
Hyperglycemia Potentiates Prothrombotic Effect of Aldosterone in a Rat Arterial Thrombosis Model

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Chemicals
2.3. Bilateral Adrenalectomy
2.4. STZ-Induced Diabetes
2.5. ALDO Infusion and Drug Administration
2.6. Arterial Thrombosis Induction
2.7. Bleeding Time
2.8. ALDO and Corticosterone Level
2.9. Blood Morphology and Plasma Hemostatic Parameters
2.10. Nitric Oxide Level
2.11. Oxidative Stress Parameters
2.12. Hemostasis, NO, and Oxidative Stress Evaluation in HUVECs
2.13. Statistical Analysis
3. Results
3.1. General Characteristic of Rats
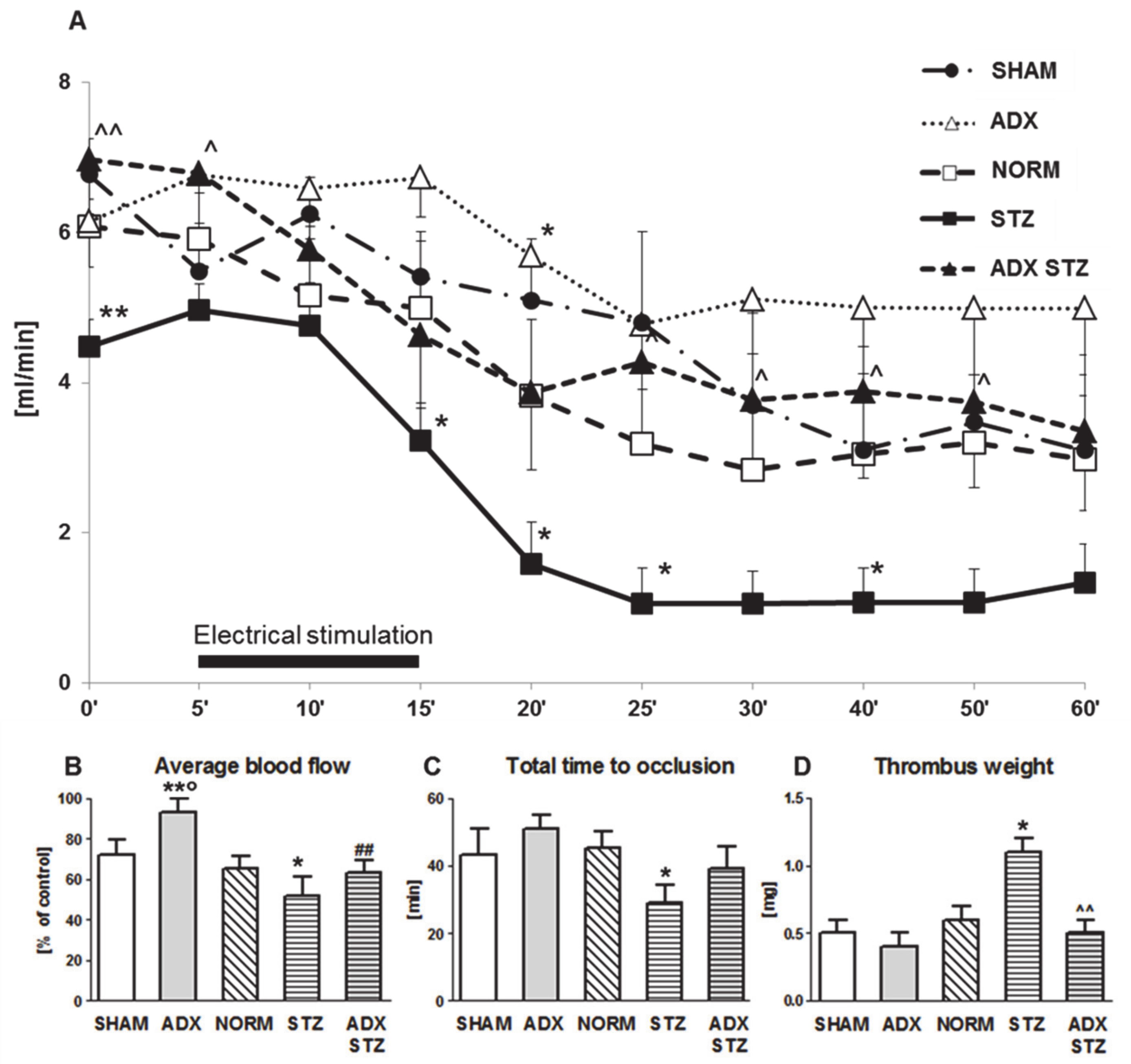
3.2. Arterial Thrombosis in ADX, STZ, and ADX STZ Rats
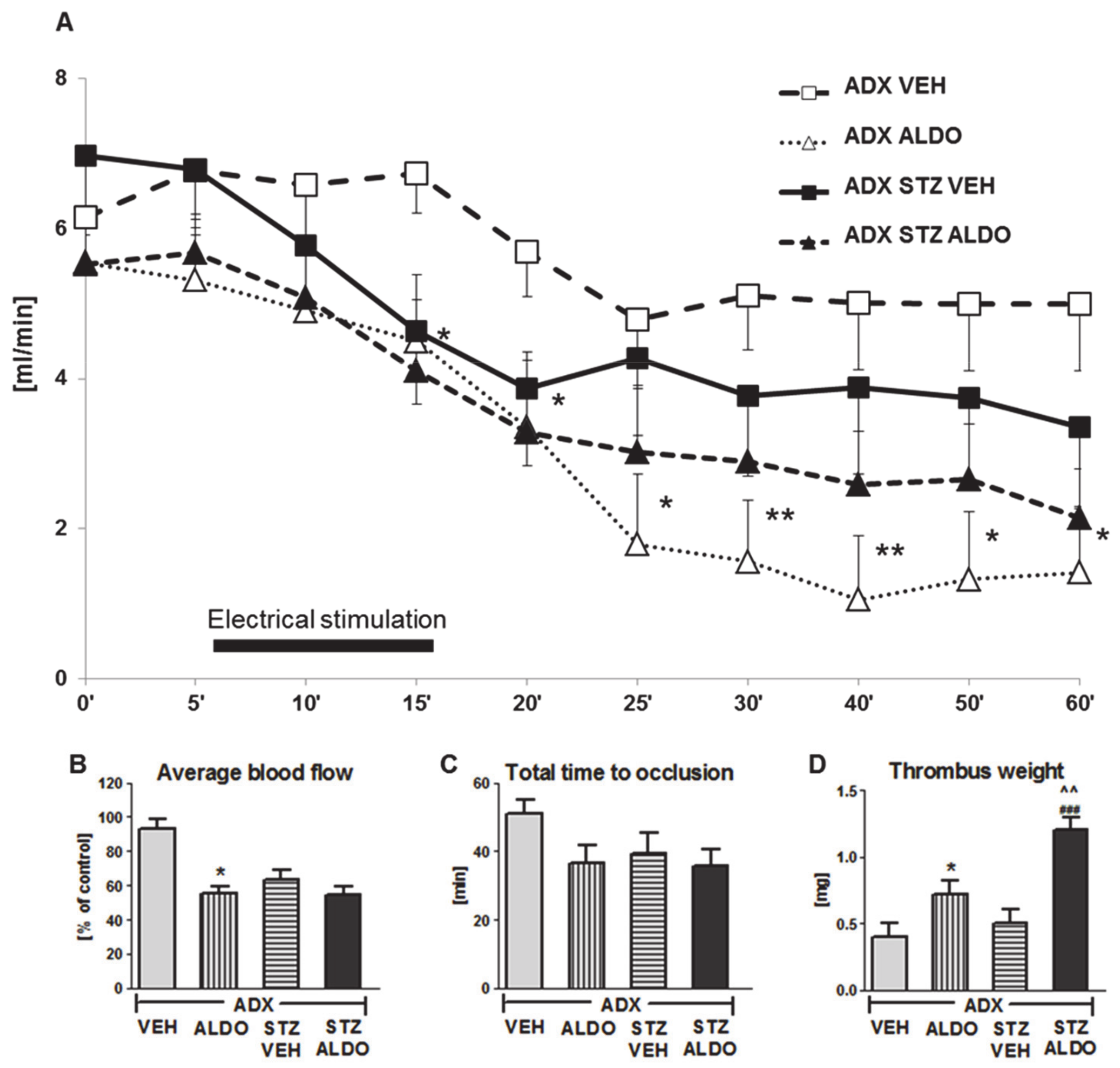
3.3. The Effect of Acute ALDO Infusion on Arterial Thrombosis in ADX and ADX STZ Rats
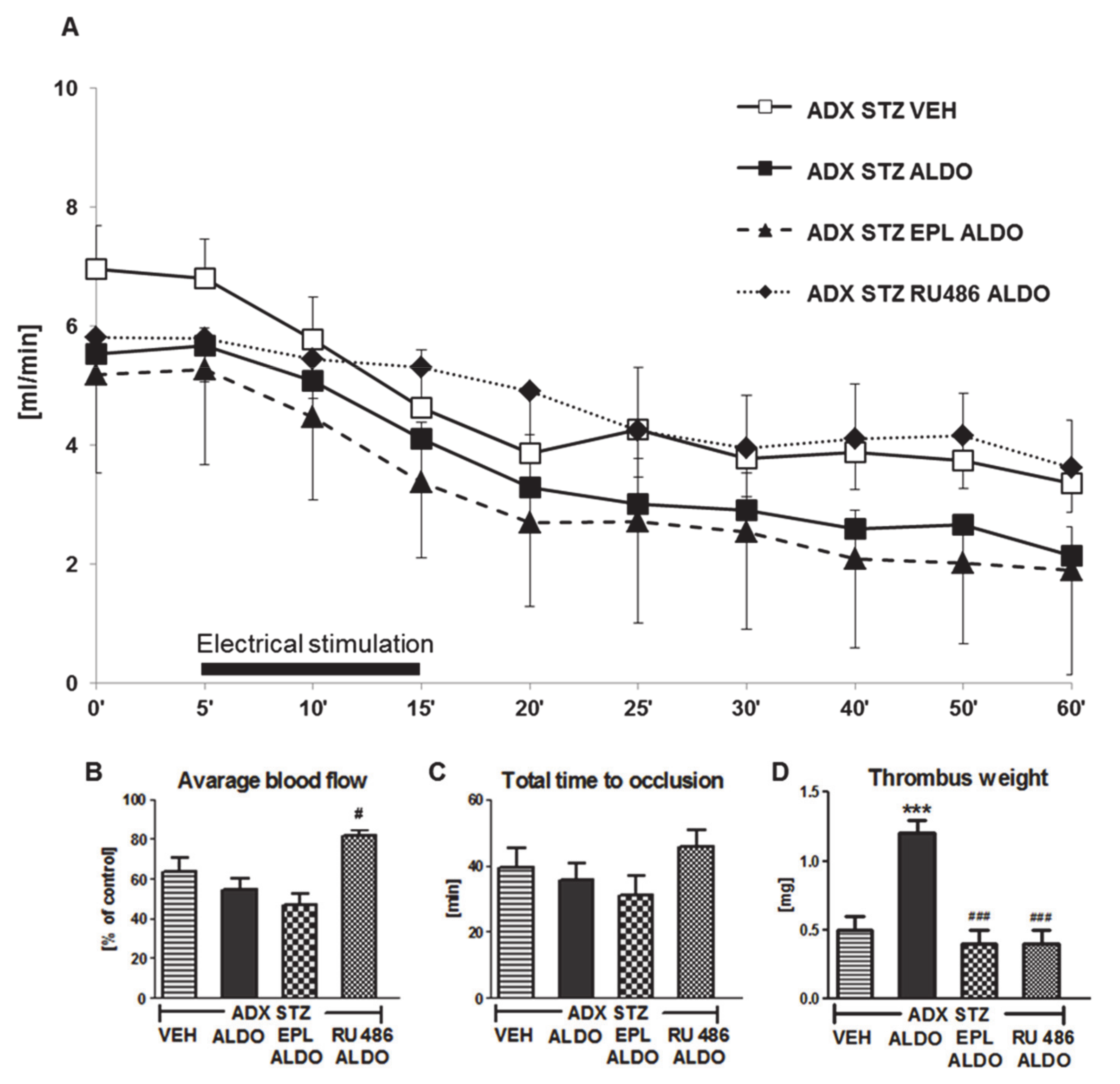
3.4. The Role of MR and GR in the Prothrombotic Effect of ALDO in ADX STZ Rats
3.5. The Effect of ALDO on Hemostasis, NO Bioavailability, and Oxidative Stress in HUVECs under Hyperglycemic Conditions
4. Discussion
4.1. The Effect of ADX on Arterial Thrombosis
4.2. Enhanced Arterial Thrombosis in STZ-Induced Diabetic Rats
4.3. Reduced Arterial Thrombosis in Adrenalectomized STZ-Induced Diabetic Rats
4.4. Acute ALDO Infusion Enhanced Arterial Thrombosis in Adrenalectomized STZ-Induced Diabetic Rats
4.5. Effect of ALDO on Hemostasis
4.6. Oxidative Stress and NO Bioavailability in the Prothrombotic Action of ALDO
4.7. Deleterious Effects of ALDO on HUVECs under Hyperglycemic Conditions
4.8. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ivanes, F.; Susen, S.; Mouquet, F.; Pigny, P.; Cuilleret, F.; Sautière, K.; Collet, J.P.; Beygui, F.; Hennache, B.; Ennezat, P.V.; et al. Aldosterone, mortality, and acute ischaemic events in coronary artery disease patients outside the setting of acute myocardial infarction or heart failure. Eur. Heart J. 2012, 33, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davel, A.P.; Anwar, I.J.; Jaffe, I.Z. The endothelial mineralocorticoid receptor: Mediator of the switch from vascular health to disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concistrè, A.; Petramala, L.; Bisogni, V.; Mezzadri, M.; Olmati, F.; Saracino, V.; Oliviero, G.; Bonvicini, M.; Tonnarini, G.; Iannucci, G.; et al. Subclinical atherosclerosis due to increase of plasma aldosterone concentrations in essential hypertensive individuals. J. Hypertens. 2019, 37, 2232–2239. [Google Scholar]
- Brown, N.J. Aldosterone and vascular inflammation. Hypertension 2008, 51, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, S.; Virdis, A.; Mattei, P.; Salvetti, A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993, 21, 929–933. [Google Scholar] [CrossRef] [Green Version]
- Sawathiparnich, P.; Kumar, S.; Vaughan, D.E.; Brown, N.J. Spironolactone abolishes the relationship between aldosterone and plasminogen activator inhibitor-1 in humans. J. Clin. Endocrinol. Metab. 2002, 87, 448–452. [Google Scholar] [CrossRef]
- Chander, P.N.; Rocha, R.; Ranaudo, J.; Singh, G.; Zuckerman, A.; Stier, C.T., Jr. Aldosterone plays a pivotal role in the pathogenesis of thrombotic microangiopathy in SHRSP. J. Am. Soc. Nephrol. 2003, 14, 1990–1997. [Google Scholar] [CrossRef] [Green Version]
- Bodary, P.F.; Sambaziotis, C.; Wickenheiser, K.J.; Rajagopalan, S.; Pitt, B.; Eitzman, D.T. Aldosterone promotes thrombosis formation after arterial injury in mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromotowicz, A.; Szemraj, J.; Stankiewicz, A.; Zakrzeska, A.; Mantur, M.; Jaroszewicz, E.; Rogowski, F.; Chabielska, E. Study of the mechanisms of aldosterone prothrombotic effect in rats. J. Renin Angiotensin Aldosterone Syst. 2011, 12, 430–439. [Google Scholar] [CrossRef]
- Gromotowicz-Poplawska, A.; Marcinczyk, N.; Misztal, T.; Golaszewska, A.; Aleksiejczuk, M.; Rusak, T.; Chabielska, E. Rapid effects of aldosterone on platelets, coagulation, and fibrinolysis lead to experimental thrombosis augmentation. Vascul. Pharmacol. 2019, 122–123, 106598. [Google Scholar] [CrossRef]
- Heylen, E.; Huang, A.; Sun, D.; Kaley, G. Nitric oxide-mediated dilation of arterioles to intraluminal administration of aldosterone. J. Cardiovasc. Pharmacol. 2009, 54, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Michea, L.; Delpiano, A.M.; Hitschfeld, C.; Lobos, L.; Lavandero, S.; Marusic, E.T. Eplerenone blocks nongenomic effects of aldosterone on the Na+/H+ exchanger, intracellular Ca2+ levels, and vasoconstriction in mesenteric resistance vessels. Endocrinology 2005, 146, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, G.; Cirillo, P.; Ragni, M.; Conte, S.; Uccello, G.; Golino, P. Reactive oxygen species induce a procoagulant state in endothelial cells by inhibiting tissue factor pathway inhibitor. J. Thromb. Thrombolysis 2015, 40, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Lagrange, J.; Li, Z.; Fassot, C.; Bourhim, M.; Louis, H.; Nguyen Dinh Cat, A.; Parlakian, A.; Wahl, D.; Lacolley, P.; Jaisser, F.; et al. Endothelial mineralocorticoid receptor activation enhances endothelial protein C receptor and decreases vascular thrombosis in mice. FASEB J. 2014, 28, 2062–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCurley, A.; Jaffe, I.Z. Mineralocorticoid receptors in vascular function and disease. Mol. Cell. Endocrinol. 2012, 350, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Creager, M.A.; Lüscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Aras, R.; Sowers, J.R.; Arora, R. The proinflammatory and hypercoagulable state of diabetes mellitus. Rev. Cardiovasc. Med. 2005, 6, 84–97. [Google Scholar]
- Srinivasan, S.; Hatley, M.E.; Bolick, D.T.; Palmer, L.A.; Edelstein, D.; Brownlee, M.; Hedrick, C.C. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 2004, 47, 1727–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenberg, N.K.; Stevanovic, R.; Agarwal, A.; Lansang, M.C.; Price, D.A.; Laffel, L.M.; Williams, G.H.; Fisher, N.D. Plasma aldosterone concentration in the patient with diabetes mellitus. Kidney Int. 2004, 65, 1435–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallo, F.; Federspil, G.; Veglio, F.; Mulatero, P. The metabolic syndrome in primary aldosteronism. Curr. Hypertens. Rep. 2007, 9, 106–111. [Google Scholar] [CrossRef] [PubMed]
- O‘Keefe, J.H.; Abuissa, H.; Pitt, B. Eplerenone improves prognosis in postmyocardial infarction diabetic patients with heart failure: Results from EPHESUS. Diabetes Obes. Metab. 2008, 10, 492–497. [Google Scholar] [CrossRef]
- Zakrzeska, A.; Gromotowicz-Poplawska, A.; Szemraj, J.; Szoka, P.; Kisiel, W.; Purta, T.; Kasacka, I.; Chabielska, E. Eplerenone reduces arterial thrombosis in diabetic rats. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, A.; Vogt, C.; Fraccarollo, D.; Widder, J.; Flierl, U.; Hildemann, S.K.; Ertl, G.; Bauersachs, J. Eplerenone improves vascular function and reduces platelet activation in diabetic rats. J. Physiol. Pharmacol. 2010, 61, 45–52. [Google Scholar]
- Gromotowicz-Poplawska, A.; Kloza, M.; Aleksiejczuk, M.; Marcinczyk, N.; Szemraj, J.; Kozlowska, H.; Chabielska, E. Nitric oxide as a modulator in platelet- and endothelium-dependent antithrombotic effect of eplerenone in diabetic rats. J. Physiol. Pharmacol. 2019, 70, 187–198. [Google Scholar]
- Gromotowicz-Poplawska, A.; Stankiewicz, A.; Kramkowski, K.; Gradzka, A.; Wojewodzka-Zelezniakowicz, M.; Dzieciol, J.; Szemraj, J.; Chabielska, E. The acute prothrombotic effect of aldosterone in rats is partially mediated via angiotensin II receptor type 1. Thromb. Res. 2016, 138, 114–120. [Google Scholar] [CrossRef]
- Moraes, L.A.; Paul-Clark, M.J.; Rickman, A.; Flower, R.J.; Goulding, N.J.; Perretti, M. Ligand-specific glucocorticoid receptor activation in human platelets. Blood 2005, 106, 4167–4175. [Google Scholar] [CrossRef] [Green Version]
- Girod, J.P.; Brotman, D.J. Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovasc. Res. 2004, 64, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, M.D.; Wolf, I.M.; Sanchez, E.R.; Witchel, S.F.; DeFranco, D.B. Glucocorticoid receptor physiology. Rev. Endocr. Metab. Disord. 2007, 8, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Ohkura, N.; Ishida, N. Adrenal gland-dependent augmentation of plasminogen activator inhibitor-1 expression in streptozotocin-induced diabetic mice. J. Thromb. Haemost. 2006, 4, 1566–1574. [Google Scholar] [CrossRef]
- Gaeggeler, H.P.; Gonzalez-Rodriguez, E.; Jaeger, N.F.; Loffing-Cueni, D.; Norregaard, R.; Loffing, J.; Horisberger, J.D.; Rossier, B.C. Mineralocorticoid versus Glucocorticoid Receptor Occupancy Mediating Aldosterone-Stimulated Sodium Transport in a Novel Renal Cell Line. J. Am. Soc. Nephrol. 2005, 16, 878–891. [Google Scholar] [CrossRef] [Green Version]
- Funder, J.W.; Mihailidou, A.S. Aldosterone and mineralocorticoid receptors: Clinical studies and basic biology. Mol. Cell. Endocrinol. 2009, 301, 2–6. [Google Scholar] [CrossRef]
- Wojewodzka-Zelezniakowicz, M.; Kisiel, W.; Kramkowski, K.; Gromotowicz-Poplawska, A.; Zakrzeska, A.; Stankiewicz, A.; Kolodziejczyk, P.; Szemraj, J.; Ladny, J.R.; Chabielska, E. Quinapril decreases antifibrinolytic and prooxidative potential of propofol in arterial thrombosis in hypertensive rats. J. Renin Angiotensin Aldosterone Syst. 2016, 17, 1470320316647239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, W.A.; Bostwick, J.S.; Ogletree, M.L.; Stewart, A.B.; Steinbacher, T.E.; Hua, J.; Price, L.A.; Wong, P.C.; Rehfuss, R.P. Biomarker optimization to track the antithrombotic and hemostatic effects of clopidogrel in rats. J. Pharmacol. Exp. Ther. 2007, 322, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojewodzka-Zelezniakowicz, M.; Gromotowicz-Poplawska, A.; Kisiel, W.; Konarzewska, E.; Szemraj, J.; Ladny, J.R.; Chabielska, E. Angiotensin-converting enzyme inhibitors attenuate propofol-induced pro-oxidative and antifibrinolytic effect in human endothelial cells. J. Renin Angiotensin Aldosterone Syst. 2017, 18, 1470320316687197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelze, M.; Knorr, M.; Schuhmacher, S.; Heeren, T.; Otto, C.; Schulz, E.; Reifenberg, K.; Wenzel, P.; Münzel, T.; Daiber, A. Vascular dysfunction in streptozotocin-induced experimental diabetes strictly depends on insulin deficiency. J. Vasc. Res. 2011, 48, 275–284. [Google Scholar] [CrossRef]
- Guarini, S. A highly reproducible model of arterial thrombosis in rats. J. Pharmacol. Toxicol. Methods 1996, 35, 101–105. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawamura, S.; Katoh, S.; Takenaka, T. Experimental model of carotid artery thrombosis in rats and the thrombolytic activity of YM866, a novel modified tissue-type plasminogen activator. Jpn. J. Pharmacol. 1993, 63, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smokovitis, A.; Kokolis, N.; Rekkas, C.; Ploumis, T. Variable response of tissue parameters of the fibrinolytic system to unilateral or bilateral adrenalectomy and unilateral or bilateral adrenal demedullation. J. Endocrinol. Invest. 1991, 14, 815–820. [Google Scholar] [CrossRef]
- Petramala, L.; Iacobellis, G.; Carnevale, R.; Marinelli, C.; Zinnamosca, L.; Concistrè, A.; Galassi, M.; Iannucci, G.; Lucia, P.; Pignatelli, P.; et al. Enhanced Soluble Serum CD40L and Serum P-Selectin Levels in Primary Aldosteronism. Horm. Metab. Res. 2016, 48, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Nakamura, S.; Ma, L.; Nakamura, I.; Donnert, E.; Freeman, M.; Vaughan, D.E.; Fogo, A.B. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. 2000, 58, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Petramala, L.; Pignatelli, P.; Carnevale, R.; Zinnamosca, L.; Marinelli, C.; Settevendemmie, A.; Concistrè, A.; Tonnarini, G.; De Toma, G.; Violi, F.; et al. Oxidative stress in patients affected by primary aldosteronism. J. Hypertens. 2014, 32, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- DeLano, F.A.; Schmid-Schönbein, G.W. Enhancement of glucocorticoid and mineralocorticoid receptor density in the microcirculation of the spontaneously hypertensive rat. Microcirculation 2004, 11, 69–78. [Google Scholar]
- Kalman, B.A.; Spencer, R.L. Rapid corticosteroid-dependent regulation of mineralocorticoid receptor protein expression in rat brain. Endocrinology 2002, 143, 4184–4195. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Ozawa, H.; Matsuda, K.I.; Lu, H.; De Kloet, E.R.; Kawata, M. Changes in the expression of corticotrophin-releasing hormone, mineralocorticoid receptor and glucocorticoid receptor mRNAs in the hypothalamic paraventricular nucleus induced by fornix transection and adrenalectomy. J. Neuroendocrinol. 2007, 19, 229–238. [Google Scholar] [CrossRef]
- Takeda, Y.; Yoneda, T.; Demura, M.; Miyamori, I.; Mabuchi, H. Cardiac aldosterone production in genetically hypertensive rats. Hypertension 2000, 36, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Känel, R.; Dimsdale, J.E. Effects of sympathetic activation by adrenergic infusions on hemostasis in vivo. Eur. J. Haematol. 2000, 65, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Belougne-Malfatti, E.; Aguejouf, O.; Doutremepuich, F.; Doutremepuich, C. Action of neurotransmitters: Acetylcholine, serotonin, and adrenaline in an experimental arterial thrombosis induced by oxygen free radicals. Thromb. Res. 1997, 88, 435–439. [Google Scholar] [CrossRef]
- Wallerath, T.; Gödecke, A.; Molojavyi, A.; Li, H.; Schrader, J.; Förstermann, U. Dexamethasone lacks effect on blood pressure in mice with a disrupted endothelial NO synthase gene. Nitric Oxide 2004, 10, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Ullian, M.E. The role of corticosteriods in the regulation of vascular tone. Cardiovasc. Res. 1999, 41, 55–64. [Google Scholar] [CrossRef]
- Majoor, C.J.; Sneeboer, M.M.; de Kievit, A.; Meijers, J.C.; van der Poll, T.; Lutter, R.; Bel, E.H.; Kamphuisen, P.W. The influence of corticosteroids on hemostasis in healthy subjects. J. Thromb. Haemost. 2016, 14, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Yoshimoto, T.; Hirata, Y. Endothelial dysfunction is related to aldosterone excess and raised blood pressure. Endocr. J. 2009, 56, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Yamashiro, K.; Tsujikawa, A.; Ishida, S.; Usui, T.; Kaji, Y.; Honda, Y.; Ogura, Y.; Adamis, A.P. Platelets accumulate in the diabetic retinal vasculature following endothelial death and suppress blood-retinal barrier breakdown. Am. J. Pathol. 2003, 163, 253–259. [Google Scholar] [CrossRef]
- Yamada, K.; Niki, H.; Nagai, H.; Nishikawa, M.; Nakagawa, H. Serotonin potentiates high-glucose-induced endothelial injury: The role of serotonin and 5-HT(2A) receptors in promoting thrombosis in diabetes. J. Pharmacol. Sci. 2012, 119, 243–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Brahmbhatt, S.; Gupta, A.; Sharma, A.C. Duration of streptozotocin-induced diabetes differentially affects p38-mitogen-activated protein kinase (MAPK) phosphorylation in renal and vascular dysfunction. Cardiovasc. Diabetol. 2005, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Winocour, P.D.; Lopes-Virella, M.; Laimins, M.; Colwell, J.A. Effect of insulin treatment in streptozocin-induced diabetic rats on in vitro platelet function and plasma von Willebrand factor activity and factor VIII-related antigen. J. Lab. Clin. Med. 1985, 106, 319–325. [Google Scholar]
- Albrecht, W.; de Gasparo, M.; Märki, F. Role of the adrenals in the development of streptozotocin (STR)-induced diabetes in male albino rats. Horm. Metab. Res. 1984, 16 (Suppl. 1), 71–76. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, R.; Kikuchi, T.; Mori, Y.; Aoki, K.; Kaburagi, Y.; Yasuda, K.; Sekihara, H. Aldosterone stimulates gene expression of hepatic gluconeogenic enzymes through the glucocorticoid receptor in a manner independent of the protein kinase B cascade. Endocr. J. 2004, 51, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Aso, Y.; Okumura, K.I.; Yoshida, N.; Tayama, K.; Takemura, Y.; Inukai, T. Enhancement of fibrinolysis in poorly controlled, hospitalized type 2 diabetic patients by short-term metabolic control: Association with a decrease in plasminogen activator inhibitor 1. Exp. Clin. Endocrinol. Diabetes 2004, 112, 175–180. [Google Scholar] [CrossRef]
- Charloux, A.; Gronfier, C.; Lonsdorfer-Wolf, E.; Piquard, F.; Brandenberger, G. Aldosterone release during the sleep-wake cycle in humans. Am. J. Physiol. 1999, 276, E43–E49. [Google Scholar] [CrossRef]
- Thohan, V.; Torre-Amione, G.; Koerner, M.M. Aldosterone antagonism and congestive heart failure: A new look at an old therapy. Curr. Opin. Cardiol. 2004, 19, 301–308. [Google Scholar] [CrossRef]
- Vasan, R.S.; Evans, J.C.; Larson, M.G.; Wilson, P.W.; Meigs, J.B.; Rifai, N.; Benjamin, E.J.; Levy, D. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N. Engl. J. Med. 2004, 351, 33–41. [Google Scholar] [CrossRef]
- Sato, A.; Funder, J.W. High glucose stimulates aldosterone-induced hypertrophy via type I mineralocorticoid receptors in neonatal rat cardiomyocytes. Endocrinology 1996, 137, 4145–4153. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Schmuck, S.; Chorazcyzewski, J.Z.; Gros, R.; Feldman, R.D. Aldosterone regulates vascular reactivity: Short-term effects mediated by phosphatidylinositol 3-kinase-dependent nitric oxide synthase activation. Circulation 2003, 108, 2400–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockyer, S.; Kambayashi, J. Demonstration of flow and platelet dependency in a ferric chloride-induced model of thrombosis. J. Cardiovasc. Pharmacol. 1999, 33, 718–725. [Google Scholar] [CrossRef]
- Hansen, H.R.; Wolfs, J.L.; Bruggemann, L.; Sommeijer, D.W.; Bevers, E.; Hauer, A.D.; Kuiper, J.; Spek, C.A.; Spronk, H.M.; Reitsma, P.H.; et al. Hyperglycemia accelerates arterial thrombus formation and attenuates the antithrombotic response to endotoxin in mice. Blood Coagul. Fibrinolysis 2007, 18, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Furukoji, E.; Marutsuka, K.; Hatakeyama, K.; Yamamoto, H.; Tamura, S.; Ikeda, Y.; Sumiyoshi, A.; Asada, Y. Increased vascular wall thrombogenicity combined with reduced blood flow promotes occlusive thrombus formation in rabbit femoral artery. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2420–2424. [Google Scholar] [CrossRef]
- Rossier, M.F.; Python, M.; Maturana, A.D. Contribution of mineralocorticoid and glucocorticoid receptors to the chronotropic and hypertrophic actions of aldosterone in neonatal rat ventricular myocytes. Endocrinology 2010, 151, 2777–2787. [Google Scholar] [CrossRef] [Green Version]
- Karolczak, K.; Kubalczyk, P.; Glowacki, R.; Pietruszynski, R.; Watala, C. Aldosterone modulates blood homocysteine and cholesterol in coronary artery disease patients - a possible impact on atherothrombosis? Physiol. Res. 2018, 67, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Ahrendt, S.A.; Ryan, C.K.; Francis, C.W.; Hruban, R.H.; Hu, Y.C.; Hostetter, G.; Harvey, J.; Taubman, M.B. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin. Cancer Res. 2007, 13, 2870–2875. [Google Scholar] [CrossRef] [Green Version]
- Kerachian, M.A.; Cournoyer, D.; Harvey, E.J.; Chow, T.Y.; Neagoe, P.E.; Sirois, M.G.; Séguin, C. Effect of high-dose dexamethasone on endothelial haemostatic gene expression and neutrophil adhesion. J. Steroid Biochem. Mol. Biol. 2009, 116, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Jia, R.; Bao, Y. Aldosterone up-regulates production of plasminogen activator inhibitor-1 by renal mesangial cells. J. Biochem. Mol. Biol. 2007, 40, 180–188. [Google Scholar] [CrossRef]
- Chun, T.Y.; Pratt, J.H. Aldosterone increases plasminogen activator inhibitor-1 synthesis in rat cardiomyocytes. Mol. Cell. Endocrinol. 2005, 239, 55–61. [Google Scholar] [CrossRef]
- Enomoto, S.; Yoshiyama, M.; Omura, T.; Matsumoto, R.; Kusuyama, T.; Kim, S.; Izumi, Y.; Akioka, K.; Iwao, H.; Takeuchi, K.; et al. Effects of eplerenone on transcriptional factors and mRNA expression related to cardiac remodelling after myocardial infarction. Heart 2005, 91, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Niwano, H.; Takahashi, H.; Tatewaki, W.; Wada, K.; Seki, Y.; Shibata, A. Behaviour of tissue plasminogen activator, plasminogen activator inhibitor 1 and their complex in various disease states. Blood Coagul. Fibrinolysis 1992, 3, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Yang, D.H.; Oh, D.Y.; Baick, S.H.; Kim, S.K.; Kim, S.J.; Hong, S.Y. Plasma t-PA and PAl-1 antigen concentrations in non-insulin dependent diabetic patients: Effects of treatment modality on fibrinolysis. Korean J. Intern. Med. 1992, 7, 81–86. [Google Scholar] [CrossRef]
- Farquharson, C.A.; Struthers, A.D. Aldosterone induces acute endothelial dysfunction in vivo in humans: Evidence for an aldosterone-induced vasculopathy. Clin. Sci. 2002, 103, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thum, T.; Schmitter, K.; Fleissner, F.; Wiebking, V.; Dietrich, B.; Widder, J.D.; Jazbutyte, V.; Hahner, S.; Ertl, G.; Bauersachs, J. Impairment of endothelial progenitor cell function and vascularization capacity by aldosterone in mice and humans. Eur. Heart J. 2011, 32, 1275–1286. [Google Scholar] [CrossRef]
- Davies, J.I.; Band, M.; Morris, A.; Struthers, A.D. Spironolactone impairs endothelial function and heart rate variability in patients with type 2 diabetes. Diabetologia 2004, 47, 1687–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jiménez-Altayó, F. The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 2021, 162, 615–635. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Maiello, M.; Boeri, D.; Podesta, F.; Cagliero, E.; Vichi, M.; Odetti, P.; Adezati, L.; Lorenzi, M. Increased expression of tissue plasminogen activator and its inhibitor and reduced fibrinolytic potential of human endothelial cells cultured in elevated glucose. Diabetes 1992, 41, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, D.; Ceriello, A.; Paolisso, G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996, 19, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| SHAM (n = 7) | ADX (n = 7) | NORM (n = 9) | STZ (n = 12) | ADX STZ (n = 10) | |
|---|---|---|---|---|---|
| Body weight [g] | 405 ± 12 | 404 ± 15 | 415 ± 7 | 308 ± 5 ** | 346 ± 20 # |
| Glucose level [mg/dl] | 74 ± 4 | 70 ± 5 | 74 ± 3 | 269 ± 43 ** | 163 ± 18 # ^ |
| Survival rate [%] | 100 | 58 | 100 | 83 | 56 |
| Aldosterone level [pg/mL] | 678 ± 40 | n/d | 429 ± 58 | 786 ± 110 * | n/d |
| Corticosterone level [pg/mL] | 430 ± 46 | n/d | 388 ± 60 | 480 ± 100 | n/d |
| WBC [103/mm3] | 3.6 ± 0.3 | 4.0 ± 0.3 | 3.7 ± 0.4 | 5.3 ± 0.7 * | 5.8 ± 1.3 |
| RBC [106/mm3] | 7.2 ± 0.2 | 6.9 ± 0.3 | 7.6 ± 0.2 | 9.4 ± 0.6 ** | 7.7 ± 0.4 ^ |
| PLT [103/mm3] | 912 ± 36 | 1034 ± 45 | 881 ± 44 | 665 ± 102 * | 939 ± 102 ^ |
| HGB [g/dl] | 14.6 ± 0.4 | 13.8 ± 0.3 | 15.2 ± 0.1 | 17.9 ± 0.9 ** | 14.7 ± 0.8 ^ |
| HCT [%] | 42.9 ± 1.3 | 38.6 ± 1.2 ° | 48.7 ± 1.3 | 52.9 ± 0.6 ** | 43.4 ± 2.7 ^ |
| BT [s] | 88 ± 4 | 100 ± 11 | 99 ± 4 | 75 ± 4 ** | 80 ± 4 # |
| TF [pg/mL] | 5.3 ± 0.1 | 4.2 ± 0.1 °° | 5.4 ± 0.1 | 5.2 ± 0.1 | 5.3 ± 0.1 |
| t-PA [ng/mL] | 6.4 ± 0.1 | 8.7 ± 0.1 °° | 7.1 ± 0.1 | 5.5 ± 0.2 * | 5.7 ± 0.1 |
| PAI-1 [ng/mL] | 5.5 ± 0.1 | 4.0 ± 0.1 °° | 6.0 ± 0.1 | 7.0 ± 0.1 * | 7.2 ± 0.2 |
| NO2/NO3 [μmol/l] | 10.7 ± 0.1 | 7.4 ± 0.1 °° | 9.1 ± 0.1 | 10.7 ± 0.2 * | 11.4 ± 0.1 ^ |
| H2O2 [ng/mL] | 86.0 ± 0.1 | 111.9 ± 3.1 °° | 91.7 ± 1.3 | 76.1 ± 2.5 * | 76.3 ± 1.3 |
| MDA [pmol/mg] | 0.25 ± 0.01 | 0.17 ± 0.01 °° | 0.28 ± 0.01 | 0.36 ± 0.02 * | 0.33 ± 0.01 |
| VEH (n = 10) | ALDO (n = 12) | EPL + ALDO (n = 10) | RU486 + ALDO (n = 10) | |
|---|---|---|---|---|
| Aldosterone level [pg/mL] | n/d | 1950 ± 224 | 4192 ± 88 ** | 4054 ± 105 ** |
| BT [s] | 80 ± 4 | 66 ± 5 * | 67 ± 4 | 97 ± 5 ## |
| TF [pg/mL] | 5.3 ± 0.1 | 5.5 ± 0.1 * | 5.1 ± 0.1 ## | 5.1 ± 0.1 ## |
| t-PA [ng/mL] | 5.7 ± 0.1 | 6.6 ± 0.1 *** | 7.4 ± 0.1 ### | 6.7 ± 0.1 |
| PAI-1 [ng/mL] | 6.4 ± 0.2 | 7.3 ± 0.1 * | 6.0 ± 0.1 ## | 5.5 ± 0.2 ## |
| NO2/NO3 [µmol/l] | 11.4 ± 0.1 | 10.4 ± 0.3 * | 12.8 ± 0.1 ### | 9.6 ± 0.1 |
| H2O2 [ng/mL] | 76.3 ± 13 | 98 ± 2.4 *** | 75.0 ± 1.1 ### | 66.2 ± 2.7 ### |
| MDA [pmol/mg] | 0.33 ± 0.01 | 0.29 ± 0.1 ** | 0.5 ± 0.02 ### | 0.63 ± 0.01 ### |
| TF [pg/mL] | t-PA [ng/mL] | PAI-1 [ng/mL] | NO2/NO3 [µmol/l] | eNOS [2-∆∆ct] | H2O2 [ng/mL] | MDA [pmol/mg] | |
|---|---|---|---|---|---|---|---|
| NORM | 4.03 ± 0.02 | 8.11 ± 0.04 | 3.76 ± 0.02 | 10.61 ± 0.14 | 0.904 ± 0.003 | 105.0 ± 0.6 | 0.320 ± 0.004 |
| NORM VEH | 4.09 ± 0.03 | 8.08 ± 0.03 | 3.80 ± 0.01 | 10.81 ± 0.02 | 0.896 ± 0.009 | 105.8 ± 0.3 | 0.322 ± 0.001 |
| NORM ALDO | 4.96 ± 0.02 ^^ | 5.83 ± 0.02 ^^ | 6.10 ± 0.04 ^^ | 9.41 ± 0.13 ^^ | 0.791 ± 0.002 ^^ | 143.4 ± 0.7 ^^^ | 0.366 ± 0.002 ^ |
| GLU | 5.17 ± 0.01 ** | 6.86 ± 0.02 ** | 5.40 ± 0.04 *** | 8.57 ± 0.17 ** | 0.728 ± 0.004 ** | 121.7 ± 0.1 ** | 0.404 ± 0.004 ** |
| GLU VEH | 5.18 ± 0.06 | 6.84 ± 0.04 | 5.36 ± 0.04 | 8.47 ± 0.11 | 0.723 ± 0.004 | 121.4 ± 0.1 | 0.406 ± 0.007 |
| GLU ALDO | 5.88 ± 0.03 ## °° | 5.10 ± 0.02 ## ° | 10.55 ± 0.1 ## °° | 7.18 ± 0.04 ## °° | 0.613 ± 0.003 ## °° | 149.6 ± 0.3 ## ° | 0.527 ± 0.004 ## °°° |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gromotowicz-Poplawska, A.; Szoka, P.; Zakrzeska, A.; Kolodziejczyk, P.; Marcinczyk, N.; Szemraj, J.; Tutka, P.; Chabielska, E. Hyperglycemia Potentiates Prothrombotic Effect of Aldosterone in a Rat Arterial Thrombosis Model. Cells 2021, 10, 471. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020471
Gromotowicz-Poplawska A, Szoka P, Zakrzeska A, Kolodziejczyk P, Marcinczyk N, Szemraj J, Tutka P, Chabielska E. Hyperglycemia Potentiates Prothrombotic Effect of Aldosterone in a Rat Arterial Thrombosis Model. Cells. 2021; 10(2):471. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020471
Chicago/Turabian StyleGromotowicz-Poplawska, Anna, Piotr Szoka, Agnieszka Zakrzeska, Patrycjusz Kolodziejczyk, Natalia Marcinczyk, Janusz Szemraj, Piotr Tutka, and Ewa Chabielska. 2021. "Hyperglycemia Potentiates Prothrombotic Effect of Aldosterone in a Rat Arterial Thrombosis Model" Cells 10, no. 2: 471. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020471