
LRRK2 Modulates the Exocyst Complex Assembly by Interacting with Sec8


 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Plasmid Constructions
2.3. Cell Lines
2.4. GST Pull-Down Assay
2.5. In-Gel Digestion and Matrix-Assisted Laser Desorption/Ionization (MALDI) Mass Spectrometry (MS) Analysis
2.6. Subcellular Fractionation of HEK293 Cells
2.7. Immunoprecipitation
2.8. Western Blot Analysis
2.9. Immunofluorescence
2.10. Disuccinimidyl Suberate (DSS) Cross-Linking
2.11. Statistical Analysis
3. Results
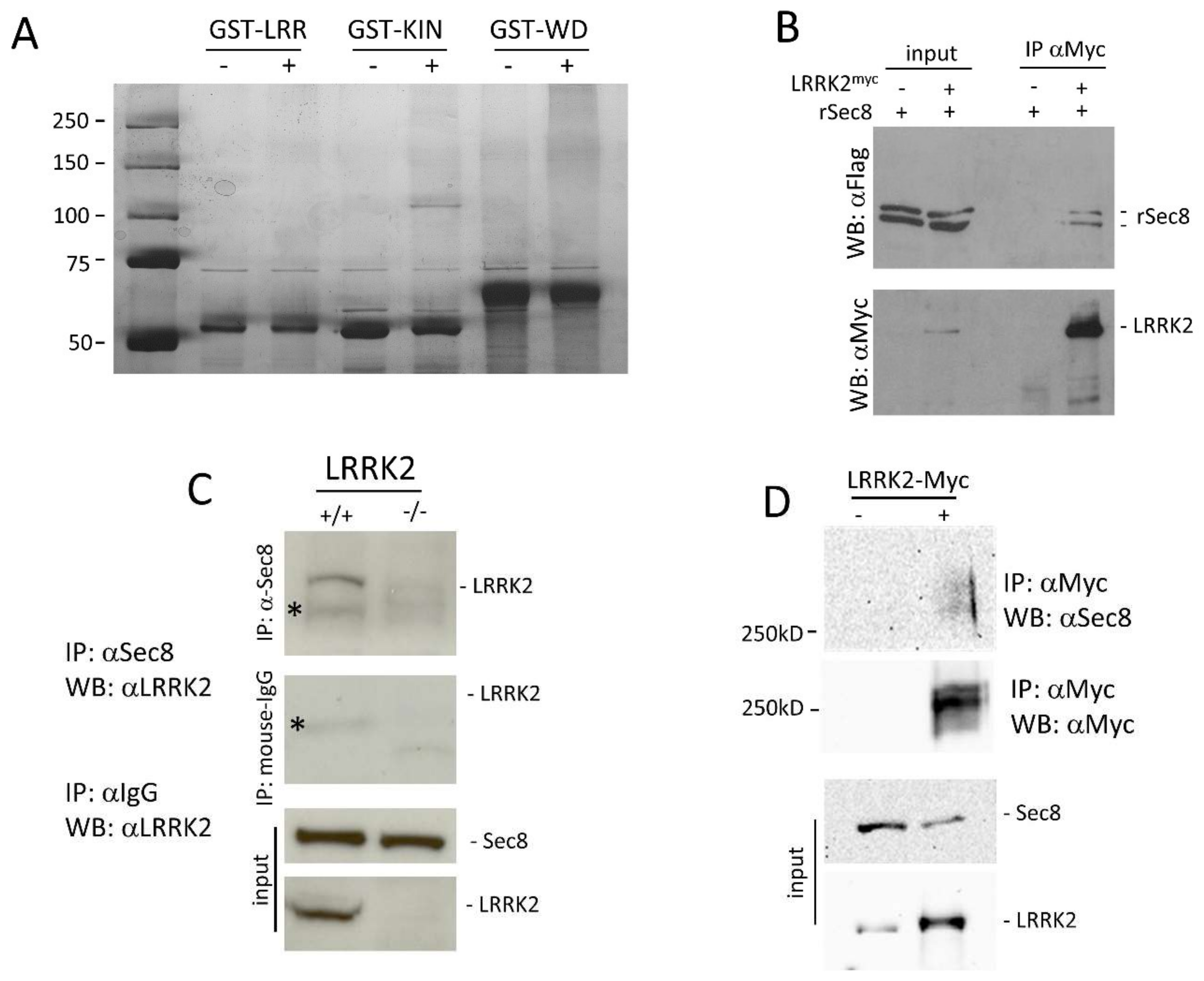
3.1. LRRK2 Interacts with Sec8
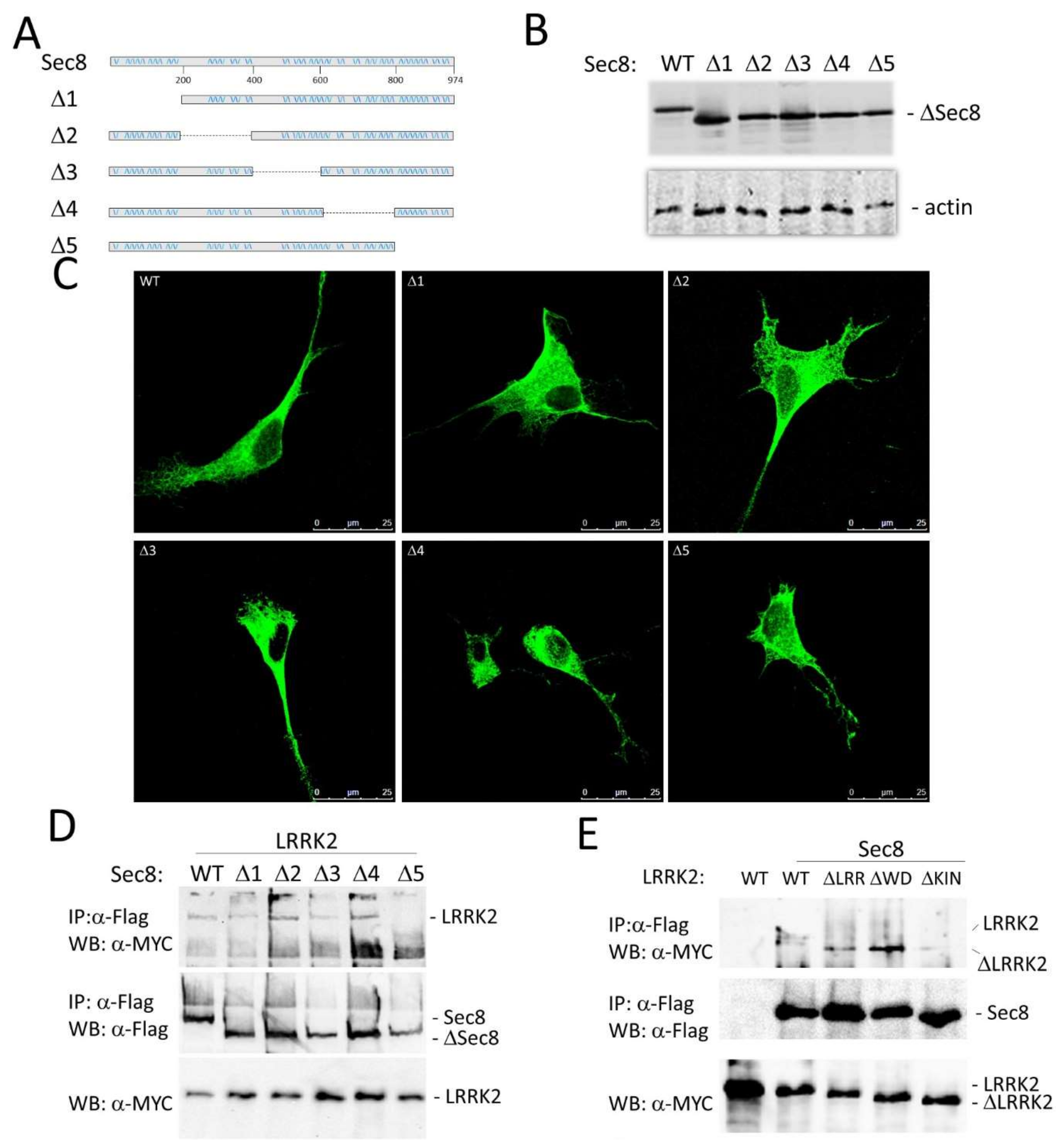
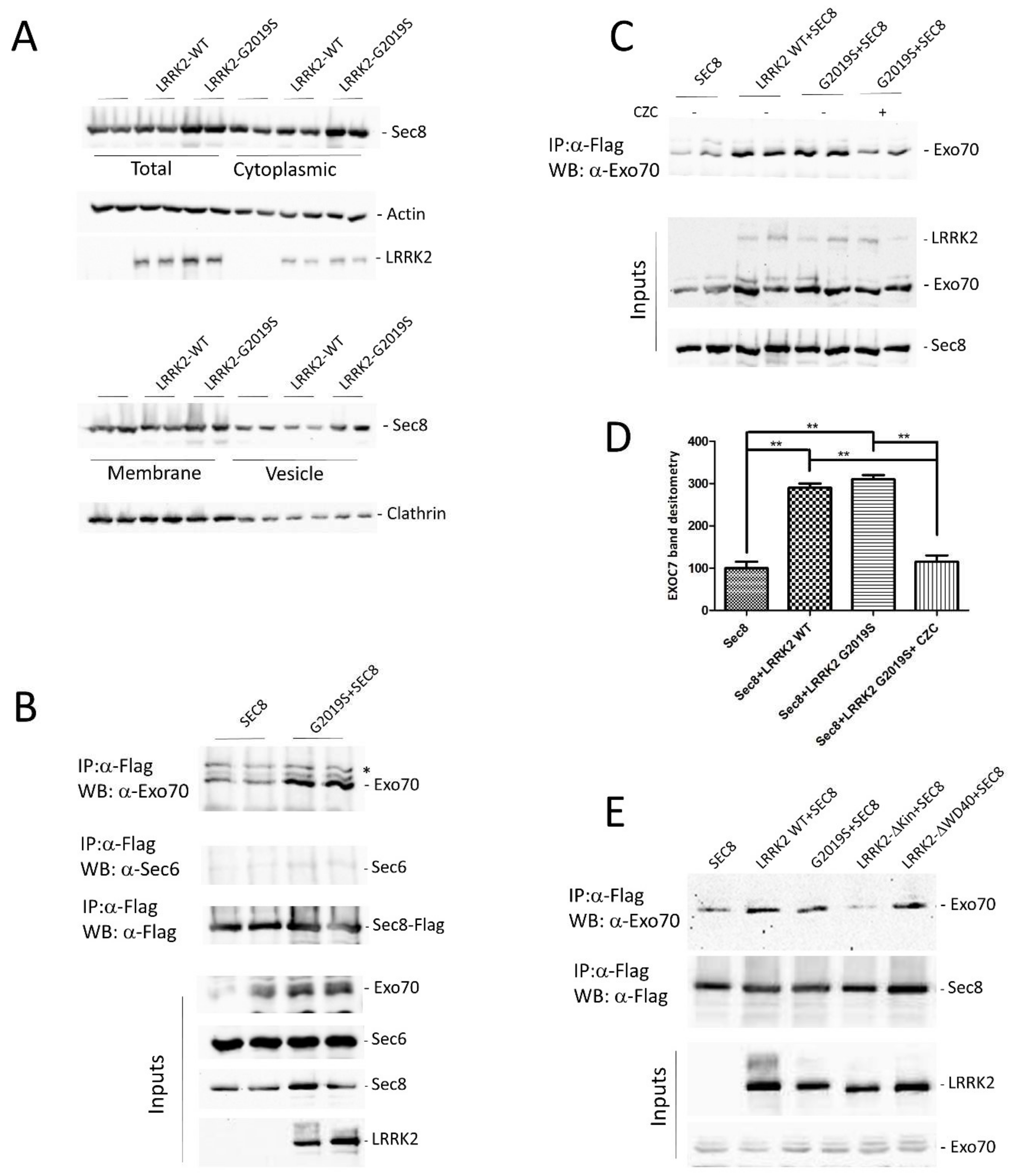
3.2. LRRK2 Modulates the Exocyst Complex Assembly
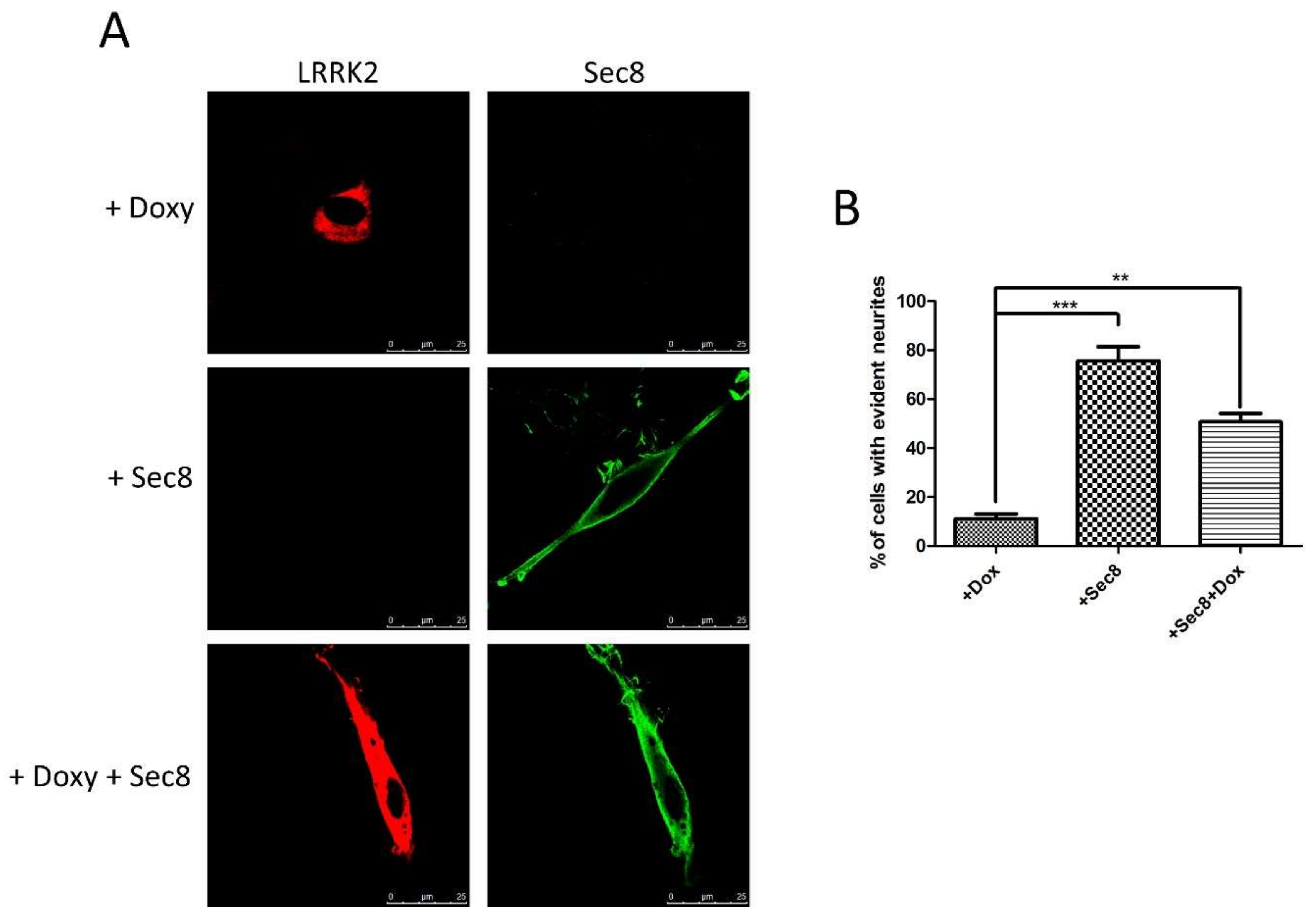
3.3. Functional Role of LRRK2/Sec8 Interaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Sanna, G.; Del Giudice, M.G.; Crosio, C.; Iaccarino, C. LRRK2 and vesicle trafficking. Biochem. Soc. Trans. 2012, 40, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Ana Clara, F.; Verstreken, P. Synaptic vesicle trafficking and Parkinson’s disease. Dev. Neurobiol. 2012, 72, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Kim, H.; Ga, I.; Oh, H.; Ho, D.H.; Kim, J.; Seo, H.; Son, I.; Seol, W. An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J. Biochem. 2015, 157, 485–495. [Google Scholar] [CrossRef]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Yu, J.; Xie, C.; Rudrabhatla, P.; Chen, X.; Wu, J.; Parisiadou, L.; Liu, G.; Sun, L.; Ma, B.; et al. Leucine-rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER-Golgi export. EMBO J. 2014, 33, 2314–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisiadou, L.; Yu, J.; Sgobio, C.; Xie, C.; Liu, G.; Sun, L.; Gu, X.L.; Lin, X.; Crowley, N.A.; Lovinger, D.M.; et al. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat. Neurosci. 2014, 17, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef]
- Godena, V.K.; Brookes-Hocking, N.; Moller, A.; Shaw, G.; Oswald, M.; Sancho, R.M.; Miller, C.C.; Whitworth, A.J.; De Vos, K.J. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 2014, 5, 5245. [Google Scholar] [CrossRef] [Green Version]
- Law, B.M.; Spain, V.A.; Leinster, V.H.; Chia, R.; Beilina, A.; Cho, H.J.; Taymans, J.M.; Urban, M.K.; Sancho, R.M.; Blanca Ramirez, M.; et al. A direct interaction between leucine-rich repeat kinase 2 and specific beta-tubulin isoforms regulates tubulin acetylation. J. Biol. Chem. 2014, 289, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Piccoli, G.; Condliffe, S.B.; Bauer, M.; Giesert, F.; Boldt, K.; De Astis, S.; Meixner, A.; Sarioglu, H.; Vogt-Weisenhorn, D.M.; Wurst, W.; et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 2011, 31, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.Y.; Li, X.; Wang, J.; Powell, J.; Wang, Q.; Zhang, Y.; Chen, Z.; Wicinski, B.; Hof, P.; Ryan, T.A.; et al. Parkinson’s disease associated LRRK2 hyperactive kinase mutant disrupts synaptic vesicle trafficking in ventral midbrain neurons. J. Neurosci. 2017, 37, 11366–11376. [Google Scholar] [CrossRef]
- Belluzzi, E.; Gonnelli, A.; Cirnaru, M.D.; Marte, A.; Plotegher, N.; Russo, I.; Civiero, L.; Cogo, S.; Carrion, M.P.; Franchin, C.; et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol. Neurodegener. 2016, 11, 1. [Google Scholar] [CrossRef]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Liu, Q.; Stankowski, J.N.; Lee, B.D.; Ko, H.S.; Lee, Y.; Grima, J.C.; Mao, X.; et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. USA 2018, 115, 1635–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassu, M.; Del Giudice, M.G.; Sanna, S.; Taymans, J.M.; Morari, M.; Brugnoli, A.; Frassineti, M.; Masala, A.; Esposito, S.; Galioto, M.; et al. Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS ONE 2017, 12, e0179082. [Google Scholar] [CrossRef] [PubMed]
- Beccano-Kelly, D.A.; Volta, M.; Munsie, L.N.; Paschall, S.A.; Tatarnikov, I.; Co, K.; Chou, P.; Cao, L.P.; Bergeron, S.; Mitchell, E.; et al. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum. Mol. Genet. 2015, 24, 1336–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matikainen-Ankney, B.A.; Kezunovic, N.; Mesias, R.E.; Tian, Y.; Williams, F.M.; Huntley, G.W.; Benson, D.L. Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. J. Neurosci. 2016, 36, 7128–7141. [Google Scholar] [CrossRef] [Green Version]
- Volta, M.; Beccano-Kelly, D.A.; Paschall, S.A.; Cataldi, S.; MacIsaac, S.E.; Kuhlmann, N.; Kadgien, C.A.; Tatarnikov, I.; Fox, J.; Khinda, J.; et al. Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. eLife 2017, 6, e28377. [Google Scholar] [CrossRef]
- Sheehan, P.; Yue, Z. Deregulation of autophagy and vesicle trafficking in Parkinson’s disease. Neurosci. Lett. 2019, 697, 59–65. [Google Scholar] [CrossRef]
- Wu, B.; Guo, W. The Exocyst at a Glance. J. Cell Sci. 2015, 128, 2957–2964. [Google Scholar] [CrossRef] [Green Version]
- Morgera, F.; Sallah, M.R.; Dubuke, M.L.; Gandhi, P.; Brewer, D.N.; Carr, C.M.; Munson, M. Regulation of exocytosis by the exocyst subunit Sec6 and the SM protein Sec1. Mol. Biol. Cell 2012, 23, 337–346. [Google Scholar] [CrossRef]
- Martin-Urdiroz, M.; Deeks, M.J.; Horton, C.G.; Dawe, H.R.; Jourdain, I. The Exocyst Complex in Health and Disease. Front. Cell Dev. Biol. 2016, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Murthy, M.; Garza, D.; Scheller, R.H.; Schwarz, T.L. Mutations in the exocyst component Sec5 disrupt neuronal membrane traffic, but neurotransmitter release persists. Neuron 2003, 37, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Pommereit, D.; Wouters, F.S. An NGF-induced Exo70-TC10 complex locally antagonises Cdc42-mediated activation of N-WASP to modulate neurite outgrowth. J. Cell Sci. 2007, 120, 2694–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koon, A.C.; Chen, Z.S.; Peng, S.; Fung, J.M.S.; Zhang, X.; Lembke, K.M.; Chow, H.K.; Frank, C.A.; Jiang, L.; Lau, K.F.; et al. Drosophila Exo70 Is Essential for Neurite Extension and Survival under Thermal Stress. J. Neurosci. 2018, 38, 8071–8086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lira, M.; Arancibia, D.; Orrego, P.R.; Montenegro-Venegas, C.; Cruz, Y.; Garcia, J.; Leal-Ortiz, S.; Godoy, J.A.; Gundelfinger, E.D.; Inestrosa, N.C.; et al. The Exocyst Component Exo70 Modulates Dendrite Arbor Formation, Synapse Density, and Spine Maturation in Primary Hippocampal Neurons. Mol. Neurobiol. 2019, 56, 4620–4638. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Guo, W. Rabs and the exocyst in ciliogenesis, tubulogenesis and beyond. Trends Cell Biol. 2011, 21, 383–386. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, G.A.; Hildebrand, J.D.; Soriano, P. The secretory protein Sec8 is required for paraxial mesoderm formation in the mouse. Dev. Biol. 1997, 192, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Riefler, G.M.; Balasingam, G.; Lucas, K.G.; Wang, S.; Hsu, S.C.; Firestein, B.L. Exocyst complex subunit sec8 binds to postsynaptic density protein-95 (PSD-95): A novel interaction regulated by cypin (cytosolic PSD-95 interactor). Biochem. J. 2003, 373, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Sans, N.; Prybylowski, K.; Petralia, R.S.; Chang, K.; Wang, Y.X.; Racca, C.; Vicini, S.; Wenthold, R.J. NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat. Cell Biol. 2003, 5, 520–530. [Google Scholar] [CrossRef]
- Chernyshova, Y.; Leshchyns’ka, I.; Hsu, S.C.; Schachner, M.; Sytnyk, V. The neural cell adhesion molecule promotes FGFR-dependent phosphorylation and membrane targeting of the exocyst complex to induce exocytosis in growth cones. J. Neurosci. 2011, 31, 3522–3535. [Google Scholar] [CrossRef]
- Tanaka, T.; Goto, K.; Iino, M. Diverse Functions and Signal Transduction of the Exocyst Complex in Tumor Cells. J. Cell. Physiol. 2017, 232, 939–957. [Google Scholar] [CrossRef]
- Lyons, P.D.; Peck, G.R.; Kettenbach, A.N.; Gerber, S.A.; Roudaia, L.; Lienhard, G.E. Insulin stimulates the phosphorylation of the exocyst protein Sec8 in adipocytes. Biosci. Rep. 2009, 29, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, C.; Mura, M.E.; Esposito, S.; Carta, F.; Sanna, G.; Turrini, F.; Carri, M.T.; Crosio, C. Bcl2-A1 interacts with pro-caspase-3: Implications for amyotrophic lateral sclerosis. Neurobiol. Dis. 2011, 43, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Migheli, R.; Del Giudice, M.G.; Spissu, Y.; Sanna, G.; Xiong, Y.; Dawson, T.M.; Dawson, V.L.; Galioto, M.; Rocchitta, G.; Biosa, A.; et al. LRRK2 affects vesicle trafficking, neurotransmitter extracellular level and membrane receptor localization. PLoS ONE 2013, 8, e77198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaccarino, C.; Crosio, C.; Vitale, C.; Sanna, G.; Carri, M.T.; Barone, P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum. Mol. Genet. 2007, 16, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, C.; Grindstaff, K.K.; Wright, J.R.; Nelson, W.J. Sec6/8 complexes on trans-Golgi network and plasma membrane regulate late stages of exocytosis in mammalian cells. J. Cell Biol. 2001, 155, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Vega, I.E.; Hsu, S.C. The exocyst complex associates with microtubules to mediate vesicle targeting and neurite outgrowth. J. Neurosci. 2001, 21, 3839–3848. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.C.; Ting, A.E.; Hazuka, C.D.; Davanger, S.; Kenny, J.W.; Kee, Y.; Scheller, R.H. The mammalian brain rsec6/8 complex. Neuron 1996, 17, 1209–1219. [Google Scholar] [CrossRef] [Green Version]
- Civiero, L.; Cirnaru, M.D.; Beilina, A.; Rodella, U.; Russo, I.; Belluzzi, E.; Lobbestael, E.; Reyniers, L.; Hondhamuni, G.; Lewis, P.A.; et al. Leucine-rich repeat kinase 2 interacts with p21-activated kinase 6 to control neurite complexity in mammalian brain. J. Neurochem. 2015, 135, 1242–1256. [Google Scholar] [CrossRef]
- Rassu, M.; Biosa, A.; Galioto, M.; Fais, M.; Sini, P.; Greggio, E.; Piccoli, G.; Crosio, C.; Iaccarino, C. Levetiracetam treatment ameliorates LRRK2 pathological mutant phenotype. J. Cell. Mol. Med. 2019, 23, 8505–8510. [Google Scholar] [CrossRef]
- Anitei, M.; Ifrim, M.; Ewart, M.A.; Cowan, A.E.; Carson, J.H.; Bansal, R.; Pfeiffer, S.E. A role for Sec8 in oligodendrocyte morphological differentiation. J. Cell Sci. 2006, 119, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Babbey, C.M.; Bacallao, R.L.; Dunn, K.W. Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. American journal of physiology. Ren. Physiol. 2010, 299, F495–F506. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Yadav, S.; DeVault, L.; Nung Jan, Y.; Sherwood, D.R. RAB-10-Dependent Membrane Transport Is Required for Dendrite Arborization. PLoS Genet. 2015, 11, e1005484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Mehta, S.Q.; Pichaud, F.; Bellen, H.J.; Quiocho, F.A. Sec15 interacts with Rab11 via a novel domain and affects Rab11 localization in vivo. Nat. Struct. Mol. Biol. 2005, 12, 879–885. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Xi, F.; Zhang, X.; Zhang, J.; Guo, W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 2007, 26, 4053–4065. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zuo, X.; Yue, P.; Guo, W. Phosphatidylinositol 4,5-bisphosphate mediates the targeting of the exocyst to the plasma membrane for exocytosis in mammalian cells. Mol. Biol. Cell 2007, 18, 4483–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleskot, R.; Cwiklik, L.; Jungwirth, P.; Zarsky, V.; Potocky, M. Membrane targeting of the yeast exocyst complex. Biochim. Biophys. Acta 2015, 1848, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Roumanie, O.; Wu, H.; Molk, J.N.; Rossi, G.; Bloom, K.; Brennwald, P. Rho GTPase regulation of exocytosis in yeast is independent of GTP hydrolysis and polarization of the exocyst complex. J. Cell Biol. 2005, 170, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Heider, M.R.; Gu, M.; Duffy, C.M.; Mirza, A.M.; Marcotte, L.L.; Walls, A.C.; Farrall, N.; Hakhverdyan, Z.; Field, M.C.; Rout, M.P.; et al. Subunit connectivity, assembly determinants and architecture of the yeast exocyst complex. Nat. Struct. Mol. Biol. 2016, 23, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Brown, M.Q.; van de Ven, W.; Zhang, Z.M.; Wu, B.; Young, M.C.; Synek, L.; Borchardt, D.; Harrison, R.; Pan, S.; et al. Endosidin2 targets conserved exocyst complex subunit EXO70 to inhibit exocytosis. Proc. Natl. Acad. Sci. USA 2016, 113, E41–E50. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fais, M.; Sanna, G.; Galioto, M.; Nguyen, T.T.D.; Trần, M.U.T.; Sini, P.; Carta, F.; Turrini, F.; Xiong, Y.; Dawson, T.M.; et al. LRRK2 Modulates the Exocyst Complex Assembly by Interacting with Sec8. Cells 2021, 10, 203. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020203
Fais M, Sanna G, Galioto M, Nguyen TTD, Trần MUT, Sini P, Carta F, Turrini F, Xiong Y, Dawson TM, et al. LRRK2 Modulates the Exocyst Complex Assembly by Interacting with Sec8. Cells. 2021; 10(2):203. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020203
Chicago/Turabian StyleFais, Milena, Giovanna Sanna, Manuela Galioto, Thi Thanh Duyen Nguyen, Mai Uyên Thi Trần, Paola Sini, Franco Carta, Franco Turrini, Yulan Xiong, Ted M. Dawson, and et al. 2021. "LRRK2 Modulates the Exocyst Complex Assembly by Interacting with Sec8" Cells 10, no. 2: 203. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020203