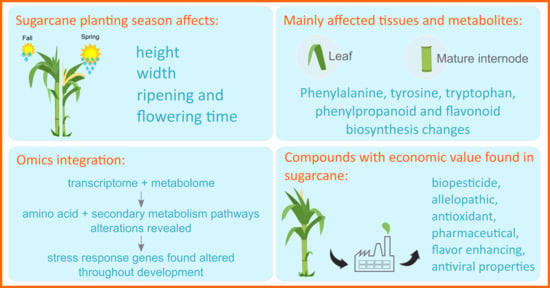
:
1. Introduction
2. Materials and Methods
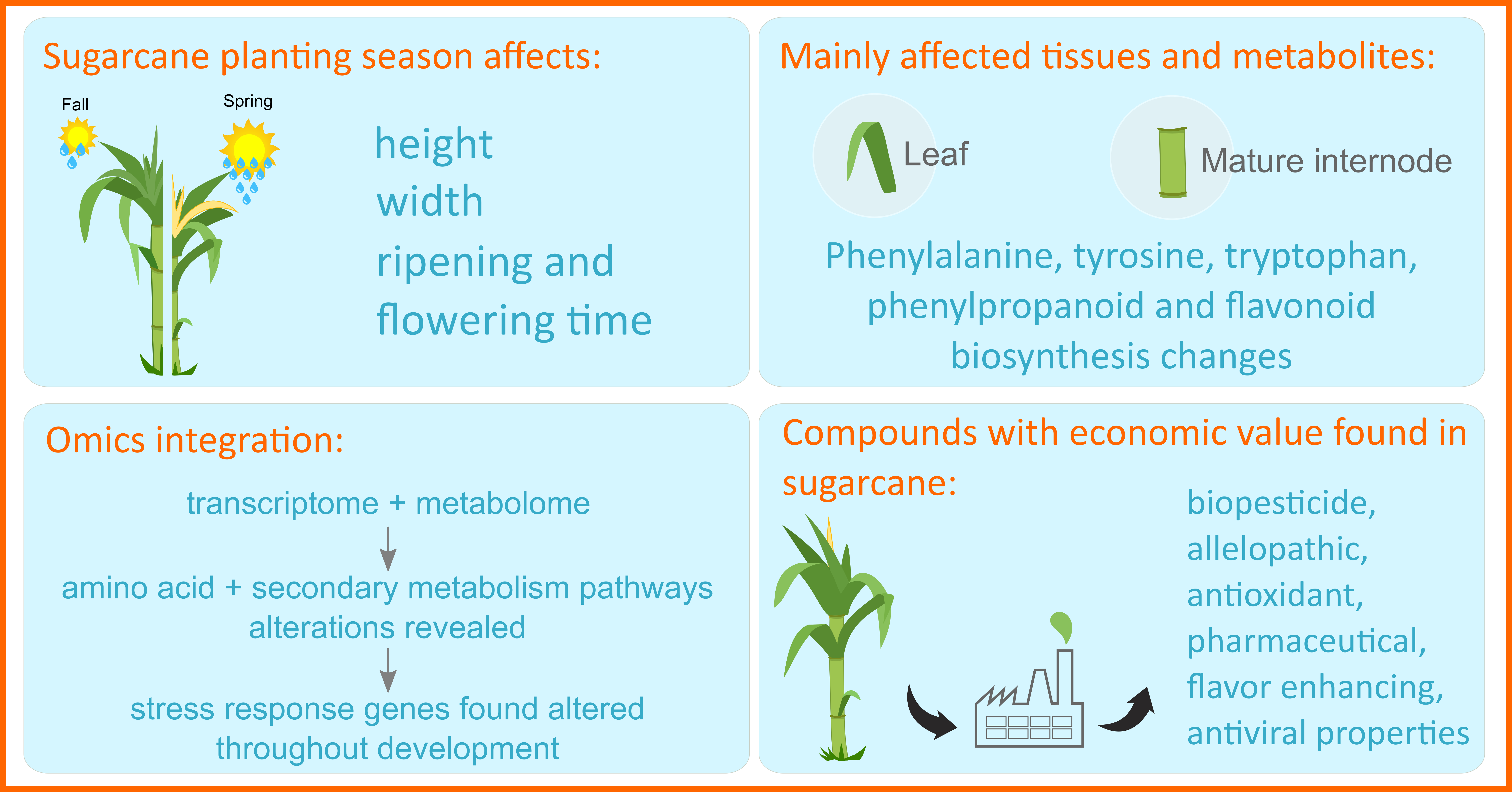
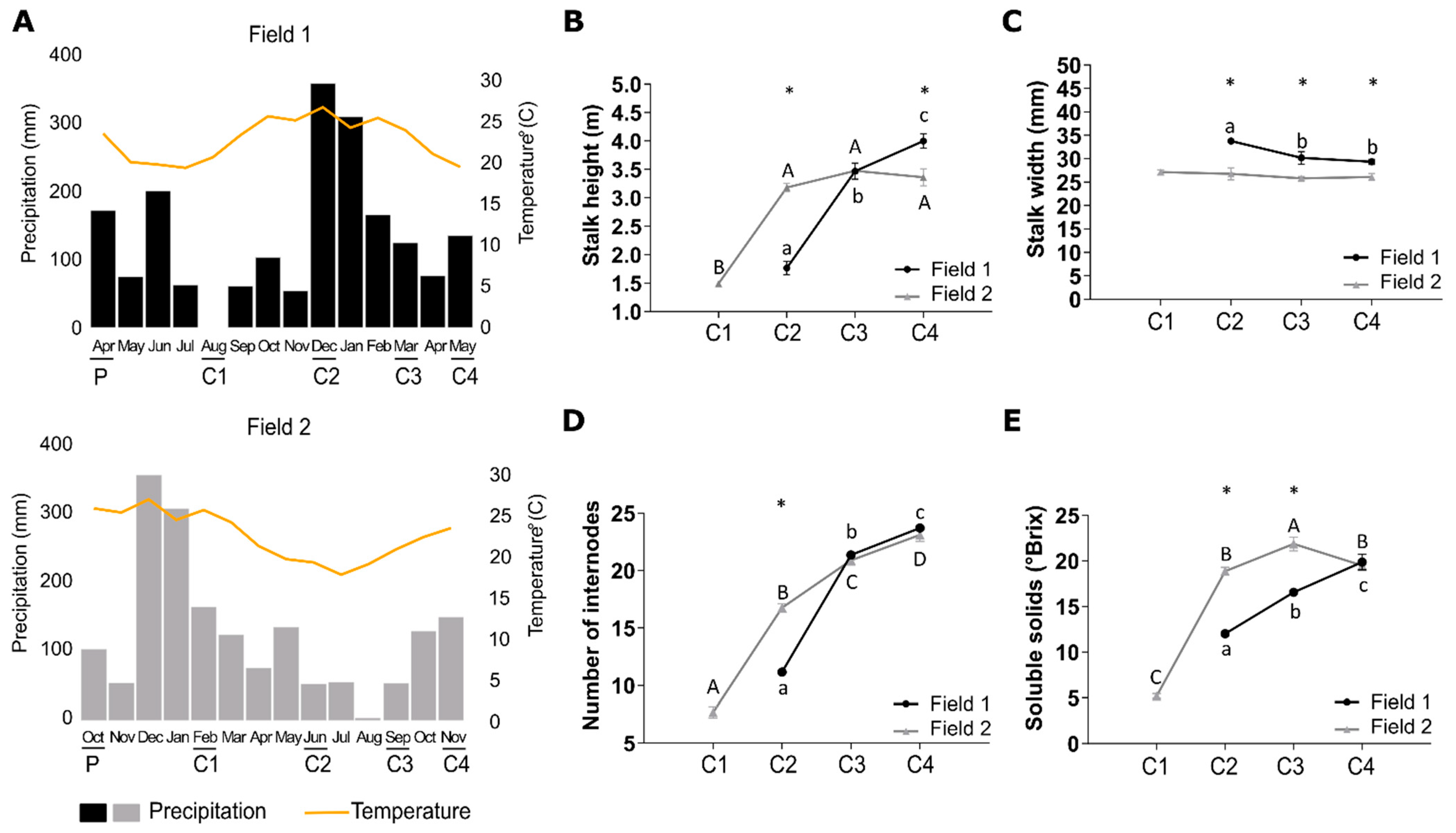
2.1. Plant Material and Experimental Design
2.2. Morphological, Technological and Physiological Data Measurements
2.3. Transcriptomic Analysis
2.3.1. RNA Extraction and Oligoarray Analysis
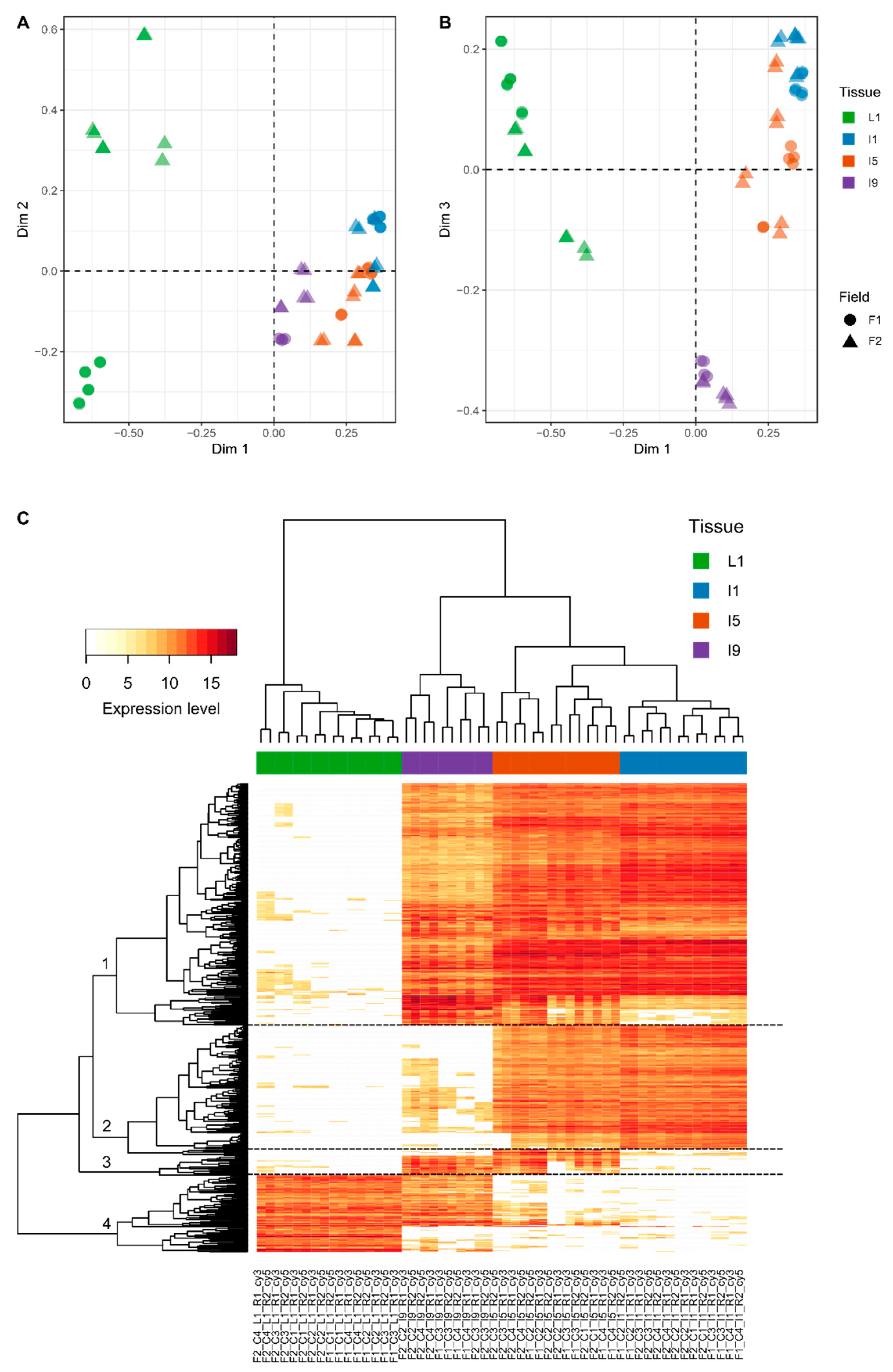
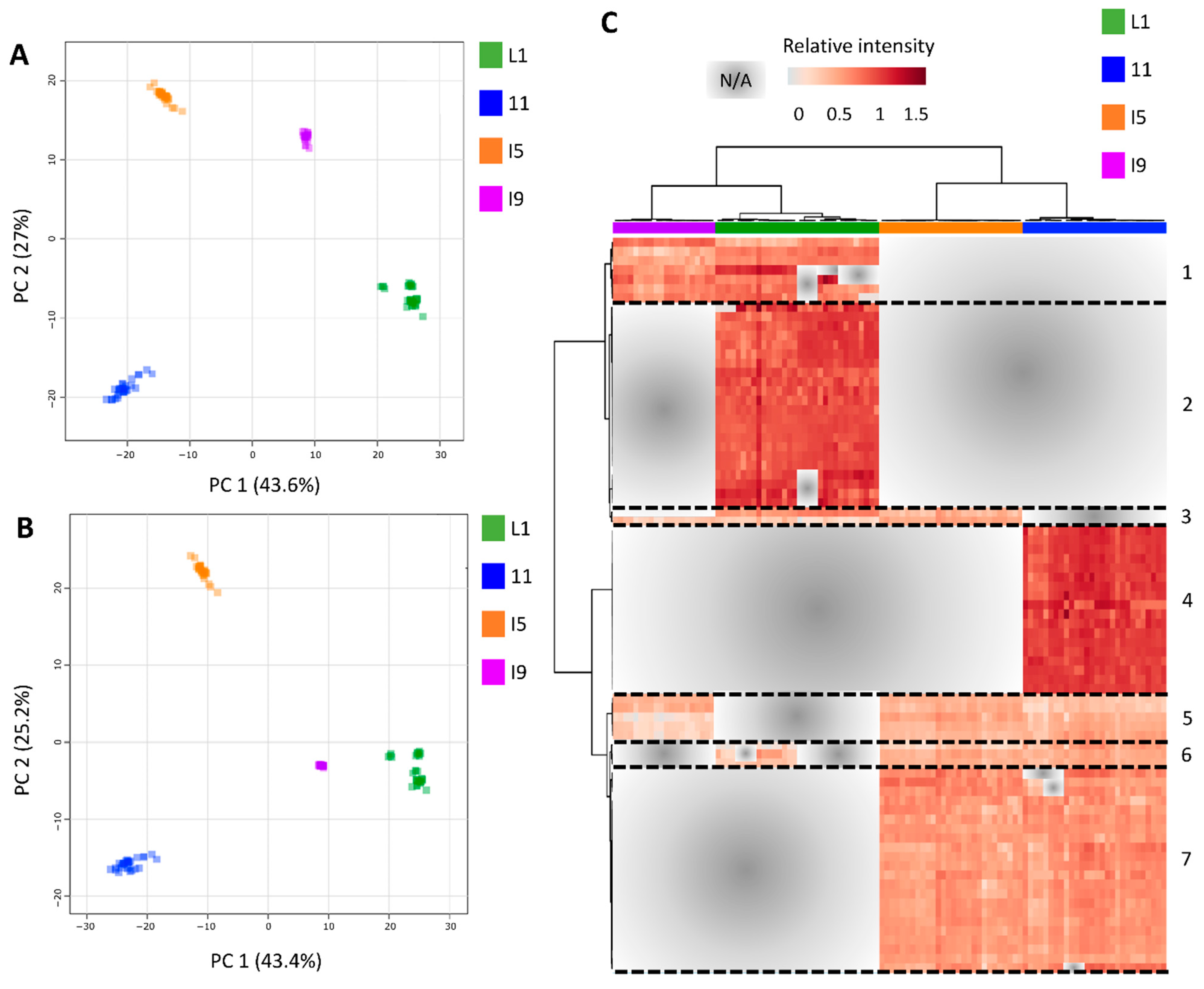
2.3.2. Multidimensional Scaling (MDS), Heatmap, GO Terms Enrichment, and Differentially Expressed Genes
2.4. Untargeted Metabolomics Analyses
2.4.1. Samples Used and Global Metabolite Extractions
2.4.2. HPLC-MS Analysis, Data Acquisition, Data Processing, and Metabolite Identification
2.5. The Integration between Two Data Modalities: Metabolomics and Transcriptomics
2.5.1. Metabolic Pathway Activities
2.5.2. The Implementation of the Multi-Omics Factor Analysis (MOFA) Tool
2.6. Statistical Analysis
3. Results
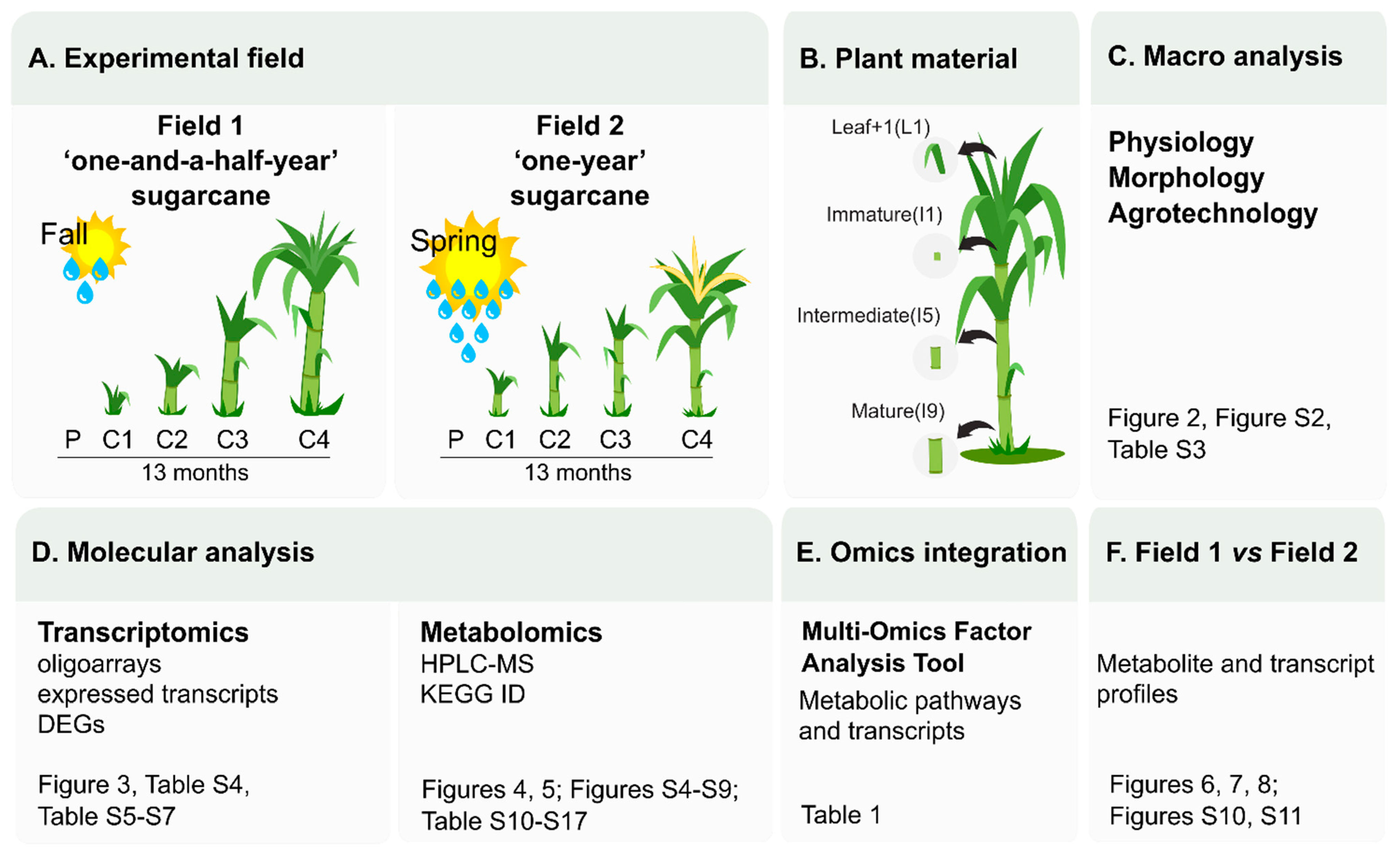
3.1. Different Climatic Conditions Influence Sugarcane Development and Ripening
3.2. Most of the Variation within Transcriptomics Profiles Is Attributed to the Differences between the Distinct Anatomical Tissues
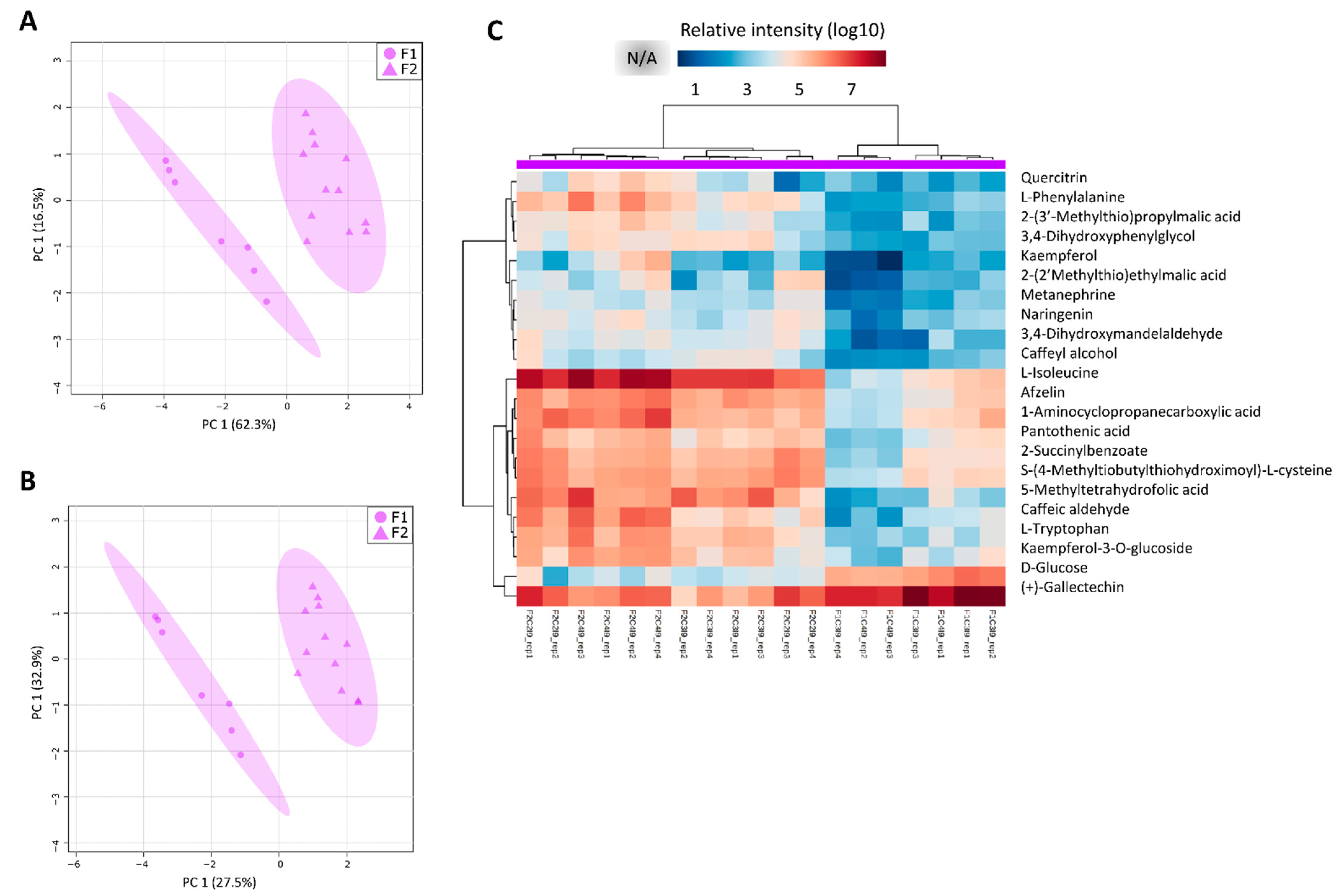
3.3. Most of the Variation within Metabolomic Profiles Is Attributed to the Differences between the Distinct Anatomical Tissues
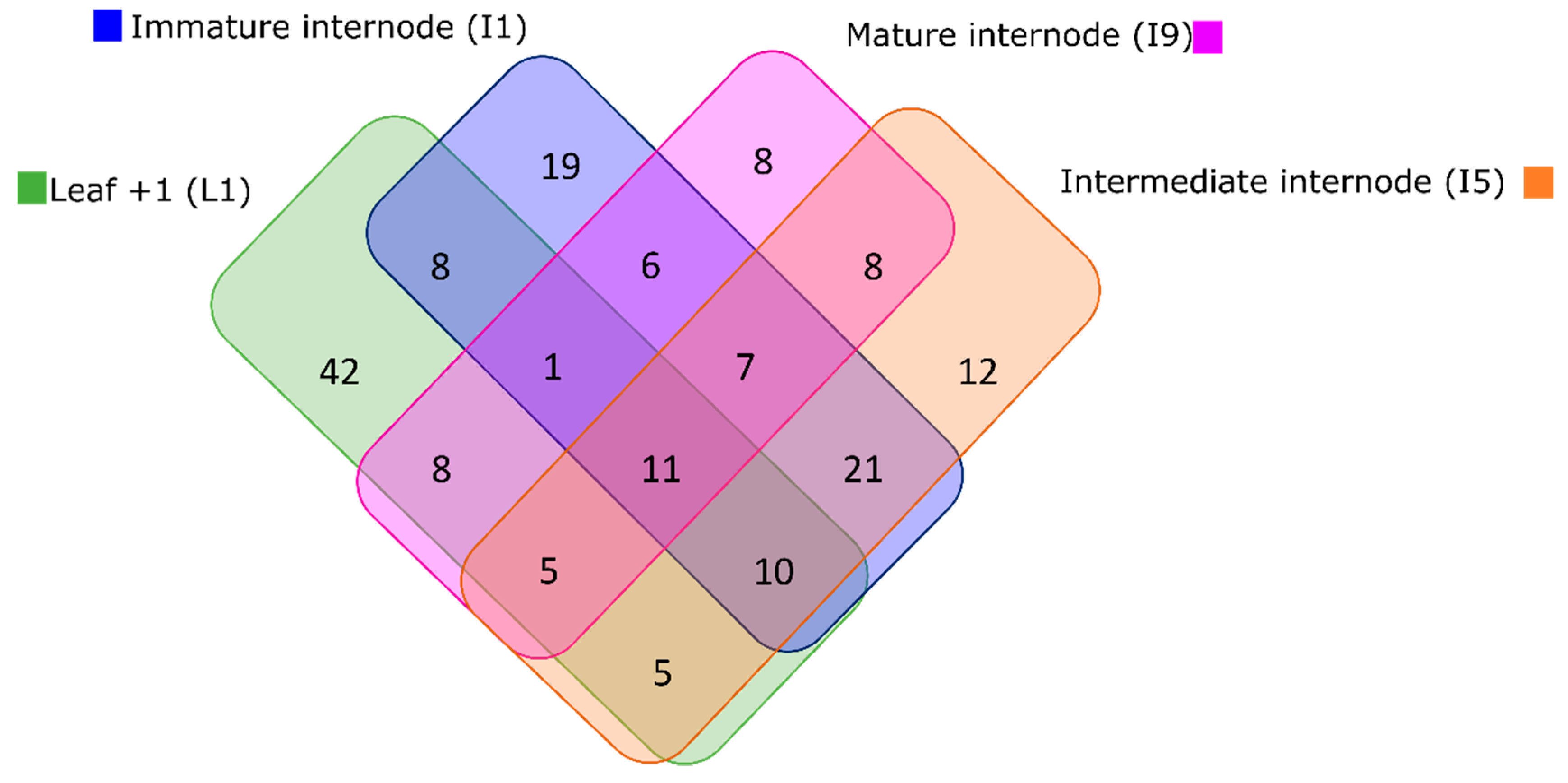
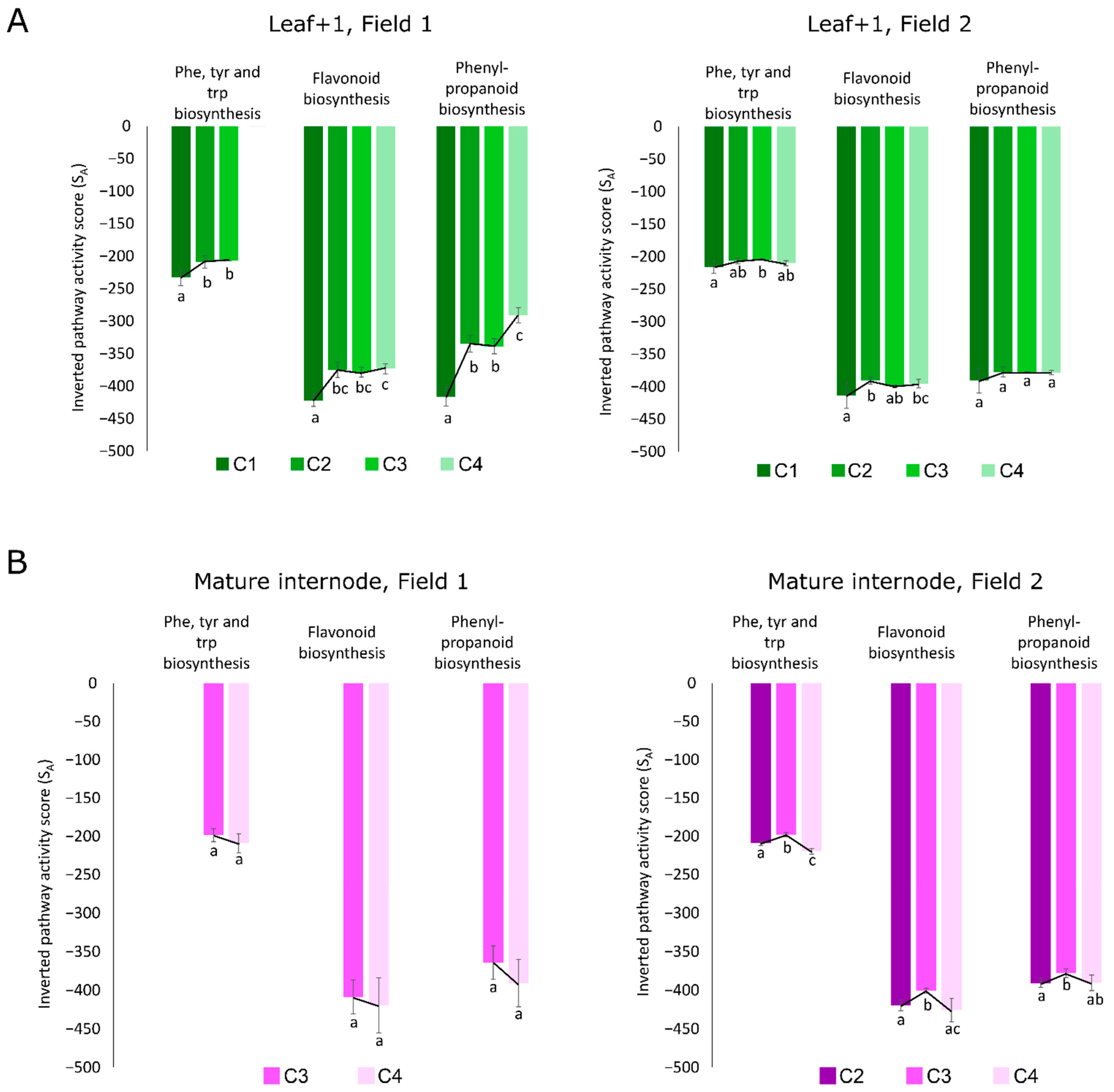
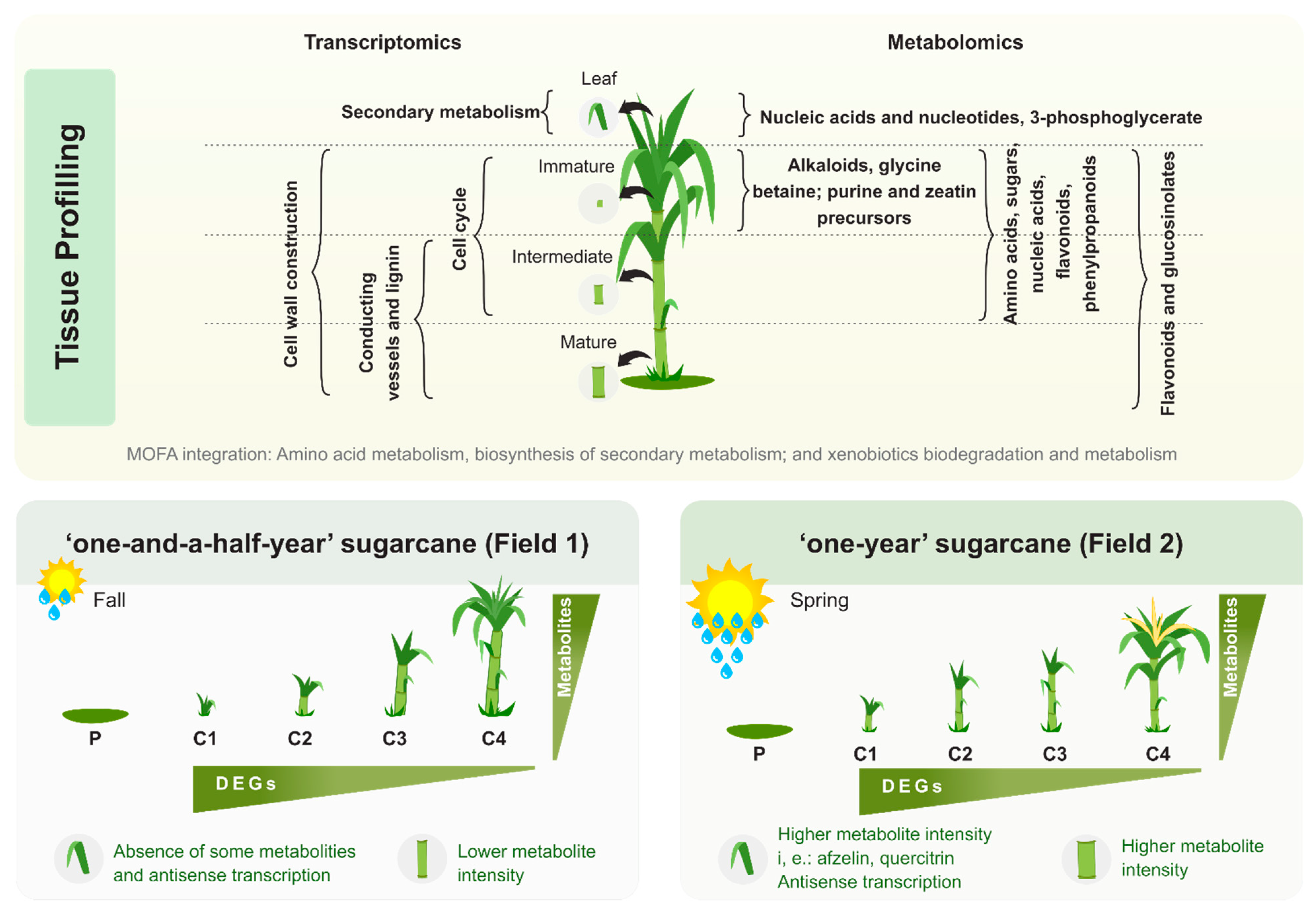
3.4. Multi-Omics Integration Highlighted Three Main Metabolic Categories in All Four Tissues: Amino Acid Metabolism, Biosynthesis of Secondary Metabolites, and Xenobiotics Biodegradation and Metabolism
3.4.1. Leaf +1 (L1)
3.4.2. Immature Internode (I1)
3.4.3. Intermediate Internode (I5)
3.4.4. Mature Internode (I9)
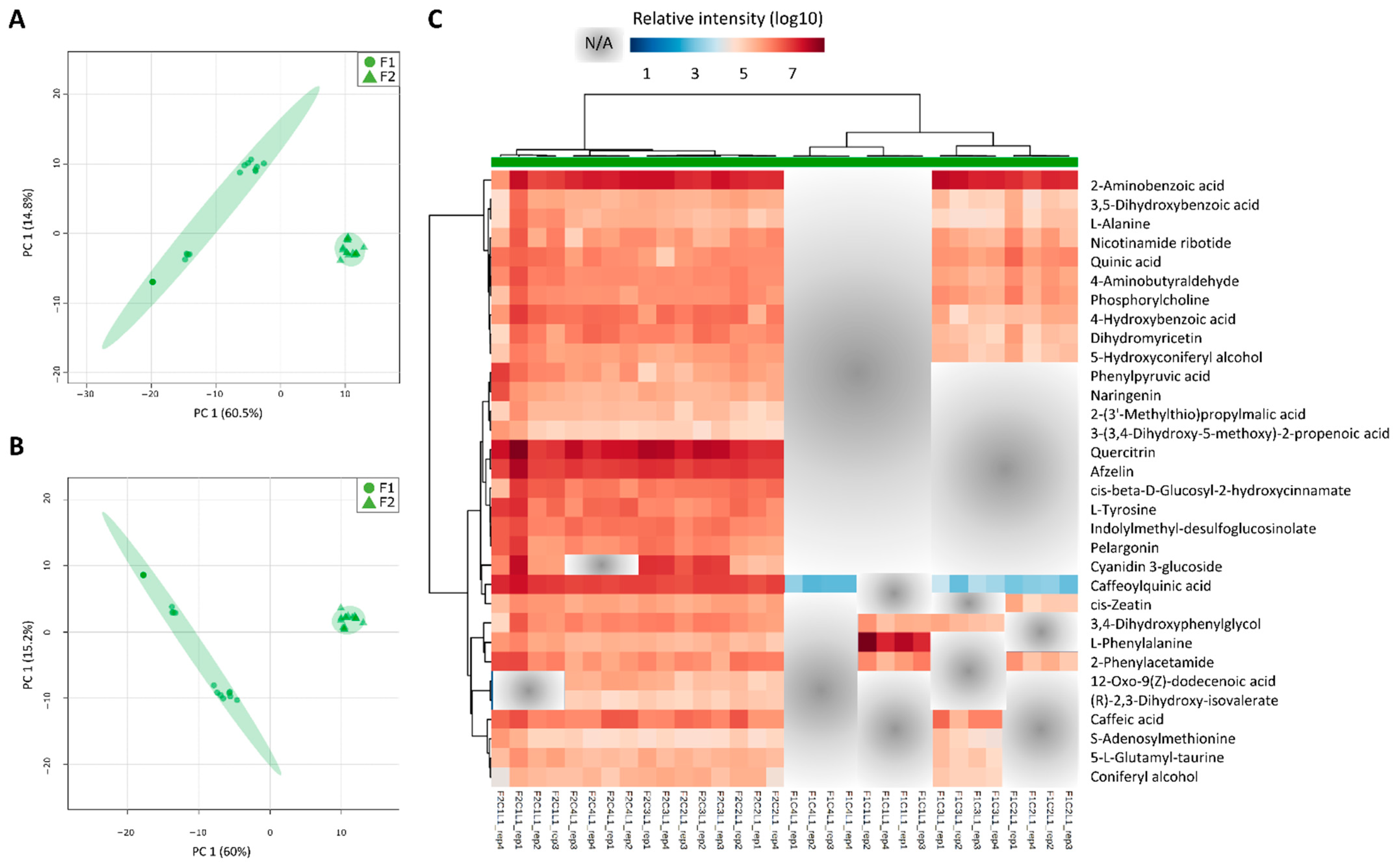
3.5. The Two Planting Conditions Imposed Differences on Mainly the Leaves and Mature Internodes, and Some Phenylpropanoids Were Detected Only in “One-Year” Sugarcane Leaves
4. Discussion
4.1. Low Precipitation and Low Temperatures, Two Factors Known to Affect Sugarcane Development, Directs Carbon Flow towards Culm Thickening Instead of Culm Elongation Affecting Ripening
4.2. Molecular Profiles of Sugarcane Tissues Reflected Their Roles in the Plant and the Higher Number of Metabolites Identified Indicates an Improved Method for Conducting Sugarcane Metabolomics Studies
4.3. Economically Valuable Compounds That Affect Plant Growth and Productivity; Inhibits SARS-CoV-2 and Are Intermediates of Glucosinolates Were Identified
4.4. Integration of Transcriptomics and Metabolomics Revealed Alterations in Metabolic Pathways Related to Development and Abiotic Stress in Plants
4.5. Leaves from “One-Year” Sugarcane Present Phenylpropanoids Not Detected in “One-and-a-Half-Year” Sugarcane That May Be Related to the Drying Off and Maturation Detected Early in F2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldemberg, J. Ethanol for a Sustainable Energy Future. Science 2007, 315, 808–810. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.B.; Demirbas, A. Gasoline, Diesel, and Ethanol Biofuels from Grasses and Plants; Cambridge University Press: Cambridge, UK, 2010; ISBN 978-0-511-77915-2. [Google Scholar]
- Lukas, E.N. Green Economy for Sustainable Development and Poverty Eradication. MJSS 2015, 6, 434. [Google Scholar] [CrossRef]
- Cortez, L.A.B. Sugarcane Bioethanol—R&D for Productivity and Sustainability; Editora Edgard Blücher: São Paulo, Brazil, 2014; ISBN 978-85-212-0822-8. [Google Scholar]
- Mekonnen, Z. Observed and Projected Reciprocate Effects of Agriculture and Climate Change: Implications on Ecosystems and Human Livelihoods. In Climate Change and Global Warming; Amini, A., Ed.; IntechOpen: London, UK, 2019; ISBN 978-1-78985-813-6. [Google Scholar]
- Wagner de Oliveira, M.; Cláudio Inácio da Silveira, L.; Bosco de Oliveira, A.; Henrique Pereira Barbosa, M.; Gomes Pereira, M.; Bezerra Albino Oliveira, T. Sugarcane Production Systems in Small Rural Properties. In Multifunctionality and Impacts of Organic Agriculture [Working Title]; IntechOpen: London, UK, 2019. [Google Scholar]
- Cardozo, N.P.; Sentelhas, P.C. Climatic Effects on Sugarcane Ripening under the Influence of Cultivars and Crop Age. Sci. Agric. 2013, 70, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Krzyzaniak, Y.; Negrel, J.; Lemaitre-Guillier, C.; Clément, G.; Mouille, G.; Klinguer, A.; Trouvelot, S.; Héloir, M.-C.; Adrian, M. Combined Enzymatic and Metabolic Analysis of Grapevine Cell Responses to Elicitors. Plant Physiol. Biochem. 2018, 123, 141–148. [Google Scholar] [CrossRef]
- Matilla, M.A. Metabolic Responses of Plants Upon Different Plant–Pathogen Interactions. In Plant Metabolites and Regulation Under Environmental Stress; Elsevier: Amsterdam, The Netherlands, 2018; pp. 195–214. ISBN 978-0-12-812689-9. [Google Scholar]
- Bhatla, S.C. Plant Growth Regulators: An Overview. In Plant Physiology, Development and Metabolism; Springer Singapore: Singapore, 2018; pp. 559–568. ISBN 9789811320224. [Google Scholar]
- Gatehouse, A.M.R. Engineering of Crops for Improved Agronomic Traits. In Handbook of Plant Biotechnology; Christou, P., Klee, H., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2004; p. kc018. ISBN 978-0-471-85199-8. [Google Scholar]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense COOLAIR Mediates the Coordinated Switching of Chromatin States at FLC during Vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [Green Version]
- Caldana, C.; Fernie, A.R.; Willmitzer, L.; Steinhauser, D. Unraveling Retrograde Signaling Pathways: Finding Candidate Signaling Molecules via Metabolomics and Systems Biology Driven Approaches. Front. Plant Sci. 2012, 3, 281. [Google Scholar] [CrossRef] [Green Version]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Manechini, J.R.V.; da Costa, J.B.; Pereira, B.T.; Carlini-Garcia, L.A.; Xavier, M.A.; de Landell, M.G.A.; Pinto, L.R. Unraveling the Genetic Structure of Brazilian Commercial Sugarcane Cultivars through Microsatellite Markers. PLoS ONE 2018, 13, e0195623. [Google Scholar] [CrossRef]
- Souza, G.M.; Van Sluys, M.-A.; Lembke, C.G.; Lee, H.; Margarido, G.R.A.; Hotta, C.T.; Gaiarsa, J.W.; Diniz, A.L.; de Oliveira, M.M.; de Ferreira, S.S.; et al. Assembly of the 373k Gene Space of the Polyploid Sugarcane Genome Reveals Reservoirs of Functional Diversity in the World’s Leading Biomass Crop. Gigascience 2019, 8, giz129. [Google Scholar] [CrossRef]
- Papini-Terzi, F.S.; Rocha, F.R.; Vêncio, R.Z.; Felix, J.M.; Branco, D.S.; Waclawovsky, A.J.; Del Bem, L.E.; Lembke, C.G.; Costa, M.D.; Nishiyama, M.Y.; et al. Sugarcane Genes Associated with Sucrose Content. BMC Genom. 2009, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, J.R.; Hossain, A.H.M.S.; Dahab, M.H.; Wyseure, G.C.L. SODCOM: A Solar Driven Computational Model of Crop Growth. Comput. Electron. Agric. 1994, 11, 293–308. [Google Scholar] [CrossRef]
- Balsalobre, T.W.A.; Mancini, M.C.; da Pereira, G.S.; Anoni, C.O.; Barreto, F.Z.; Hoffmann, H.P.; de Souza, A.P.; Garcia, A.A.F.; Carneiro, M.S. Mixed Modeling of Yield Components and Brown Rust Resistance in Sugarcane Families. Agron. J. 2016, 108, 1824–1837. [Google Scholar] [CrossRef] [Green Version]
- Consecana Manual de Instruções. 2006. Available online: http://www.oricana.com.br/novosite/manual_consecana.pdf (accessed on 3 December 2021).
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric Method for Determination of Sugars and Related Substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Yang, Y.H. Normalization for CDNA Microarray Data: A Robust Composite Method Addressing Single and Multiple Slide Systematic Variation. Nucleic Acids Res. 2002, 30, e15. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. TopGO: Enrichment Analysis for Gene Ontology, Version 2.3.6 R Package. 2019. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 28 October 2021).
- Salem, M.A.; Jüppner, J.; Bajdzienko, K.; Giavalisco, P. Protocol: A Fast, Comprehensive and Reproducible One-Step Extraction Method for the Rapid Preparation of Polar and Semi-Polar Metabolites, Lipids, Proteins, Starch and Cell Wall Polymers from a Single Sample. Plant Methods 2016, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.R.; Jourdan, F.; Salvanha, D.M.; Letisse, F.; Jamin, E.L.; Guidetti-Gonzalez, S.; Labate, C.A.; Vêncio, R.Z.N. ProbMetab: An R Package for Bayesian Probabilistic Annotation of LC–MS-Based Metabolomics. Bioinformatics 2014, 30, 1336–1337. [Google Scholar] [CrossRef] [Green Version]
- Aggio, R.B.M.; Ruggiero, K.; Villas-Bôas, S.G. Pathway Activity Profiling (PAPi): From the Metabolite Profile to the Metabolic Pathway Activity. Bioinformatics 2010, 26, 2969–2976. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards More Transparent and Integrative Metabolomics Analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A Web Server for Metabolomic Data Analysis and Interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—a Framework for Unsupervised Integration of Multi-Omics Data Sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, E.B.; Cavalcanti, P.P.; Nogueira, D.A. ExpDes: An R Package for ANOVA and Experimental Designs. AM 2014, 5, 2952–2958. [Google Scholar] [CrossRef] [Green Version]
- Lembke, C.G.; Nishiyama, M.Y.; Sato, P.M.; de Andrade, R.F.; Souza, G.M. Identification of Sense and Antisense Transcripts Regulated by Drought in Sugarcane. Plant Mol. Biol. 2012, 79, 461–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wishart, D.S. Metabolomic Data Processing, Analysis, and Interpretation Using MetaboAnalyst. Curr. Protoc. Bioinform. 2011, 34, 14.10.1–14.10.48. [Google Scholar] [CrossRef]
- Brieger, F.O. Início Da Safra. Como Determinar a Maturação. Bol. Inf. Copereste 1968, 4, 1–3. [Google Scholar]
- Wagih, M.E.; Ala, A.; Musa, Y. Evaluation of Sugarcane Varieties for Maturity Earliness and Selection for Efficient Sugar Accumulation. Sugar Tech 2004, 6, 297–304. [Google Scholar] [CrossRef]
- Prabu, G.; Kawar, P.G.; Pagariya, M.C.; Prasad, D.T. Identification of Water Deficit Stress Upregulated Genes in Sugarcane. Plant Mol. Biol. Rep. 2011, 29, 291–304. [Google Scholar] [CrossRef]
- Ebrahim, M.K.; Zingsheim, O.; El-Shourbagy, M.N.; Moore, P.H.; Komor, E. Growth and Sugar Storage in Sugarcane Grown at Temperatures below and above Optimum. J. Plant Physiol. 1998, 153, 593–602. [Google Scholar] [CrossRef]
- Dominy, C.; Haynes, R.; van Antwerpen, R. Loss of Soil Organic Matter and Related Soil Properties under Long-Term Sugarcane Production on Two Contrasting Soils. Biol. Fertil. Soils 2002, 36, 350–356. [Google Scholar] [CrossRef]
- Robertson, M.J.; Wood, A.W.; Muchow, R.C. Growth of Sugarcane under High Input Conditions in Tropical Australia. I. Radiation Use, Biomass Accumulation and Partitioning. Field Crop. Res. 1996, 48, 11–25. [Google Scholar] [CrossRef]
- de Medeiros Barbosa, V.C.F. Sugarcane; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 978-0-12-802239-9. [Google Scholar]
- Carr, M.K.V.; Knox, J.W. The Water Relations and Irrigation Requirements of Sugar Cane (Saccharum officinarum): A Review. Exp. Agric. 2011, 47, 27–51. [Google Scholar] [CrossRef]
- Cardozo, N.P.; Sentelhas, P.C.; Panosso, A.R.; Palhares, A.L.; Ide, B.Y. Modeling Sugarcane Ripening as a Function of Accumulated Rainfall in Southern Brazil. Int. J. Biometeorol. 2015, 59, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, W.-C.; Xu, Y.-Q.; Li, G.-J.; Liao, Y.; Fu, F.-L. Differential Expression of Candidate Genes for Lignin Biosynthesis under Drought Stress in Maize Leaves. J. Appl. Genet. 2009, 50, 213–223. [Google Scholar] [CrossRef]
- da Silva, V.; da Silva, B.B.; Albuquerque, W.G.; Borges, C.J.R.; de Sousa, I.F.; Neto, J.D. Crop Coefficient, Water Requirements, Yield and Water Use Efficiency of Sugarcane Growth in Brazil. Agric. Water Manage. 2013, 128, 102–109. [Google Scholar] [CrossRef]
- Singels, A.; Donaldson, R.A.; Smit, M.A. Improving Biomass Production and Partitioning in Sugarcane: Theory and Practice. Field Crop. Res. 2005, 92, 291–303. [Google Scholar] [CrossRef]
- Marli, S.; Aparecida, F.; da Graa, J.P.; de Matos Pereira, L.; Vergnia, M. Sugarcane Responses at Water Deficit Conditions. Water Stress 2012, 255–276. [Google Scholar] [CrossRef] [Green Version]
- Hennig, L. Patterns of Beauty—Omics Meets Plant Development. Trends Plant Sci. 2007, 12, 287–293. [Google Scholar] [CrossRef]
- Bottcher, A.; Cesarino, I.; Brombini dos Santos, A.; Vicentini, R.; Mayer, J.L.S.; Vanholme, R.; Morreel, K.; Goeminne, G.; Moura, J.C.M.S.; Nobile, P.M.; et al. Lignification in Sugarcane: Biochemical Characterization, Gene Discovery, and Expression Analysis in Two Genotypes Contrasting for Lignin Content. Plant Physiol. 2013, 163, 1539–1557. [Google Scholar] [CrossRef]
- Kurosaki, F. Induction and Activation of Plant Secondary Metabolism by External Stimuli. In Drug Discovery Research in Pharmacognosy; Vallisuta, O., Ed.; InTech: London, UK, 2012; ISBN 978-953-51-0213-7. [Google Scholar]
- Tejera, N.; Ortega, E.; Rodes, R.; Lluch, C. Nitrogen Compounds in the Apoplastic Sap of Sugarcane Stem: Some Implications in the Association with Endophytes. J. Plant Physiol. 2006, 163, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Guerzoni, J.T.S.; Belintani, N.G.; Moreira, R.M.P.; Hoshino, A.A.; Domingues, D.S.; Filho, J.C.B.; Vieira, L.G.E. Stress-Induced Δ1-Pyrroline-5-Carboxylate Synthetase (P5CS) Gene Confers Tolerance to Salt Stress in Transgenic Sugarcane. Acta Physiol. Plant 2014, 36, 2309–2319. [Google Scholar] [CrossRef]
- Dvořáková, L.; Srba, M.; Opatrny, Z.; Fischer, L. Hybrid Proline-Rich Proteins: Novel Players in Plant Cell Elongation? Ann. Bot. 2012, 109, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, R.; Costantino, P.; Trovato, M. Proline Accumulation in Plants: Not Only Stress. Plant Signal. Behav. 2009, 4, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.B.; Bottcher, A.; Vicentini, R.; Sampaio Mayer, J.L.; Kiyota, E.; Landell, M.A.G.; Creste, S.; Mazzafera, P. Lignin Biosynthesis in Sugarcane Is Affected by Low Temperature. Environ. Exp. Bot. 2015, 120, 31–42. [Google Scholar] [CrossRef]
- Glassop, D.; Roessner, U.; Bacic, A.; Bonnett, G.D. Changes in the Sugarcane Metabolome with Stem Development. Are They Related to Sucrose Accumulation? Plant. Cell Physiol. 2007, 48, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Bosch, S.; Rohwer, J.M.; Botha, F.C. The Sugarcane Metabolome. Proc. S. Afr. Sug. Technol. Ass. 2003, 7, 129–133. [Google Scholar]
- Ferreira, D.A.; Martins, M.C.M.; Cheavegatti-Gianotto, A.; Carneiro, M.S.; Amadeu, R.R.; Aricetti, J.A.; Wolf, L.D.; Hoffmann, H.P.; de Abreu, L.G.F.; Caldana, C. Metabolite Profiles of Sugarcane Culm Reveal the Relationship Among Metabolism and Axillary Bud Outgrowth in Genetically Related Sugarcane Commercial Cultivars. Front. Plant Sci. 2018, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Shao, Q.; Yin, L.; Younis, A.; Zheng, B. Polyamine Function in Plants: Metabolism, Regulation on Development, and Roles in Abiotic Stress Responses. Front. Plant Sci. 2018, 9, 1945. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Fuell, C.; Parr, A.; Hill, L.; Bailey, P.; Elliott, K.; Fairhurst, S.A.; Martin, C.; Michael, A.J. A Novel Polyamine Acyltransferase Responsible for the Accumulation of Spermidine Conjugates in Arabidopsis Seed. Plant Cell 2009, 21, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Martin-Tanguy, J. Conjugated Polyamines and Reproductive Development: Biochemical, Molecular and Physiological Approaches. Physiol. Plant 1997, 100, 675–688. [Google Scholar] [CrossRef]
- Nkomo, M.; Gokul, A.; Keyster, M.; Klein, A. Exogenous P-Coumaric Acid Improves Salvia hispanica L. Seedling Shoot Growth. Plants 2019, 8, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenck, C.A.; Maeda, H.A. Tyrosine Biosynthesis, Metabolism, and Catabolism in Plants. Phytochemistry 2018, 149, 82–102. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, U.; Kliebenstein, D.J.; Lambrix, V.; Reichelt, M.; Gershenzon, J. Chapter Five Glucosinolate Hydrolysis and Its Impact on Generalist and Specialist Insect Herbivores. In Recent Advances in Phytochemistry; Elsevier: Amsterdam, The Netherlands, 2003; Volume 37, pp. 101–125. ISBN 978-0-08-044277-8. [Google Scholar]
- Rongai, D.; Cerato, C.; Lazzeri, L. A Natural Fungicide for the Control of Erysiphe Betae and Erysiphe Cichoracearum. Eur. J. Plant Pathol. 2009, 124, 613–619. [Google Scholar] [CrossRef]
- Borges, A.; Abreu, A.C.; Ferreira, C.; Saavedra, M.J.; Simões, L.C.; Simões, M. Antibacterial Activity and Mode of Action of Selected Glucosinolate Hydrolysis Products against Bacterial Pathogens. J. Food Sci. Technol. 2015, 52, 4737–4748. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, R.D.L.; Dhingra, O.D.; Lima, A.O.; Jham, G.N.; Berhow, M.A.; Holloway, R.K.; Vaughn, S.F. Glucosinolate Content and Nematicidal Activity of Brazilian Wild Mustard Tissues against Meloidogyne Incognita in Tomato. Plant Soil 2011, 341, 155–164. [Google Scholar] [CrossRef]
- Halkier, B.A.; Gershenzon, J. BIOLOGY AND BIOCHEMISTRY OF GLUCOSINOLATES. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and Isothiocyanates in Health and Disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef]
- Johnson, I.T. Glucosinolates: Bioavailability and Importance to Health. Int. J. Vitam. Nutr. Res. 2002, 72, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Claros Cuadrado, J.L.; Pinillos, E.O.; Tito, R.; Mirones, C.S.; Gamarra Mendoza, N.N. Insecticidal Properties of Capsaicinoids and Glucosinolates Extracted from Capsicum Chinense and Tropaeolum Tuberosum. Insects 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittstock, U.; Halkier, B.A. Glucosinolate Research in the Arabidopsis Era. Trends Plant Sci. 2002, 7, 263–270. [Google Scholar] [CrossRef]
- Das, S.K.; Mahanta, S.; Tanti, B.; Tag, H.; Hui, P.K. Identification of Phytocompounds from Houttuynia Cordata Thunb. as Potential Inhibitors for SARS-CoV-2 Replication Proteins through GC–MS/LC–MS Characterization, Molecular Docking and Molecular Dynamics Simulation. Mol. Divers 2021, 31, 179–193. [Google Scholar] [CrossRef]
- Kandeel, M.; Abdelrahman, A.H.M.; Oh-Hashi, K.; Ibrahim, A.; Venugopala, K.N.; Morsy, M.A.; Ibrahim, M.A.A. Repurposing of FDA-Approved Antivirals, Antibiotics, Anthelmintics, Antioxidants, and Cell Protectives against SARS-CoV-2 Papain-like Protease. J. Biomol. Struct. Dyn. 2021, 39, 5129–5136. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Omarini, A.B.; Li, B. Virtual Screening for Functional Foods against the Main Protease of SARS-CoV-2. J. Food Biochem. 2020, 44, e13481. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Gupta, V.K.; Eldesoky, G.E.; Wabaidur, S.M.; Patil, S.A.; Islam, M.A. Identification of Potential Anti-TMPRSS2 Natural Products through Homology Modelling, Virtual Screening and Molecular Dynamics Simulation Studies. J. Biomol. Struct. Dyn. 2021, 39, 6660–6675. [Google Scholar] [CrossRef]
- Gao, W.; Sun, H.-X.; Xiao, H.; Cui, G.; Hillwig, M.L.; Jackson, A.; Wang, X.; Shen, Y.; Zhao, N.; Zhang, L.; et al. Combining Metabolomics and Transcriptomics to Characterize Tanshinone Biosynthesis in Salvia Miltiorrhiza. BMC Genom. 2014, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Shin, T.H.; Lee, D.Y.; Lee, H.-S.; Park, H.J.; Jin, M.S.; Paik, M.-J.; Manavalan, B.; Mo, J.-S.; Lee, G. Integration of Metabolomics and Transcriptomics in Nanotoxicity Studies. BMB Rep. 2018, 51, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Udhane, S.S.; Legeza, B.; Marti, N.; Hertig, D.; Diserens, G.; Nuoffer, J.-M.; Vermathen, P.; Flück, C.E. Combined Transcriptome and Metabolome Analyses of Metformin Effects Reveal Novel Links between Metabolic Networks in Steroidogenic Systems. Sci. Rep. 2017, 7, 8652. [Google Scholar] [CrossRef] [Green Version]
- Izawa, T.; Mihara, M.; Suzuki, Y.; Gupta, M.; Itoh, H.; Nagano, A.J.; Motoyama, R.; Sawada, Y.; Yano, M.; Hirai, M.Y.; et al. Os-GIGANTEA Confers Robust Diurnal Rhythms on the Global Transcriptome of Rice in the Field. Plant Cell 2011, 23, 1741–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogel, K.-H.; Voll, L.M.; Schafer, P.; Jansen, C.; Wu, Y.; Langen, G.; Imani, J.; Hofmann, J.; Schmiedl, A.; Sonnewald, S.; et al. Transcriptome and Metabolome Profiling of Field-Grown Transgenic Barley Lack Induced Differences but Show Cultivar-Specific Variances. Proc. Natl. Acad. Sci. USA 2010, 107, 6198–6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, A.L.; da Silva, D.I.R.; Lembke, C.G.; Costa, M.D.-B.L.; ten-Caten, F.; Li, F.; Vilela, R.D.; Menossi, M.; Ware, D.; Endres, L.; et al. Amino Acid and Carbohydrate Metabolism Are Coordinated to Maintain Energetic Balance during Drought in Sugarcane. Int. J. Mol. Sci. 2020, 21, 9124. [Google Scholar] [CrossRef]
- Hildebrandt, T.M. Synthesis versus Degradation: Directions of Amino Acid Metabolism during Arabidopsis Abiotic Stress Response. Plant Mol. Biol. 2018, 98, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Fernie, A.R. The Use of Metabolomics to Dissect Plant Responses to Abiotic Stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef] [Green Version]
- Dare, A.P.; Yauk, Y.-K.; Tomes, S.; McGhie, T.K.; Rebstock, R.S.; Cooney, J.M.; Atkinson, R.G. Silencing a Phloretin-Specific Glycosyltransferase Perturbs Both General Phenylpropanoid Biosynthesis and Plant Development. Plant J. 2017, 91, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Ferrandino, A.; Lovisolo, C. Abiotic Stress Effects on Grapevine (Vitis vinifera L.): Focus on Abscisic Acid-Mediated Consequences on Secondary Metabolism and Berry Quality. Environ. Exp. Bot. 2014, 103, 138–147. [Google Scholar] [CrossRef]
- Davin, L.B.; Lewis, N.G. Phenylpropanoid Metabolism: Biosynthesis of Monolignols, Lignans and Neolignans, Lignins and Suberins. In Phenolic Metabolism in Plants; Stafford, H.A., Ibrahim, R.K., Eds.; Springer US: Boston, MA, USA, 1992; ISBN 978-1-4613-6517-4. [Google Scholar]
- Kasirajan, L.; Hoang, N.V.; Furtado, A.; Botha, F.C.; Henry, R.J. Transcriptome Analysis Highlights Key Differentially Expressed Genes Involved in Cellulose and Lignin Biosynthesis of Sugarcane Genotypes Varying in Fiber Content. Sci. Rep. 2018, 8, 11612. [Google Scholar] [CrossRef] [Green Version]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of Oxidative and Drought Tolerance in Arabidopsis by Overaccumulation of Antioxidant Flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef]
- He, F.; Li, D.; Wang, D.; Deng, M. Extraction and Purification of Quercitrin, Hyperoside, Rutin, and Afzelin from Zanthoxylum Bungeanum Maxim Leaves Using an Aqueous Two-Phase System: ATPS of Z. Bungeanum Leaves Flavonoids. J. Food Sci. 2016, 81, C1593–C1602. [Google Scholar] [CrossRef]
- Shi, M.; He, N.; Li, W.; Li, C.; Kang, W. Simultaneous Determination of Myricetrin, Quercitrin and Afzelin in Leaves of Cercis Chinensis by a Fast and Effective Method of Ionic Liquid Microextraction Coupled with HPLC. Chem. Cent. J. 2018, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- de Barros, M.; Mota da Silva, L.; Boeing, T.; Somensi, L.B.; Cury, B.J.; de Moura Burci, L.; Santin, J.R.; de Andrade, S.F.; Monache, F.D.; Cechinel-Filho, V. Pharmacological Reports about Gastroprotective Effects of Methanolic Extract from Leaves of Solidago Chilensis (Brazilian Arnica) and Its Components Quercitrin and Afzelin in Rodents. Naunyn-Schmiedeberg’s Arch. Pharm. 2016, 389, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Treml, J.; Šmejkal, K. Flavonoids as Potent Scavengers of Hydroxyl Radicals: Flavonoids versus Hydroxyl Radical. Compr. Rev. Food Sci. Food Saf. 2016, 15, 720–738. [Google Scholar] [CrossRef]
- Min, D.-Y.; Chang, H.-M.; Jameel, H.; Lucia, L.; Wang, Z.-G. The Structure of Lignin of Corn Stover and Its Changes Induced by Mild Sodium Hydroxide Treatment. BioResources 2014, 9, 2405–2414. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chantreau, M.; Sibout, R.; Hawkins, S. Plant Cell Wall Lignification and Monolignol Metabolism. Front. Plant Sci. 2013, 4, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassard, J.-E.; Ullmann, P.; Bernier, F.; Werck-Reichhart, D. Phenolamides: Bridging Polyamines to the Phenolic Metabolism. Phytochemistry 2010, 71, 1808–1824. [Google Scholar] [CrossRef]
- Goujon, T.; Sibout, R.; Pollet, B.; Maba, B.; Nussaume, L.; Bechtold, N.; Lu, F.; Ralph, J.; Mila, I.; Barrière, Y.; et al. [No Title Found]. Plant Mol. Biol. 2003, 51, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.C.; Doran-Peterson, J. Pectin-Rich Biomass as Feedstock for Fuel Ethanol Production. Appl. Microbiol. Biotechnol. 2012, 95, 565–575. [Google Scholar] [CrossRef] [Green Version]
- de Souza, A.P.; Leite, D.C.C.; Pattathil, S.; Hahn, M.G.; Buckeridge, M.S. Composition and Structure of Sugarcane Cell Wall Polysaccharides: Implications for Second-Generation Bioethanol Production. Bioenergy Res. 2013, 6, 564–579. [Google Scholar] [CrossRef]
- Dunstan, W.R. Cyanogenesis in Plants. Part II.—The Great Millet, Sorghum vulgare. Proc. R. Soc. Lond. 1902, 70, 153–154. [Google Scholar] [CrossRef]
- de Rosa Júnior, V.E.; Nogueira, F.T.S.; Mazzafera, P.; Landell, M.G.; Arruda, P. Sugarcane Dhurrin: Biosynthetic Pathway Regulation and Evolution; International Society Sugar Cane Technologists (ISSCT): Durban, South Africa, 2007. [Google Scholar]
- Hotta, C.T.; Nishiyama, M.Y.; Souza, G.M. Circadian Rhythms of Sense and Antisense Transcription in Sugarcane, a Highly Polyploid Crop. PLoS ONE 2013, 8, e71847. [Google Scholar] [CrossRef]
- Ferreira, S.S.; Hotta, C.T.; de Poelking, V.G.C.; Leite, D.C.C.; Buckeridge, M.S.; Loureiro, M.E.; Barbosa, M.H.P.; Carneiro, M.S.; Souza, G.M. Co-Expression Network Analysis Reveals Transcription Factors Associated to Cell Wall Biosynthesis in Sugarcane. Plant Mol. Biol. 2016, 91, 15–35. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, Y.; Nakayama, N.; Saneoka, H.; Mohapatra, P.K.; Fujita, K. Differences in the Responses of Stem Diameter and Pod Thickness to Drought Stress during the Grain Filling Stage in Soybean Plants. Acta Physiol. Plant. 2009, 31, 271–277. [Google Scholar] [CrossRef]
- Okuyama, L.A.; Federizzi, L.C.; Barbosa Neto, J.F. Plant Traits to Complement Selection Based on Yield Components in Wheat. Cienc. Rural 2005, 35, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, M.C. Morpho-Agronomic Characterization and Evaluation for Drought Tolerance of 50 Selected Philippine Traditional Rice (Oryza sativa L.) Varieties. Philipp. J. Crop. Sci. 2015, 39, 116–117. [Google Scholar]
- Tzin, V.; Galili, G. New Insights into the Shikimate and Aromatic Amino Acids Biosynthesis Pathways in Plants. Mol. Plant 2010, 3, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Agati, G.; Azzarello, E.; Pollastri, S.; Tattini, M. Flavonoids as Antioxidants in Plants: Location and Functional Significance. Plant Sci. 2012, 196, 67–76. [Google Scholar] [CrossRef]
- Rao, M.J.; Xu, Y.; Tang, X.; Huang, Y.; Liu, J.; Deng, X.; Xu, Q. CsCYT75B1, a Citrus CYTOCHROME P450 Gene, Is Involved in Accumulation of Antioxidant Flavonoids and Induces Drought Tolerance in Transgenic Arabidopsis. Antioxidants 2020, 9, 161. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Transcriptomics Data Modality | Metabolomics Data Modality | ||
|---|---|---|---|---|
| GO ID | GO Term | KEGG Map ID | KEGG Map Name | |
| L1 | GO:0044281 | small molecule metabolic process | map00940 | Phenylpropanoid biosynthesis |
| GO:0006520 | cellular amino acid metabolic process | map00941 | Flavonoid biosynthesis | |
| GO:0043436 | oxoacid metabolic process | map00360 | Phenylalanine metabolism | |
| GO:0006082 | organic acid metabolic process | map00640 | Cyanoamino acid metabolism | |
| GO:0019752 | carboxylic acid metabolic process | map00966 | Glucosinolate biosynthesis | |
| GO:1901566 | organonitrogen compound biosynthetic process | map00030 | Pentose phosphate pathway | |
| GO:0008652 | cellular amino acid biosynthetic process | map00400 | Phenylalanine, tyrosine and tryptophan metabolism | |
| GO:1901605 | alpha-amino acid metabolic process | map00380 | Tryptophan metabolism | |
| GO:0044283 | small molecule biosynthetic process | map00362 | Benzoate degradation | |
| GO:1901607 | alpha-amino acid biosynthetic process | map00960 | Tropane, piperidine and pyridine alkaloid biosynthesis | |
| GO:0016311 | Dephosphorylation | map00350 | Tyrosine metabolism | |
| GO:0051186 | cofactor metabolic process | map00623 | Toluene degradation | |
| GO:0017144 | drug metabolic process | map00627 | Aminobenzoate degradation | |
| GO:0055086 | nucleobase-containing small molecule metabolism | map00130 | Ubiquinone and other terpenoid-quinone biosynthesis | |
| I1 | GO:0010035 | response to inorganic substance | map00943 | Isoflavonoid biosynthesis |
| GO:0010077 | maintenance of inflorescence meristem identity | map00966 | Glucosinolate biosynthesis | |
| GO:0009414 | response to water deprivation | map00944 | Flavone and flavonol biosynthesis | |
| GO:0006952 | defense response | map00680 | Methane metabolism | |
| GO:0009415 | response to water | map00360 | Phenylalanine metabolism | |
| GO:0005980 | glycogen catabolic process | map00940 | Phenylpropanoid biosynthesis | |
| GO:0046398 | UDP-glucuronate metabolic process | map00998 | Biosynthesis of secondary metabolites-unclassified | |
| GO:0050832 | defense response to fungus | map00130 | Ubiquinone and other terpenoid-quinone biosynthesis | |
| GO:0006950 | response to stress | map00980 | Metabolism of xenobiotics by cytochrome P450 | |
| GO:0045944 | positive regulation of transcription by RNA polymerase II | map00524 | Neomycin, kanamycin and gentamicin biosynthesis | |
| GO:0006457 | protein folding | map00950 | Isoquinoline alkaloid biosynthesis | |
| GO:0061077 | chaperone-mediated protein folding | map00350 | Tyrosine metabolism | |
| GO:0006298 | mismatch repair | map00564 | Glycerophospholipid metabolism | |
| GO:0050896 | response to stimulus | map00965 | Betalain biosynthesis | |
| GO:0032508 | DNA duplex unwinding | map00261 | Monobactam biosynthesis | |
| GO:0051704 | multi-organism process | map00410 | beta-Alanine metabolism | |
| GO:0009620 | response to fungus | map01055 | Biosynthesis of vancomycin group antibiotics | |
| GO:0032392 | DNA geometric change | map00480 | Glutathione metabolism | |
| I5 | GO:0005975 | carbohydrate metabolic process | map00908 | Zeatin biosynthesis |
| GO:0051186 | cofactor metabolic process | map00954 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | |
| GO:0017144 | drug metabolic process | map00564 | Glycerophospholipid metabolism | |
| GO:0042737 | drug catabolic process | map00230 | Purine metabolism | |
| GO:0098754 | Detoxification | map00640 | Propanoate metabolism | |
| GO:0009636 | response to toxic substance | map00770 | Pantothenate and CoA biosynthesis | |
| GO:0045229 | external encapsulating structure organization | map00626 | Naphthalene degradation | |
| GO:0044281 | small molecule metabolic process | map00361 | Chlorocyclohexane and chlorobenzene degradation | |
| GO:0055086 | nucleobase-containing small molecule metabolism | map00350 | Tyrosine metabolism | |
| GO:0098869 | cellular oxidant detoxification | map00643 | Styrene degradation | |
| I9 | GO:0006470 | protein dephosphorylation | map00860 | Porphyrin and chlorophyll metabolism |
| GO:0009072 | aromatic amino acid family metabolic process | map00630 | Glyoxylate and dicarboxylate metabolism | |
| GO:0016311 | dephosphorylation | map00998 | Biosynthesis of secondary metabolites-unclassified | |
| GO:0005975 | carbohydrate metabolic process | map00040 | Pentose and glucuronate interconversions | |
| GO:0009073 | aromatic amino acid family biosynthetic process | map00680 | Methane metabolism | |
| GO:0016053 | organic acid biosynthetic process | map00380 | Tryptophan metabolism | |
| GO:0046394 | carboxylic acid biosynthetic process | map00523 | Polyketide sugar unit biosynthesis | |
| GO:0044281 | small molecule metabolic process | map00340 | Histidine metabolism | |
| GO:0017144 | drug metabolic process | map00261 | Monobactam biosynthesis | |
| GO:0006520 | cellular amino acid metabolic process | map00983 | Drug metabolism-other enzymes | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijma, M.; Lembke, C.G.; Diniz, A.L.; Santini, L.; Zambotti-Villela, L.; Colepicolo, P.; Carneiro, M.S.; Souza, G.M. Planting Season Impacts Sugarcane Stem Development, Secondary Metabolite Levels, and Natural Antisense Transcription. Cells 2021, 10, 3451. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123451
Wijma M, Lembke CG, Diniz AL, Santini L, Zambotti-Villela L, Colepicolo P, Carneiro MS, Souza GM. Planting Season Impacts Sugarcane Stem Development, Secondary Metabolite Levels, and Natural Antisense Transcription. Cells. 2021; 10(12):3451. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123451
Chicago/Turabian StyleWijma, Maryke, Carolina Gimiliani Lembke, Augusto Lima Diniz, Luciane Santini, Leonardo Zambotti-Villela, Pio Colepicolo, Monalisa Sampaio Carneiro, and Glaucia Mendes Souza. 2021. "Planting Season Impacts Sugarcane Stem Development, Secondary Metabolite Levels, and Natural Antisense Transcription" Cells 10, no. 12: 3451. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123451