
Promising Anti-Mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma

 ,
,
Abstract
:1. Introduction
2. Re-Sensitization to Chemotherapy through Caspase-Independent Apoptosis via ROS
3. Re-Sensitization to Chemotherapy through Caspase-Dependent Apoptosis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2ME | 2-methoxyestradiol |
| ROS | Reactive Oxygen Species |
| c-JNK | c-Jun N-terminal Kinase |
| MKK4/7 | Mitogen-activated protein Kinase Kinase 4/7 |
| MM | Multiple Myeloma |
| AIF | Apoptosis Inducing Factor |
| EndoG | Endonuclease G |
| AML | Acute Myeloid Leukemia |
| CNS | Central Nervous System |
| OXPHOS | Oxidative Phosphorilation |
| Nrf-2 | Nuclear factor erythroid 2-Related Factor 2 |
| VEGF | Vascular Endothelial Growh Factor |
| Bcl-2 | B-cell Lymphoma 2 |
| Evo | Evodiamine |
| Cdc2 | Cyclin-dependent protein kinase |
| IAPs | Inhibitors of Apoptosis |
| NF-kB | Nuclear Factor kappa light chain enhancer of activated B cells |
| HSP70B’ | Heat Shock Protein 70B’ |
| HSP40 | Heat Shock Protein 40 |
| HO-1 | Heme Oxygenase 1 |
| WM | Waldenstrom’s Macroglobulinemia |
| DLBLC | Diffuse Large B Cell Lymphoma |
| UPS14 | Ubiquitin carboxyl-terminal hydrolase 14 |
| MCL | Mantle cell Lymphoma |
| ATP | Adenosine triphosphate |
| NAD | Nicotinamide adenine dinucleotide |
| CML | Chronic Myeloid Leukemia |
| RNS | Reactive Nitrogen Specie |
| TLR4 | Toll-like receptor 4 |
| SMAC/DIABLO | Second Mitochondria-derived Activator of Caspases/Direct Inhibitor of Apoptosis-Binding protein with low isoelectric point |
| BAx | Bcl-2 Associated X |
| HCC | Hepato cellular carcinoma |
| CLL | Chronic Lymphocytic Leukemia |
| APAF-1 | Apoptotic Protease Activating Factor-1 |
| CTCL | Cutaneous T Cell Lymphoma |
| HL | Hodgkin Lymphoma |
| APL | Acute Promyelocytic Leukemia |
| NSCLC | Non-Small Cell Lung Cancer |
| TRAIL | TNF-Related Apoptosis-Inducing Ligand |
References
- Aragona, P.; Postorino, E.I.; Rania, L.; Innao, V.; Wylegala, E.; Pisani, A.; Puzzolo, D.; Micali, A.; Allegra, A.; Nowinska, A.; et al. Corneal Structural Changes in Nonneoplastic and Neoplastic Monoclonal Gammopathies. Investig. Opthalmol. Vis. Sci. 2016, 57, 2657. [Google Scholar] [CrossRef] [Green Version]
- Allegra, A.; Penna, G.; Innao, V.; Greve, B.; Maisano, V.; Russo, S.; Musolino, C. Vaccination of multiple myeloma: Current strategies and future prospects. Crit. Rev. Oncol. 2015, 96, 339–354. [Google Scholar] [CrossRef]
- Innao, V.; Allegra, A.; Pulvirenti, N.; Allegra, A.; Musolino, M. Therapeutic potential of antagomiRs in haematological and oncological neoplasms. Eur. J. Cancer Care 2020, 29, 2. [Google Scholar] [CrossRef]
- Conticello, C.; Romano, A.; Del Fabro, V.; Martino, E.; Calafiore, V.; Sapienza, G.; Leotta, V.; Parisi, M.; Markovic, U.; Garibaldi, B.; et al. Feasibility, Tolerability and Efficacy of Carfilzomib in Combination with Lenalidomide and Dexamethasone in Relapsed Refractory Myeloma Patients: A Retrospective Real-Life Survey of the Sicilian Myeloma Network. J. Clin. Med. 2019, 8, 877. [Google Scholar] [CrossRef] [Green Version]
- Holstein, S.A.; Suman, V.J.; McCarthy, P.L. Update on the role of lenaliomide in patients with multiple myeloma. Ther. Adv. Hematol. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delforge, M.; Ludwig, H. How I manage the toxicities of myeloma drugs. Blood 2017, 129, 2359–2367. [Google Scholar] [CrossRef] [PubMed]
- Innao, V.; Allegra, A.G.; Russo, S.; Gerace, D.; Vaddinelli, D.; Alonci, A.; Musolino, C. Standardisation of minimal residual disease in multiple myeloma. Eur. J. Cancer Care 2017, 26, e12732. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Oeck, S.; West, A.P.; Mangalhara, K.C.; Sainz, A.G.; Newman, L.E.; Zhang, X.-O.; Wu, L.; Yan, Q.; Bosenberg, M.; et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 2019, 1, 1209–1218. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Pulvirenti, N.; Pugliese, M.; Musolino, C. Antitumorigenic action of nelfinavir: Effects on multiple myeloma and hematologic malignancies (Review). Oncol. Rep. 2020, 43, 6. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Gerace, D.; Vaddinelli, D.; Allegra, A.G.; Musolino, C. Nanobodies and Cancer: Current Status and New Perspectives. Cancer Investig. 2018, 36, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.-Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Li, G.; Podar, K.; Hideshima, T.; Mitsiades, C.; Schlossman, R.; Munshi, N.; Richardson, P.; Cotter, F.; Anderson, K. Targeting mitochondria to overcome conventional and bortezomib/proteasome inhibitor PS-341 resistance in multiple my-eloma (MM). Cells 2004, 104, 8. [Google Scholar]
- Besse, L.; Besse, A.; Mendez-Lopez, M.; Vasickova, K.; Sedlackova, M.; Vanhara, P.; Kraus, M.; Bader, J.; Ferreira, R.; Castel-lano, R.; et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mito-chondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019, 104, 9. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Lee, J.H.; Lahuerta, J.J.; Morgan, G.; Richardson, P.G.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2011, 26, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Song, I.; Kim, H.; Lee, S.; Jeong, S.; Kim, N.; Ko, K.; Rhee, B.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Song, I.-S.; Jeong, Y.J.; Jeong, S.H.; Heo, H.J.; Kim, H.K.; Lee, S.R.; Ko, T.H.; Youm, J.B.; Kim, N.; Ko, K.S.; et al. Combination treatment with 2-methoxyestradiol overcomes bortezomib resistance of multiple myeloma cells. Exp. Mol. Med. 2013, 45, e50. [Google Scholar] [CrossRef] [Green Version]
- Pal, P.; Hales, K.; Hales, D.B. The pro-apoptotic actions of 2-methoxyestradiol against ovarian cancer involve catalytic activation of PKCδ signaling. Oncotarget 2020, 11, 3646–3659. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Castillejos-López, M.; Romero-García, S.; Aguayo, A.S.; Herrera, I.; Garcia-Martin, M.O.; Torres-Espíndola, L.M.; Negrete-García, M.C.; Olvera, A.C.; Huerta-Cruz, J.C.; et al. Antitumor Therapy under Hypoxic Microenvironment by the Combination of 2-Methoxyestradiol and Sodium Dichloroacetate on Human Non-Small-Cell Lung Cancer. Oxid. Med. Cell. Longev. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Lebelo, M.T.; Joubert, A.M.; Visagie, M.H. Sulphamoylated Estradiol Analogue Induces Reactive Oxygen Species Generation to Exert Its Antiproliferative Activity in Breast Cancer Cell Lines. Molecules 2020, 25, 4337. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Jung, H.; Min, S.; Kang, J.; Jang, D.; Shin, S.; Kim, J.; Lee, S.; Sung, C.; Lee, W.; et al. Targeting antioxidant enzymes enhances the therapeutic efficacy of the BCL-X L inhibitor ABT-263 in KRAS-mutant colorectal cancers. Cancer Lett. 2021, 497, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Pan, W.; Wang, X.P.; Chen, T.S. Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. J. Cell. Physiol. 2012, 227, 3778–3786. [Google Scholar] [CrossRef]
- Papanikolaou, X.; Johnson, S.; Garg, T.; Tian, E.; Tytarenko, R.; Zhang, Q.; Stein, C.; Barlogie, B.; Epstein, J.; Heuck, C. Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget 2014, 5, 4118–4128. [Google Scholar] [CrossRef]
- Zhan, X.; Yu, W.; Franqui-Machin, R.; Bates, M.; Nadiminti, K.; Cao, H.; Amendt, B.; Jethava, Y.; Frech, I.; Zhan, F.T.G. Research Paper Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget 2017, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, R.; Zhu, Y.; Zhong, S.; Qian, J.; Yang, D.; Jurczyszyn, A.; Beksac, M.; Gu, C.; Yang, Y. Dihydroartemisinin Induces Growth Arrest and Overcomes Dexamethasone Resistance in Multiple Myeloma. Front. Oncol. 2020, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Zhang, E.; Chen, J.; Huang, X.; Yan, H.; Cao, W.; Qu, J.; Gu, H.; Xu, R.; et al. Dihydroartemisinin Modulates Apoptosis and Autophagy in Multiple Myeloma through the P38/MAPK and Wnt/β−Catenin Signaling Pathways. Oxid. Med. Cell. Longev. 2020, 2020, 6096391. [Google Scholar] [CrossRef]
- Wu, X.H.; Zhou, H.J.; Lee, J. Dihydroartemisinin inhibits angiogenesis induced by multiple myeloma RPMI8226 cells under hypoxic conditions via downregulation of vascular endothelial growth factor expression and suppression of vascular endo-thelial growth factor secretion. Anticancer Drugs 2006, 17, 7. [Google Scholar] [CrossRef]
- Qiu, C.; Gao, L.; Yan, K.; Cui, Y.; Zhang, Y. A promising antitumor activity of evodiamine incorporated in hydroxypropyl-beta- cyclodextrin: Pro-apoptotic activity in human hepatoma HepG2 cells. Chem. Cent. J. 2016, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wu, L.-J.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Evodiamine induces tumor cell death through different pathways: Apoptosis and necrosis. Acta Pharmacol. Sin. 2004, 25, 1. [Google Scholar]
- Wang, R.; Deng, D.; Shao, N.; Xu, Y.; Xue, L.; Peng, Y.; Liu, Y.; Zhi, F. Evodiamine activates cellular apoptosis through sup-pressing PI3K/AKT and activating MAPK in glioma. Oncotargets Ther. 2018, 11, 1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Liu, Y.; Zhao, J.; Wang, J.; Li, Y.; Pang, Y.; Chen, J.; Wang, J. Evodiamine inactivates NF-kappaB and potentiates the antitumor effects of gemcitabine on tongue cancer both in vitro and in vivo. Oncotargets Ther. 2018, 12, 257. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.; Jiang, S.; Li, C. Evodiamine Selectively Inhibits Multiple Myeloma Cell Growth by Triggering Activation of Intrinsic Apoptosis Pathway. Oncotargets Ther. 2019, 12, 11383–11391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Hu, C. Evodiamine: A novel anti-cancer alkaloid from Evodia retaecarpa. Molecules 2009, 14, 1852. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Chitta, K.; Paulus, A.; Akhtar, S.; Blake, M.K.K.; Caulfield, T.R.; Novak, A.J.; Ansell, S.M.; Advani, P.; Ailawadhi, S.; Asher, C.-K.; et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenström macroglobulinaemia tumour cells. Br. J. Haematol. 2015, 169, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Sun, Y.; Wang, J.; He, Q.; Chen, X.; Lan, X.; Chen, J.; Dou, Q.P.; Shi, X.; Liu, J. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J. Exp. Clin. Cancer Res. 2019, 38, 414–453. [Google Scholar] [CrossRef]
- Zhang, X.; Espinosa, B.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Linder, S. Oxidative Stress Induced by the Deubiquitinase Inhibitor b-AP15 Is Associated with Mitochondrial Impairment. Oxid. Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Zhang, X.; Pellegrini, P.; Saei, A.; Hillert, E.; Mazurkiewicz, M.; Olofsson, M.; Zubarev, R.; D’Arcy, P.; Linder, S. The deubiq-uitinase inhibitor b-AP15 induces strong proteotoxic stress and mito- chondrial damage. Biochem. Pharmacol. 2018, 156, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Didier, R.; Mallavialle, A.; Ben Jouira, R.; Domdom, M.; Tichet, M.; Auberger, P.; Luciano, F.; Ohanna, M.; Tartare-Deckert, S.; Deckert, M. Targeting the proteasome-associated deubiquitinating enzyme USP14 impairs melanoma cell survival and over-comes resistance to MAPK-targeting therapies. Mol. Cancer Ther. 2018, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Erdal, H.; Berndtsson, M.; Castro, J.; Brunk, U.; Shoshan, B.; Linder, S. Induction of lysosomal membrane perme- abilization by compounds that activate p53-independent apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, P.; Linder, S. Proteasome deubiquitinases as novel targets for cancer therapy. Int J Biochem Cell Biol. 2012, 44, 1729–1738. [Google Scholar] [CrossRef]
- Kropp, K.N.; Maurer, S.; Rothfelder, K.; Schmied, B.J.; Clar, K.L.; Schmidt, M.; Strunz, B.; Kopp, H.-G.; Steinle, A.; Grünebach, F.; et al. The novel deubiquitinase inhibitor b-AP15 induces direct and NK cell-mediated antitumor effects in human mantle cell lymphoma. Cancer Immunol. Immunother. 2018, 67, 935–947. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, X.; Wang, B.; Yu, B.; Ge, J. Deubiquitinase inhibitor b-AP15 activates endoplasmic reticulum (ER) stress and inhibits Wnt/Notch1 signaling pathway leading to the reduction of cell survival in hepatocellular carcinoma cells. Eur. J. Pharmacol. 2018, 825, 10–18. [Google Scholar] [CrossRef]
- Cai, J.; Xia, X.; Liao, Y.; Liu, N.; Guo, Z.; Chen, J.; Yang, L.; Long, H.; Yang, Q.; Zhang, X.; et al. A novel deubiquitinase inhibitor b-AP15 triggers apoptosis in both androgen receptor-dependent and -independent prostate cancers. Oncotarget 2017, 8, 63232–63246. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.; Olofsson, M.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiqui-tin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 1–15. [Google Scholar]
- Rowinsky, E.K.; Paner, A.; Berdeja, J.G.; Paba-Prada, C.; Venugopal, P.; Porkka, K.; Gullbo, J.; Linder, S.; Loskog, A.; Richardson, P.G.; et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Investig. New Drugs 2020, 38, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soncini, D.; Minetto, P.; Martinuzzi, C.; Becherini, P.; Fenu, V.; Guolo, F.; Todoerti, K.; Calice, G.; Contini, P.; Miglino, M.; et al. Amino acid depletion triggered by ʟ-asparaginase sensitizes MM cells to carfilzomib by inducing mitochondria ROS-mediated cell death. Blood Adv. 2020, 4, 4312–4326. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Puglisi, F.; Barbato, A.; Vicario, N.; Cambria, D.; Parrinello, N.L.; Romano, A.; Conticello, C.; Forte, S.; et al. Inhibition of TLR4 Signaling Affects Mito-chondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers 2020, 12, 1999. [Google Scholar] [CrossRef]
- Konopleva, M.; Tsao, T.; Ruvolo, P.; Stiouf, I.; Estrov, Z.; Leysath, C.E.; Zhao, S.; Harris, D.; Chang, S.; Jackson, C.E.; et al. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood 2002, 99, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, I.M.; Kitada, S.; Schimmer, A.; Kim, Y.; Zapata, J.M.; Charboneau, L.; Rassenti, L.; Andreeff, M.; Bennett, F.; Sporn, M.B.; et al. The triterpenoid CDDO induces apoptosis in refractory CLL B cells. Blood 2002, 100, 2965–2972. [Google Scholar] [CrossRef]
- Stadheim, T.A.; Suh, N.; Ganju, N.; Sporn, M.B.; Eastman, A. The Novel Triterpenoid 2-Cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) Potently Enhances Apoptosis Induced by Tumor Necrosis Factor in Human Leukemia Cells. J. Biol. Chem. 2002, 277, 16448–16455. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Li, G.; Podar, K.; Hideshima, T.; Shringarpure, R.; Catley, L.; Mitsiades, C.; Munshi, N.; Tai, Y.T.; Suh, N.; et al. The bortezomib/proteasome inhibitor PS-341 and triterpenoid CDDO-Im induce synergistic anti–multiple myeloma (MM) activity and overcome bortezomib resistance. Blood 2004, 103, 3158–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.-Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome Inhibitors Trigger NOXA-Mediated Apoptosis in Melanoma and Myeloma Cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Galán, P.; Roué, G.; Villamor, N.; Campo, E.; Colomer, D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood 2007, 109, 4441–4449. [Google Scholar] [CrossRef] [Green Version]
- Hauck, P.; Chao, B.H.; Litz, J.; Krystal, G.W. Alterations in the Noxa/Mcl-1 axis determine sensitivity of small cell lung cancer to the BH3 mimetic ABT-737. Mol. Cancer Ther. 2009, 8, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, H.; Hideshima, T.; Raje, N.; Roccaro, A.M.; Shiraishi, N.; Kumar, S.; Hamasaki, M.; Ishitsuka, K.; Tai, Y.-T.; Podar, K.; et al. FTY720 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Drug Resistance. Cancer Res. 2005, 65, 7478–7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, E.; Lv, N.; Ma, L.; Yao, S.; Yan, M.; Zi, F.; Deng, G.; Liu, X.; He, J.; et al. Metformin and FTY720 Sinergisticallu Induce Apoptosis in Multiple Myeloma Cells. Cell Physiol. Biochem. 2018, 48, 2. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Li, G.; Podar, K.; Hideshima, T.; Neri, P.; He, D.; Mitsiades, N.; Richardson, P.; Chang, Y.; Schindler, J.; et al. A Novel Carbohydrate-Based Therapeutic GCS-100 Overcomes Bortezomib Resistance and Enhances Dexamethasone-Induced Apoptosis in Multiple Myeloma Cells. Cancer Res. 2005, 65, 18. [Google Scholar] [CrossRef] [Green Version]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [Green Version]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.; Lopez-Pérez, R.; Mateo, G.; Gutiérrez, N.; Atadja, P.; Pandiella, A.; San Miguel, J. The Histone Deacetylase Inhibitor LBH589 Is a Potent Antimyeloma Agent that Overcomes Drug Resistance. Cancer Res. 2006, 66, 1. [Google Scholar] [CrossRef] [Green Version]
- Bhuyan, B.K.; Fraser, T.J.; Li, L.H. Cell cycle phase specificity and biochemical effects of ellipticine on mammalian cells. Cancer Res. 1972, 32, 11. [Google Scholar]
- Tian, E.; Landowski, T.H.; Stephens, O.W.; Yaccoby, S.; Barlogie, B.; Shaughnessy, J.D. Ellipticine derivative NSC 338258 represents a potential new antineoplastic agent for the treatment of multiple myeloma. Mol. Cancer Ther. 2008, 7, 500–509. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, R.C.; Pontes, G.S.; Kostyuk, A.; Camargo, G.B.C.; Dhyani, A.; Shvydenko, T.; Shvydenko, K.; Grafov, A. Anticancer and Immunomodulatory Activities of a Novel Water-Soluble Derivative of Ellipticine. Molecules 2020, 25, 2130. [Google Scholar] [CrossRef]
- Kline, M.P.; Rajkumar, S.V.; Timm, M.M.; Kimlinger, T.K.; Haug, J.L.; Lust, J.A.; Greipp, P.R.; Kumar, S. R-(-)−gossypol (AT-101) activates programmed cell death in multiple myeloma cells. Exp. Hematol. 2008, 36, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Masood, A.; Chitta, K.; Paulus, A.; Khan, A.N.H.; Sher, T.; Ersing, N.; Miller, K.C.; Manfredi, D.; Ailawadhi, S.; Borrelo, I.; et al. Downregulation of BCL2 by AT-101 enhances the antileukaemic effect of lenalidomide both by an immune dependant and independent manner. Br. J. Haematol. 2011, 157, 59–66. [Google Scholar] [CrossRef]
- Bodet, L.; Gomez-Bougie, P.; Touzeau, C.; Dousset, C.; Descamps, G.; Maïga, S.; Avet-Loiseau, H.; Bataille, R.; Moreau, P.; Le Gouill, S.; et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood 2011, 118, 3901–3910. [Google Scholar] [CrossRef]
- Lavik, A.; Zhong, F.; Chang, M.J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A syntetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, R.; Sharma, A.; Achreja, A.; Edgar, C.L.; Wei, C.; Siddiqa, A.A.; Gupta, V.A.; Matulis, S.M.; McBrayer, S.K.; Mittal, A.; et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Ma, H.; Hassig, C.A.; Payne, J.E.; Smith, N.D.; Mapara, M.Y.; Hager, J.H.; Lentzsch, S. KD5170, a novel mercaptoketone-based histone deacetylase inhibitor, exerts antimyeloma effects by DNA damage and mitochondrial signaling. Mol. Cancer Ther. 2008, 7, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Hassig, C.A.; Symons, K.T.; Guo, X.; Nguyen, P.-M.; Annable, T.; Wash, P.L.; Payne, J.E.; Jenkins, D.A.; Bonnefous, C.; Trotter, C.; et al. KD5170, a novel mercaptoketone-based histone deacetylase inhibitor that exhibits broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 1054–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.; Oton, A.; Mapara, M.Y.; Anderson, G.; Belani, C.; Lentzsch, S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br. J. Haematol. 2007, 139, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, Y.; Yang, D.; Zhao, Y. Induction of apoptosis and antitumor effects of a small molecule inhibitor of Bcl-2 and Bcl-xl, gossypol acetate, in multiple myeloma in vitro and in vivo. Oncol. Rep. 2013, 30, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, T.; Wang, X.; Li, L. Single-cell analysis of dihydroartemisinin-induced apoptosis through reactive oxygen spe-cies-mediated caspase-8 activation and mitochondrial pathway in ASTC-a-1 cells using fluorescence imaging techniques. J. Biomed. Opt. 2010, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Gu, H.; Qu, X.; Sun, J.; Song, B.; Gao, W.; Liu, J.; Shao, Q. Involvement of the mitochondrial pathway and Bim/Bcl-2 balance in dihydroartemisinin-induced apoptosis in human breast cancer in vitro. Int. J. Mol. Med. 2012, 31, 213–218. [Google Scholar] [CrossRef]
- Lu, M.; Sun, L.; Zhou, J.; Yang, J. Dihydroartemisinin induces apoptosis in colorectal cancer cells through the mitochondria-dependent pathway. Tumor Biol. 2014, 35, 6. [Google Scholar] [CrossRef]
- Aghaei, M.; Poupel, F.; Movahedian, A.; Jafari, S.M.; Shahrestanaki, M.K. Dihydroartemisinin induces apoptosis in human bladder cancer cell lines through reactive oxygen species, mitochondrial membrane potential, and cytochrome C pathway. Int. J. Prev. Med. 2017, 8, 78. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a Mammalian Protein that Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Neri, P.; Velankar, M.; Podar, K.; Hideshima, T.; Fulciniti, M.; Tassone, P.; Raje, N.; Mitsiades, C.; Mitsiades, N.; et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM). Blood 2006, 109, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Huerta, S. Smac mimetics as new cancer therapeutics. Anticancer Drugs 2009, 20, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Nikolovska-Coleska, Z.; Lu, J.; Qiu, S.; Yang, C.-Y.; Gao, W.; Meagher, J.; Stuckey, J.; Wang, S. Design, Synthesis, and Evaluation of a Potent, Cell-Permeable, Conformationally Constrained Second Mitochondria Derived Activator of Caspase (Smac) Mimetic. J. Med. Chem. 2006, 49, 7916–7920. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Schilder, R.J.; Martin, L.P.; Levin, M.; Graham, M.A.; Weng, D.E.; Adjei, A.A. A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma. Mol. Cancer Ther. 2015, 14, 2569–2575. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, B.; Giacalone, N.; Torossian, A.; Sun, Y.; Niu, K.; Lin-Tsai, O.; Lu, B. BV6, an IAP antagonist, activates apoptosis and enhances radiosensi- tization of non-small cell lung carcinoma in vitro. J. Thorac. Oncol. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehlgans, S.; Oppermann, J.; Reichert, S.; Fulda, S.; Rödel, C.; Rödel, F. The SMAC mimetic BV6 sensitizes colorectal cancer cells to ionizing radiation by interfering with DNA repair processes and enhancing apoptosis. Radiat. Oncol. 2015, 10, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mesery, M.E.; Shaker, M.; Elgaml, A. The SMAC mimetic BV6 induces cell death and sensitizes different cell lines to TNF-α and TRAIL-induced apoptosis. Exp. Biol. Med. 2016, 241, 2015–2022. [Google Scholar] [CrossRef] [Green Version]
- Bake, V.; Roesler, S.; Eckhardt, I.; Belz, K.; Fulda, S. Synergistic interaction of Smac mimetic and IFNalpha to trigger apoptosis in acute myeloid leukemia cells. Cancer Lett. 2014, 355, 2. [Google Scholar] [CrossRef] [PubMed]
- Steinwascher, S.; Nugues, A.; Schoeneberger, H.; Fulda, S. Identificationof a novel synergistic induction of cell death by Smac mimetic and HDAC inhibitors in acute myeloid leukemia cells. Cancer Lett. 2015, 366, 1. [Google Scholar] [CrossRef] [PubMed]
- Opel, D.; Schnaiter, A.; Dodier, D.; Jovanovic, M.; Gerhardinger, A.; Idler, I.; Mertens, D.; Bullinger, L.; Stilgenbauer, S.; Fulda, S. Targeting inhibitor of apoptosis proteins by Smac mimetic elicits cell death in poor prognostic subgroups of chronic lym-phocytic leukemia. Int. J. Cancer 2015, 137, 12. [Google Scholar] [CrossRef] [PubMed]
- Cristofanon, S.; Abhari, B.; Krueger, M.; Tchoghandjian, A.; Momma, S.; Calaminus, C.; Vucic, D.; Pichler, B.; Fulda, S. Iden-tification of RIP1 as a critical mediator of Smac mimetic-mediated sensitization of glioblastoma cells for Drozitumab-induced apoptosis. Cell Death Dis. 2015, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Liese, J.; Abhari, B.; Fulda, S. Smac mimetic and oleanolic acid synergize to induce cell death in human hepatocellular carci-noma cells. Cancer Lett. 2015, 365, 1. [Google Scholar] [CrossRef]
- Marschall, V.; Fulda, S. Smac mimetic-induced upregulation of interferon-beta sensitizes glioblastoma to temozolomide-induced cell death. Cell Death Dis. 2015, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Younes, A.; Stewart, K.; Orford, K.W.; Bennett, M.; Siegel, D.; Berdeja, J.G. Phase 1 Study of CB-839, a First-in-Class, Glutaminase Inhibitor in Patients with Multiple Myeloma and Lymphoma. Blood 2015, 126, 3059. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-Independent Glutamine Metabolism via TCA Cycling for Proliferation and Survival in B Cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Erickson, J.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.; Ambrosio, A.; Dias, S.; Dang, C.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010, 18, 3. [Google Scholar] [CrossRef]
- Vaidya, A.; Ayat, N.; Buford, M.N.; Wang, H.; Shankardass, A.; Zhao, Y.; Gilmore, H.; Wang, Z.; Lu, Z. Noninvasive assessment and therapeutic monitoring of drug-resistant colorectal cancer by MR molecular imaging of extradomain-B fibronectin. Theranostics 2020, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, S.A.; Zaal, E.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Riess, J.W.; Frankel, P.; Shackelford, D.; Dunphy, M.; Badawi, R.D.; Nardo, L.; Cherry, S.R.; Lanza, I.; Reid, J.; Gonsalves, W.I.; et al. Phase 1 Trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in Patients with Advanced NSCLC (NCI 10327): Rationale and Study Design. Clin. Lung Cancer 2021, 22, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P. Management of multiple myeloma in the relapsed/refractory patient. Hematology 2017, 2017, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [Green Version]
- Jullien, M.; Touzeau, C.; Moreau, P. Monoclonal antibodies as an addition to current myeloma therapy strategies. Expert Rev. Anticancer Ther. 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Demel, I.; Bago, J.R.; Hajek, R.; Jelinek, T. Focus on monoclonal antibodies targeting B-cell maturation antigen (BCMA) in multiple myeloma: Update 2020. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]



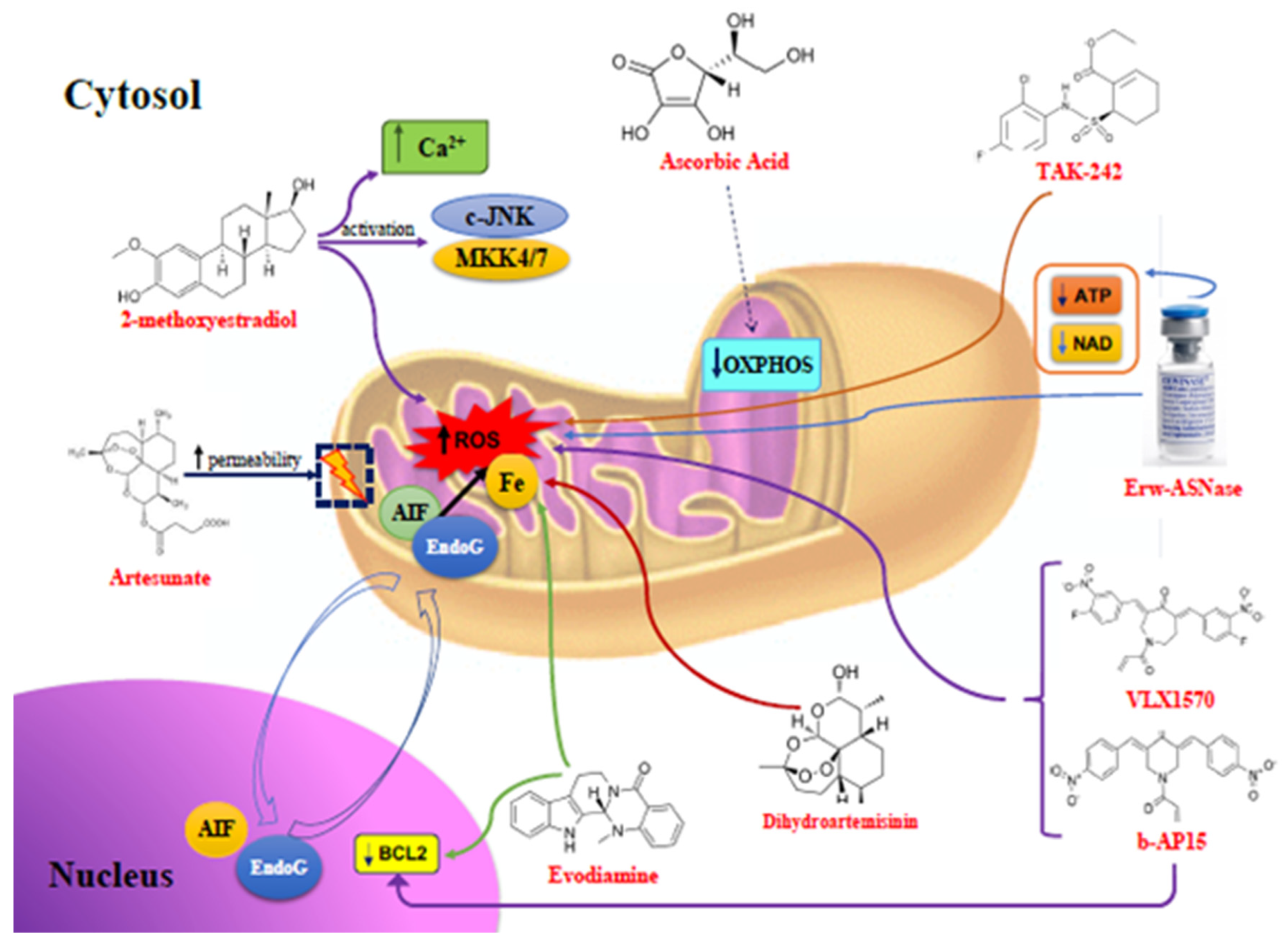{kind=link}
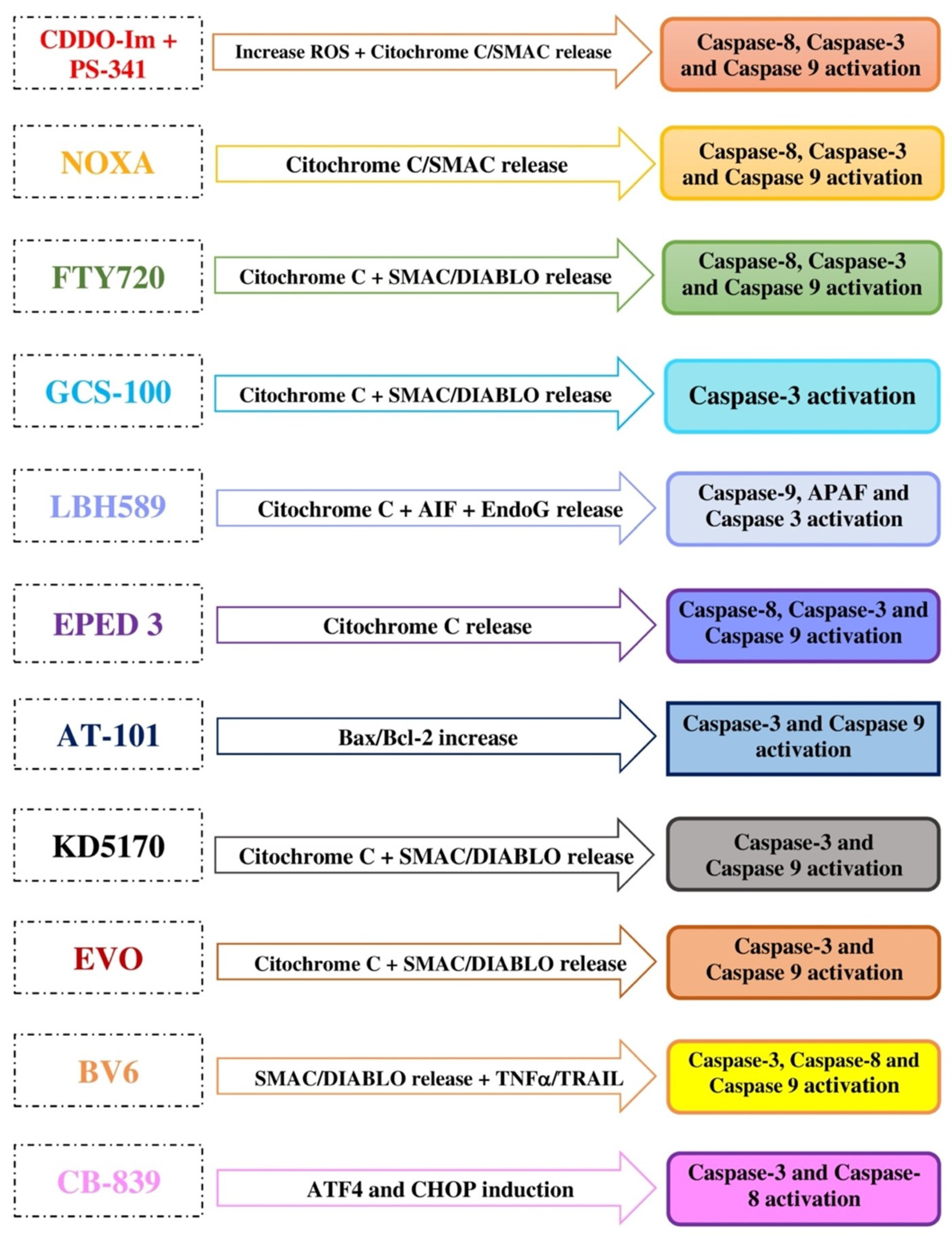{kind=link}
| Compounds | Mitochondrial Changes | Pathways Activated | Tumors |
|---|---|---|---|
| 2ME | ↑ Mitochondrial ROS and Ca2+ | c-JNK and MKK4/7 | MM, ovarian, lung, breast, and colorectal cancers |
| Artesunate | ↑ Mitochondrial ROS, loss of mitochondrial membrane integrity, release of cytochrome c, AIF, and EndoG into the cytosol | Chromatine condensation and DNA fragmentation by AIF and EndoG | MM, AML, melanoma, osteosarcoma, pancreas, breast, prostate, ovarian, renal, CNS cancers |
| Ascorbic Acid | ↓ Iron levels, inhibition of mitochondrial OXPHOS, ↓ ATP | ↓ Nrf-2, p53 upregulation, cell cycle arrest | MM, lung, pancreas, breast, cervix, urothelial cancers, and mesothelioma |
| DHA | ↑ Mitochondrial ROS, ↓ Iron levels, ↓ VEGF, loss of mitochondrial membrane integrity, release of cytochrome c into the cytosol | ↓ Bcl-2, ↑ caspases activity | MM, neuroblastoma, cervix, liver, pancreas, prostate cancers |
| Evo | ↑ Mitochondrial ROS, loss of mitochondrial membrane integrity, release of cytochrome c into the cytosol | ↓ Bcl-2, ↑ caspases 3 and 9 activity, activation of Cdc2/Cyclin B, ↑ IAPs, ↓ NF-kB, ↓ Cyclin D1 | MM, T cell leukemia, melanoma, Cervix, colorectal, lung, breast, prostate cancers |
| b-AP15 | ↑ Mitochondrial ROS, mitochondrial deformations, ↑ HSP70B’ and HSP40 | ↓ Bcl-2, ↓ Nrf-2 and HO-1 | MM, WM, DLBCL, AML, pancreas, lung cancers |
| VLX1570 | ↓ UPS14, ↑ HSP70B’ | ↓ HO-1, ↓ NF-kB, ↑ caspases activity | MM, WM, ALL, MCL, Ewings Sarcoma, ovarian cancer |
| Erw-ASNase | ↑ Mitochondrial ROS, ↓ mitochondrial ATP and NAD levels, ↓ amino acids | ↓ Nrf-2, ↓ genomic instability, ↓ DNA-repair tools | MM, ALL, AML, CML, NK/T cell lymphoma, colon and CNS cancers |
| TAK-242 | ↑ Mitochondrial ROS and RNS, mitochondrial membrane depolarization, release of cytochrome c into the cytosol | ↓ TLR4, caspase 9 activation | MM, breast, ovarian cancers |
| CDDO-Im | ↑ Mitochondrial ROS, ↓ mitochondrial glutathione, mitochondrial membrane depolarization, release of cytochrome c and SMAC/DIABLO into the cytosol | Induction of caspases 8, 3, and 9 | MM, leukemia, lymphoma |
| FTY720 | ↑ Mitochondrial ROS, mitochondrial membrane depolarization, release of cytochrome c and SMAC/DIABLO into the cytosol | ↑ proapoptotic Bax, activation of caspases 8, 9, and 3 | MM, leukemia, glioblastoma, mesothelioma, HCC, breast and bladder cancers |
| GCS-100 | Loss of mitochondrial membrane integrity, release of cytochrome c and SMAC/DIABLO into the cytosol | Induction of caspases 9, 3, and 8, ↓ NF-kB, ↓VEGF, ↓ Bcl-2 | MM, CLL, DLBCL, colorectal, pancreatic, prostate, renal cancers |
| LBH589 | ↑ Mitochondrial membrane permeability, release of cytochrome c, AIF and EndoG into the cytosol | Induction of caspases 9 and 3, ↑ APAF-1 | MM, CTCL, DLBCL, AML, HL, breast, colon, prostate, pancreatic, ovarian, esophageal squamous cell cancers |
| EPED3 | Loss of mitochondrial membrane integrity, release of cytochrome c into the cytosol | Inhibition of topoisomerase II, induction of caspases 8, 9, and 3 | MM, APL, T cell leukemia, melanoma, CNS, thyroid, breast, lung, ovarian cancers |
| AT-101 | Mitochondrial membrane depolarization, release of cytochrome c into the cytosol | ↑ Bax/Bcl2 ratio, induction of caspases 3 and 9 | MM, MCL, lung cancer |
| KD5170 | Loss of mitochondrial membrane integrity, release of cytochrome c, SMAC/DIABLO and AIF into the cytosol | Induction of caspases 3, 8, and 9, inhibition of Bcl2/Bcl-XL | MM, CTCL, MCL, CLL, colorectal, NSCLC, prostate cancer |
| BV6 | Loss of mitochondrial membrane integrity, release of cytochrome c and SMAC/DIABLO into the cytosol, induced degradation of IAPs | TRAIL-induced cell death, induction of caspases | MM, AML, CLL, glioblastoma, HCC |
| LBW242 | Loss of mitochondrial membrane integrity, release of cytochrome c and SMAC/DIABLO into the cytosol | Induction of caspases 8, 9, and 3 | MM, neuroblastoma, glioma, breast, renal cancers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Innao, V.; Rizzo, V.; Allegra, A.G.; Musolino, C.; Allegra, A. Promising Anti-Mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma. Cells 2021, 10, 439. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020439
Innao V, Rizzo V, Allegra AG, Musolino C, Allegra A. Promising Anti-Mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma. Cells. 2021; 10(2):439. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020439
Chicago/Turabian StyleInnao, Vanessa, Vincenzo Rizzo, Andrea Gaetano Allegra, Caterina Musolino, and Alessandro Allegra. 2021. "Promising Anti-Mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma" Cells 10, no. 2: 439. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020439