
A Trans-Omics Comparison Reveals Common Gene Expression Strategies in Four Model Organisms and Exposes Similarities and Differences between Them


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
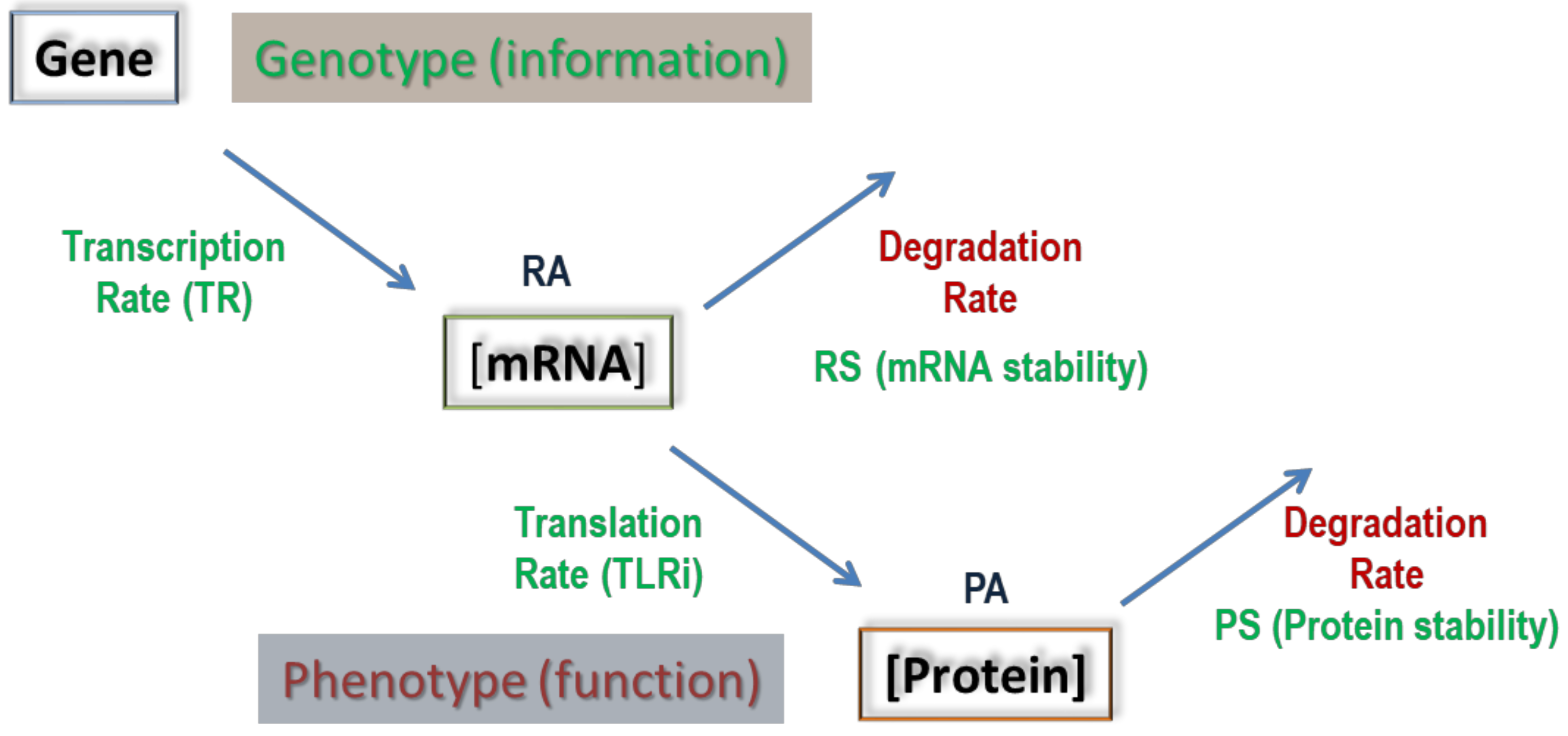
:1. Introduction
2. Materials and Methods
2.1. Selection and Features of the Original Data
2.2. Correlation Analyses
2.3. Six-Variable Profiles (6VPs)
2.4. Cluster Analyses
2.5. Gene Ontology Category Searches
2.6. Comparison of the Proximity in 6VPs among the Genes Belonging to Macromolecular Complexes and Those Belonging to GO Categories Not Forming Macromolecular Complexes
2.7. Phenogram Tree Construction
3. Results
3.1. Datasets for the Variables Selected in This Study
3.2. Comparisons and Correlations between Variables
3.3. Clustering of Genes According to the Six Variables of Gene Expression into Four Different Organisms
3.4. Detailed Analysis of the Selected Functional Groups in Eukaryotes
3.5. Trans-Organism Clusters Comparison: 6VP Phenograms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6VP | six variable profiles |
| ANOV | Aanalysis of variance |
| BMA | bayesian model averaging |
| BP | biological process |
| CC | cellular component |
| CES | common expressions strategies |
| CHX | cycloheximide |
| DTA | dynamic transcriptome analysis |
| FDR | false discovery rate |
| GFP | green fluorescent protein |
| GO | gene ontology |
| GRO | genomic run on |
| HUGO | human genome organization |
| iBAQ | intensity-based absolute quantification |
| KNN | k-nearest neighbor method |
| LB | Luria-Bertani medium |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| MIPS | Munich information center for protein sequences |
| NJ | neighbor-joining |
| PA | proteín abundance |
| PCN | protein copies per cell |
| PS | proteín stability |
| RA | mRNA abundance |
| RATE-seq | RNA approach to equilibrium sequencing |
| REVIGO | reduce + visualize gene ontology |
| RiBi | ribosome biogenesis |
| RNA-seq | RNA sequencing |
| RS | mRNA stability |
| SGD | Saccharomyces Genome Database |
| SILAC | stable isotope labeling by/with amino acids in cell culture |
| SOTA | self-organizing tree algorithm |
| TLR | translation rate |
| TLRi | individual translation rate |
| TR | transcription rate |
| UPGMA | unweighted pair group method with arithmetic mean |
References
- Crick, F.H.C. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 13, 138–163. [Google Scholar]
- Laurent, J.M.; Vogel, C.; Kwon, T.; Craig, S.A.; Boutz, D.R.; Huse, H.K.; Nozue, K.; Walia, H.; Whiteley, M.; Ronald, P.C.; et al. Protein abundances are more conserved than mRNA abundances across diverse taxa. Proteomics 2010, 10, 4209–4212. [Google Scholar] [CrossRef] [Green Version]
- García-Martínez, J.; González-Candelas, F.; Pérez-Ortín, J.E. Common gene expression strategies revealed by genome-wide analysis in yeast. Genome Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csárdi, G.; Franks, A.; Choi, D.S.; Airoldi, E.; Drummond, D.A. Accounting for Experimental Noise Reveals That mRNA Levels, Amplified by Post-Transcriptional Processes, Largely Determine Steady-State Protein Levels in Yeast. PLoS Genet. 2015, 11, e1005206. [Google Scholar] [CrossRef] [Green Version]
- Lahtvee, P.-J.; Sánchez, B.J.; Smialowska, A.; Kasvandik, S.; Elsemman, I.E.; Gatto, F.; Nielsen, J. Absolute Quantification of Protein and mRNA Abundances Demonstrate Variability in Gene-Specific Translation Efficiency in Yeast. Cell Syst. 2017, 4, 495–504.e5. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Bickel, P.J.; Biggin, M.D. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2014, 2, e270. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Chew, G.-L.; Biggin, M.D. Quantitating translational control: mRNA abundance-dependent and independent contributions and the mRNA sequences that specify them. Nucleic Acids Res. 2017, 45, 11821–11836. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Biggin, M.D. Statistics requantitates the central dogma. Science 2015, 347, 1066–1067. [Google Scholar] [CrossRef] [Green Version]
- McManus, J.; Cheng, Z.; Vogel, C. Next-generation analysis of gene expression regulation–comparing the roles of synthesis and degradation. Mol. BioSyst. 2015, 11, 2680–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Belle, A.; Tanay, A.; Bitincka, L.; Shamir, R.; O’Shea, E.K. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 13004–13009. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Ortín, J.E.; Tordera, V.; Chávez, S. Homeostasis in the Central Dogma of molecular biology: The importance of mRNA instability. RNA Biol. 2019, 16, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ortín, J.E.; Alepuz, P.M.; Moreno, J. Genomics and gene transcription kinetics in yeast. Trends Genet. 2007, 23, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.R.S.; Ghaemmaghami, S.; Ihmels, J.; Breslow, D.K.; Noble, M. Single-cell proteomic analysis of S. cerevisiae re-veals the architecture of biological noise. Nature 2006, 441, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.B.; Hirsh, A.E.; Giaever, G.; Kumm, J.; Eisen, M.B. Noise Minimization in Eukaryotic Gene Expression. PLoS Biol. 2004, 2, e137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Even, A.; Paulsson, J.; Maheshri, N.; Carmi, M.; Oshea, E.K.; Pilpel, Y.; Barkai, N. Noise in protein expression scales with natural protein abundance. Nat. Genet. 2006, 38, 636–643. [Google Scholar] [CrossRef]
- Baudrimont, A.; Jaquet, V.; Wallerich, S.; Voegeli, S.; Becskei, A. Contribution of RNA Degradation to Intrinsic and Extrinsic Noise in Gene Expression. Cell Rep. 2019, 26, 3752–3761.e5. [Google Scholar] [CrossRef] [Green Version]
- Sipiczki, M. Where does fission yeast sit on the tree of life? Genome Biol. 2000, 1, 101. [Google Scholar] [CrossRef] [Green Version]
- Dujon, B. Yeast evolutionary genomics. Nat. Rev. Genet. 2010, 11, 512–524. [Google Scholar] [CrossRef]
- Douzery, E.J.; Snell, E.A.; Bapteste, E.; Delsuc, F.; Philippe, H. The timing of eukaryotic evolution: Does a relaxed molecular clock reconcile proteins and fossils? Proc. Natl. Acad. Sci. USA 2004, 101, 15386–15391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, C.S.; Wood, V.; Fantes, P.A. An ancient yeast for young geneticists: A Primer on the Schizosaccharomyces pombe model system. Genetics 2016, 201, 403–423, Erratum in 2016, 202, 1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Sung, Y.-J.; Paik, S. Cancer Cell Line Panels Empower Genomics-Based Discovery of Precision Cancer Medicine. Yonsei Med. J. 2015, 56, 1186–1198. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G. Impute: Imputation for Microarray Data. R package version 1.64.0. 2020. Available online: https://www.bioconductor.org/packages/release/bioc/html/impute.html (accessed on 1 February 2021).
- Miller, C.; Schwalb, B.; Maier, K.; Schulz, D.; Dümcke, S.; Zacher, B.; Mayer, A.; Sydow, J.; Marcinowski, L.; Dölken, L.; et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 2011, 7, 458. [Google Scholar] [CrossRef] [PubMed]
- Neymotin, B.; Athanasiadou, R.; Gresham, D. Determination of in vivo RNA kinetics using RATE-seq. RNA 2014, 20, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Siwiak, M.; Zielenkiewicz, P. A Comprehensive, Quantitative, and Genome-Wide Model of Translation. PLoS Comput. Biol. 2010, 6, e1000865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiano, R.; Nagaraj, N.; Fröhlich, F.; Walther, T.C. Global Proteome Turnover Analyses of the Yeasts S. cerevisiae and S. pombe. Cell Rep. 2014, 9, 1959–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Y.T.; Koh, J.L.; Friesen, H.; Duffy, S.K.; Cox, M.J.; Moses, A.M.; Moffat, J.; Boone, C.; Andrews, B.J. Yeast Proteome Dynamics from Single Cell Imaging and Automated Analysis. Cell 2015, 161, 1413–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelechano, V.; Ch&vez, S.; Pérez-Ortín, J.E. A complete set of nascent transcription rates for yeast genes. PLoS ONE 2010, 5, e15442. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, M.J.; Cotobal, C.; Duncan, C.; Mata, J. Global coordination of transcriptional control and mRNA decay during cellular differentiation. Mol. Syst. Biol. 2010, 6, 380. [Google Scholar] [CrossRef]
- Eser, P.; Wachutka, L.; Maier, K.C.; Demel, C.; Boroni, M.; Iyer, S.; Cramer, P.; Gagneur, J. Determinants of RNA metabolism in the Schizosaccharomyces pombe genome. Mol. Syst. Biol. 2016, 12, 857. [Google Scholar] [CrossRef]
- Sun, M.; Schwalb, B.; Schulz, D.; Pirkl, N.; Etzold, S.; Larivière, L.; Maier, K.C.; Seizl, M.; Tresch, A.; Cramer, P. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 2012, 22, 1350–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marguerat, S.; Schmidt, A.; Codlin, S.; Chen, W.; Aebersold, R.; Bähler, J. Quantitative Analysis of Fission Yeast Transcriptomes and Proteomes in Proliferating and Quiescent Cells. Cell 2012, 151, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, D.H.; Beilharz, T.H.; Marguerat, S.; Mata, J.; Watt, S.; Schubert, F.; Preiss, T.; Bähler, J. A Network of Multiple Regulatory Layers Shapes Gene Expression in Fission Yeast. Mol. Cell 2007, 26, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, J.; Schmidt, A.; Quandt, A.; Neo, S.P.; Saraç, O.S.; Gracia, T.; Loguercio, S.; Ahrné, E.; Xia, R.L.; Tan, K.H.; et al. Extensive mass spectrometry-based analysis of the fission yeast proteome: The Schizosaccharomyces pombe PeptideAtlas. Mol Cell Proteom. 2013, 12, 1741–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, H.; Mizutani, R.; Salam, K.A.; Tano, K.; Ijiri, K.; Wakamatsu, A.; Isogai, T.; Suzuki, Y.; Akimitsu, N. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012, 22, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Pääbo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Hallström, B.M.; Lindskog, C.; Mardinoglu, A.; Pontén, F.; Nielsen, J. Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 2016, 12, 862. [Google Scholar]
- Cambridge, S.B.; Gnad, F.; Nguyen, C.; Bermejo, J.L.; Krüger, M.; Mann, M. Systems-wide Proteomic Analysis in Mammalian Cells Reveals Conserved, Functional Protein Turnover. J. Proteome Res. 2011, 10, 5275–5284. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Ostasiewicz, P.; Duś, K.; Zielińska, D.F.; Gnad, F.; Mann, M. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol. 2012, 8, 611. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Esquerré, T.; Laguerre, S.; Turlan, C.; Carpousis, A.J.; Girbal, L.; Cocaign-Bousquet, M. Dual role of transcription and transcript stability in the regulation of gene expression in Escherichia coli cells cultured on glucose at different growth rates. Nucleic Acids Res. 2013, 42, 2460–2472. [Google Scholar] [CrossRef] [PubMed]
- Esquerré, T.; Moisan, A.; Chiapello, H.; Arike, L.; Vilu, R.; Gaspin, C.; Cocaign-Bousquet, M.; Girbal, L. Genome-wide investigation of mRNA lifetime determinants in Escherichia coli cells cultured at different growth rates. BMC Genom. 2015, 16, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.-W.; Burkhardt, D.; Gross, C.; Weissman, J.S. Quantifying Absolute Protein Synthesis Rates Reveals Principles Underlying Allocation of Cellular Resources. Cell 2014, 157, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Valgepea, K.; Adamberg, K.; Seiman, A.; Vilu, R. Escherichia coli achieves faster growth by increasing catalytic and translation rates of proteins. Mol. Biosyst. 2013, 9, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Kochanowski, K.; Vedelaar, S.; Ahrné, E.; Volkmer, B.; Callipo, L.; Knoops, K.; Bauer, M.; Aebersold, R.; Heinemann, M. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016, 34, 104–110. [Google Scholar] [CrossRef]
- Keseler, I.M.; Mackie, A.; Santos-Zavaleta, A.; Billington, R.; Bonavides-Martínez, C.; Caspi, R.; Fulcher, C.; Gama-Castro, S.; Kothari, A.; Krummenacker, M.; et al. The EcoCyc database: Reflecting new knowledge aboutEscherichia coliK-12. Nucleic Acids Res. 2017, 45, D543–D550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steel, M.F.J. Model Averaging and Its Use in Economics. J. Econ. Lit. 2020, 58, 644–719. [Google Scholar] [CrossRef]
- Garcia-Donato, G.; Forte-Deltell, A. Bayesian testing, variable selection and model averaging in linear models using R with BayesVarSel. R J. 2018, 10, 155–174. [Google Scholar] [CrossRef] [Green Version]
- Brock, G.; Pihur, V.; Datta, S.; Datta, S. clValid: An R package for cluster validation. J. Statist. Softw. 2008, 25, 4. [Google Scholar] [CrossRef] [Green Version]
- Herrero, J.; Valencia, A.; Dopazo, J. A hierarchical unsupervised growing neural network for clustering gene expression patterns. Bioinformatics 2001, 17, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yu, G. Gene Ontology Semantic Similarity Analysis Using GOSemSim. Toxicity Assess. 2020, 2117, 207–215. [Google Scholar] [CrossRef]
- Resnik, P. Semantic Similarity in a Taxonomy: An Information-Based Measure and its Application to Problems of Ambiguity in Natural Language. J. Artif. Intell. Res. 1999, 11, 95–130. [Google Scholar] [CrossRef]
- Lin, D. An information-theoretic definition of similarity. In Proceedings of the 15th International Conference on Machine Learning, Madison, WI, USA, 24–27 July 1998; pp. 296–304. [Google Scholar]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformstics 2010, 27, 592–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, J.J.M.; Pyl, P.T.; Rausch, T.; Zichner, T.; Tekkedil, M.M.; Stütz, A.M.; Jauch, A.; Aiyar, R.S.; Pau, G.; Delhomme, N.; et al. The Genomic and Transcriptomic Landscape of a HeLa Cell Line. G3 Genes Genomes Genet. 2013, 3, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Fortelny, N.; Overall, C.M.; Pavlidis, P.; Freue, G.V.C. Can we predict protein from mRNA levels? Nat. Cell Biol. 2017, 547, E19–E20. [Google Scholar] [CrossRef]
- Wilhelm, M.; Hahne, H.; Savitski, M.; Marx, H.; Lemeer, S.; Bantscheff, M.; Kuster, B. Wilhelm et al. reply. Nat. Cell Biol. 2017, 547, E23. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nat. Cell Biol. 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, R.L.; Requião, R.D.; Rossetto, S.; Domitrovic, T.; Palhano, F.L. Codon stabilization coefficient as a metric to gain insights into mRNA stability and codon bias and their relationships with translation. Nucleic Acids Res. 2019, 47, 2216–2228. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.Y.; Mugler, C.F.; Heinrich, S.; Vallotton, P.; Weis, K. Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Grigull, J.; Mnaimneh, S.; Pootoolal, J.; Robinson, M.D.; Hughes, T.R. Genome-Wide Analysis of mRNA Stability Using Transcription Inhibitors and Microarrays Reveals Posttranscriptional Control of Ribosome Biogenesis Factors. Mol. Cell. Biol. 2004, 24, 5534–5547. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Ortín, J.E.; Alepuz, P.; Chávez, S.; Choder, M. Eukaryotic mRNA Decay: Methodologies, Pathways, and Links to Other Stages of Gene Expression. J. Mol. Biol. 2013, 425, 3750–3775. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, N.; Luo, X.; Capaldi, A.; Buchan, J.R. Alterations of signaling pathways in response to chemical perturbations used to measure mRNA decay rates in yeast. RNA 2020, 26, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Heck, A.M.; Wilusz, J.; Wilusz, C.J. Metabolic labeling and recovery of nascent RNA to accurately quantify mRNA stability. Methods 2017, 120, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudrimont, A.; Voegeli, S.; Viloria, E.C.; Stritt, F.; Lenon, M.; Wada, T.; Jaquet, V.; Becskei, A. Multiplexed gene control reveals rapid mRNA turnover. Sci. Adv. 2017, 3, e1700006. [Google Scholar] [CrossRef] [Green Version]
- Furlan, M.; De Pretis, S.; Pelizzola, M. Dynamics of transcriptional and post-transcriptional regulation. Brief. Bioinform. 2020, bbaa389. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef]
- Presnyak, V.; Alhusaini, N.; Chen, Y.-H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon Optimality Is a Major Determinant of mRNA Stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harigaya, Y.; Parker, R. Analysis of the association between codon optimality and mRNA stability in Schizosaccharomyces pombe. BMC Genom. 2016, 17, 895. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.C.; Karp, P.D.; Keseler, I.M.; Krummenacker, M.; Siegele, D.A. What we can learn about Escherichia coli through application of Gene Ontology. Trends Microbiol. 2009, 17, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Krebs, J.E.; Goldstein, E.S.; Kilpatrick, S.T. Lewin’s Genes XII, 12th ed.; Jones & Burlett Learning: Burlington, MA, USA, 2018. [Google Scholar]
- Kristensen, A.R.; Gsponer, J.; Foster, L.J. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol. 2013, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, C.H. The Tree of Life Viewed Through the Contents of Genomes. Toxic. Assess. 2009, 532, 141–161. [Google Scholar] [CrossRef]
- García-Martínez, J.; Aranda, A.; Pérez-Ortín, J.E. Genomic Run-On Evaluates Transcription Rates for All Yeast Genes and Identifies Gene Regulatory Mechanisms. Mol. Cell 2004, 15, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, H.-J.; Donnard, E.; Gustafsson, H.T.; Garber, M.; Rando, O.J. Transcriptome-wide Analysis of Roles for tRNA Modifications in Translational Regulation. Mol. Cell 2017, 68, 978–992.e4. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192–205.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.; Joachim, A.O.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell 2017, 169, 326–337.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Agarwala, S.D.; Mumbach, M.R.; Jovanovic, M.; Mertins, P.; Shishkin, A.; Tabach, Y.; Mikkelsen, T.S.; Satija, R.; Ruvkun, G.; et al. High-Resolution Mapping Reveals a Conserved, Widespread, Dynamic mRNA Methylation Program in Yeast Meiosis. Cell 2013, 155, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, C.L.; Storey, J.D.; Tibshirani, R.J.; Herschlag, D.; Brown, P.O. Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. USA 2002, 99, 5860–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, C.H.; Fitz-Gibbon, S.T. Using Homolog Groups to Create a Whole-Genomic Tree of Free-Living Organisms: An Update. J. Mol. Evol. 2002, 54, 539–547. [Google Scholar] [CrossRef]
- Tekaia, F.; Lazcano, A.; Dujon, B. The Genomic Tree as Revealed from Whole Proteome Comparisons. Genome Res. 1999, 9, 550–557. [Google Scholar]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Sci. 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briones, C.; Manrubia, S.C.; Lázaro, E.; Lazcano, A.; Amils, R. Reconstructing evolutionary relationships from functional data: A consistent classification of organisms based on translation inhibition response. Mol. Phylogenet. Evol. 2005, 34, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, P.; Dessimoz, C. Gene Ontology: Pitfalls, Biases, and Remedies. In The Gene Ontology Handbook; Methods in Molecular Biology; Dessimoz, C., Škunca, N., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1446. [Google Scholar]
- Gerstein, M.B.; Rozowsky, J.; Yan, K.-K.; Wang, D.; Cheng, C.; Brown, J.B.; Davis, C.A.; Hillier, L.W.; Sisu, C.; Li, J.J.; et al. Comparative analysis of the transcriptome across distant species. Nat. Cell Biol. 2014, 512, 445–448. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forés-Martos, J.; Forte, A.; García-Martínez, J.; Pérez-Ortín, J.E. A Trans-Omics Comparison Reveals Common Gene Expression Strategies in Four Model Organisms and Exposes Similarities and Differences between Them. Cells 2021, 10, 334. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020334
Forés-Martos J, Forte A, García-Martínez J, Pérez-Ortín JE. A Trans-Omics Comparison Reveals Common Gene Expression Strategies in Four Model Organisms and Exposes Similarities and Differences between Them. Cells. 2021; 10(2):334. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020334
Chicago/Turabian StyleForés-Martos, Jaume, Anabel Forte, José García-Martínez, and José E. Pérez-Ortín. 2021. "A Trans-Omics Comparison Reveals Common Gene Expression Strategies in Four Model Organisms and Exposes Similarities and Differences between Them" Cells 10, no. 2: 334. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020334