
The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells

 , ,
, ,
Abstract
:1. Pancreatic Cancer
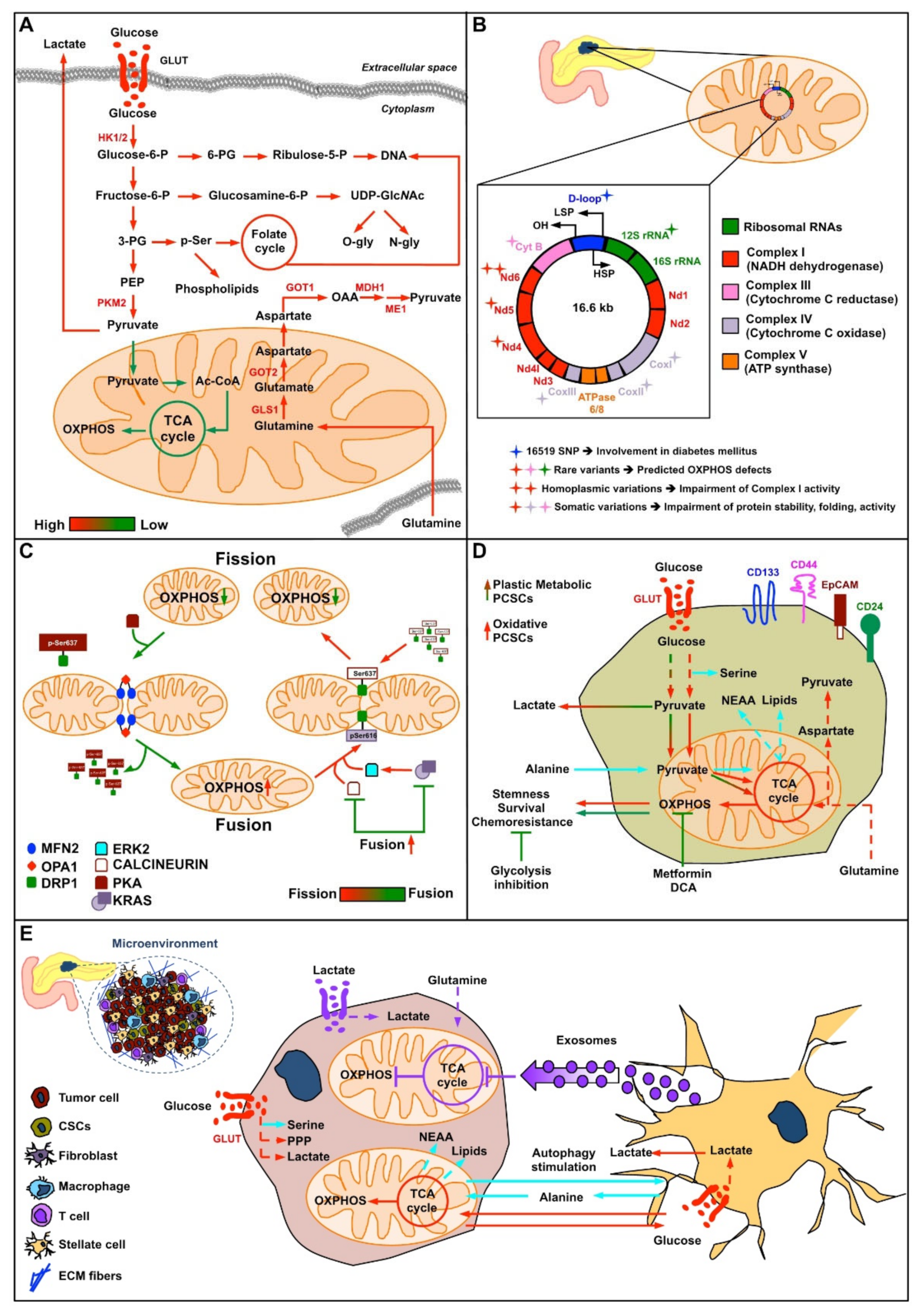
2. Roles of Mitochondria in the Origin of Pancreatic Cancer
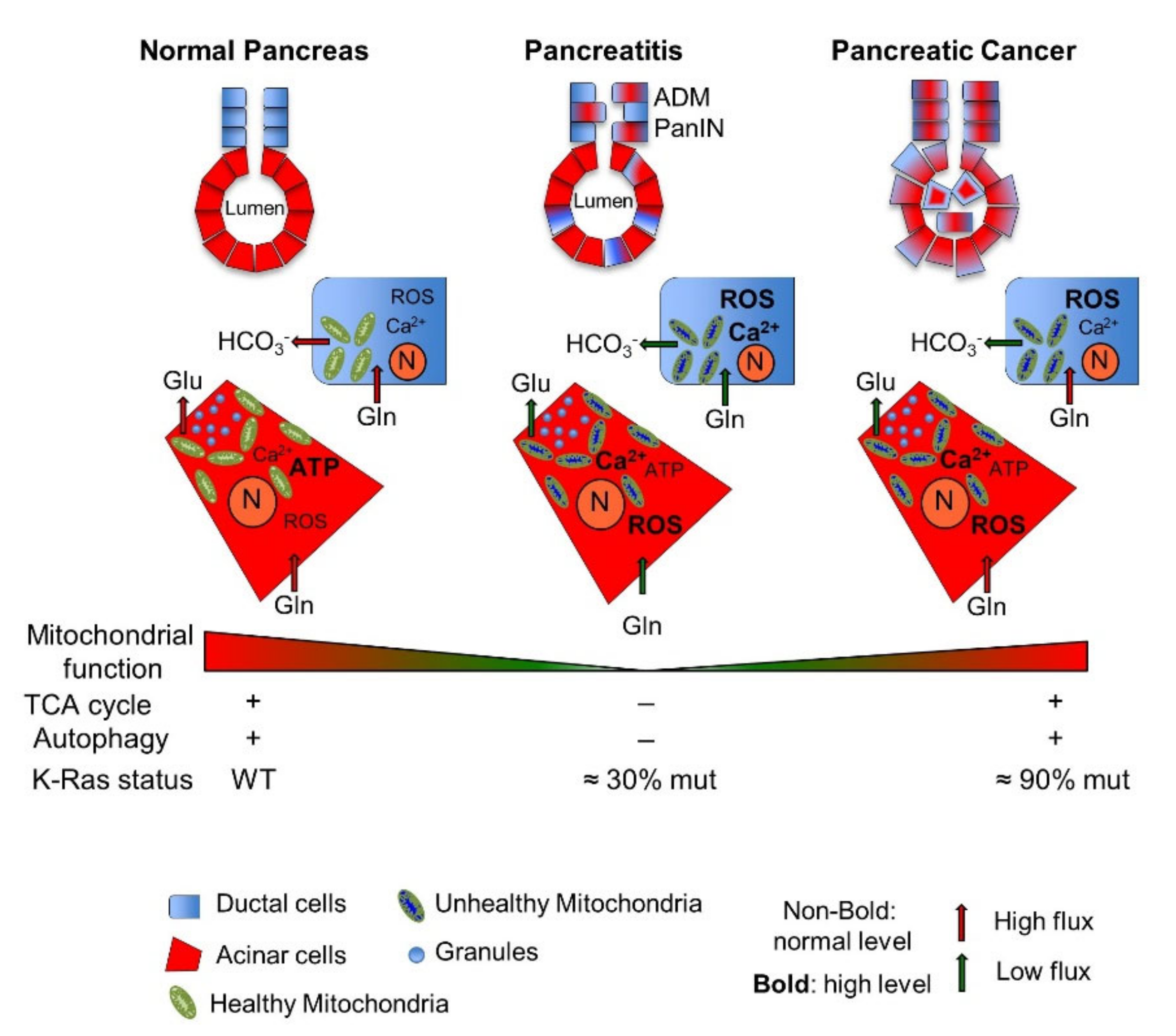
2.1. Pancreatic Mitochondrial Dysfunction: From Pathological Pancreatic Conditions to Pancreatic Cancer
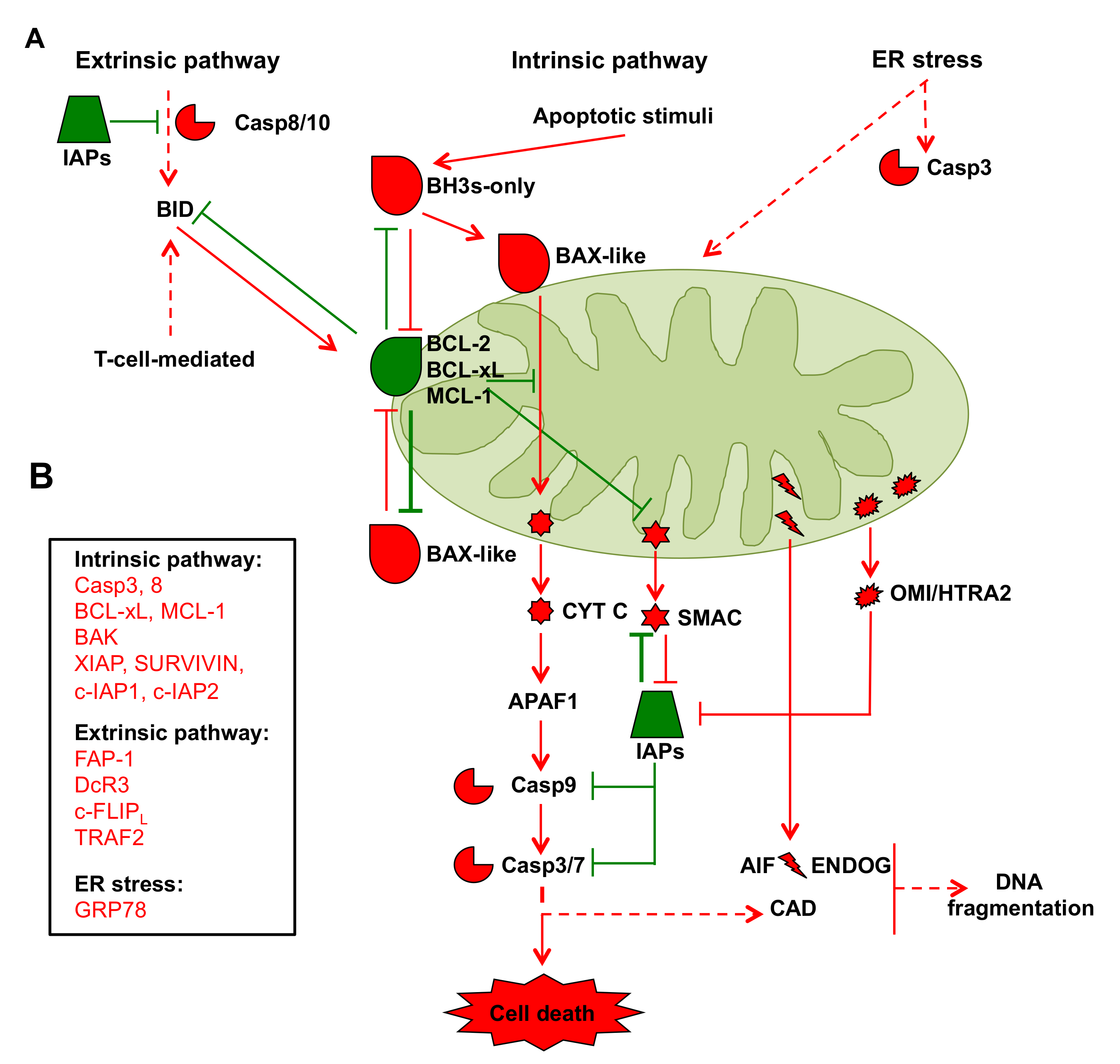
2.2. Mitochondria in Cancer Cell Apoptosis
2.3. mtDNA in Pancreatic Cancer Cells
2.4. Mitochondrial Dynamics in Pancreatic Cancer
2.5. Pancreatic Cancer Stem Cells
2.6. Cross-Talk between Pancreatic Cancer Cells and Stromatic Cells: Mitochondrial Metabolism in Perspective
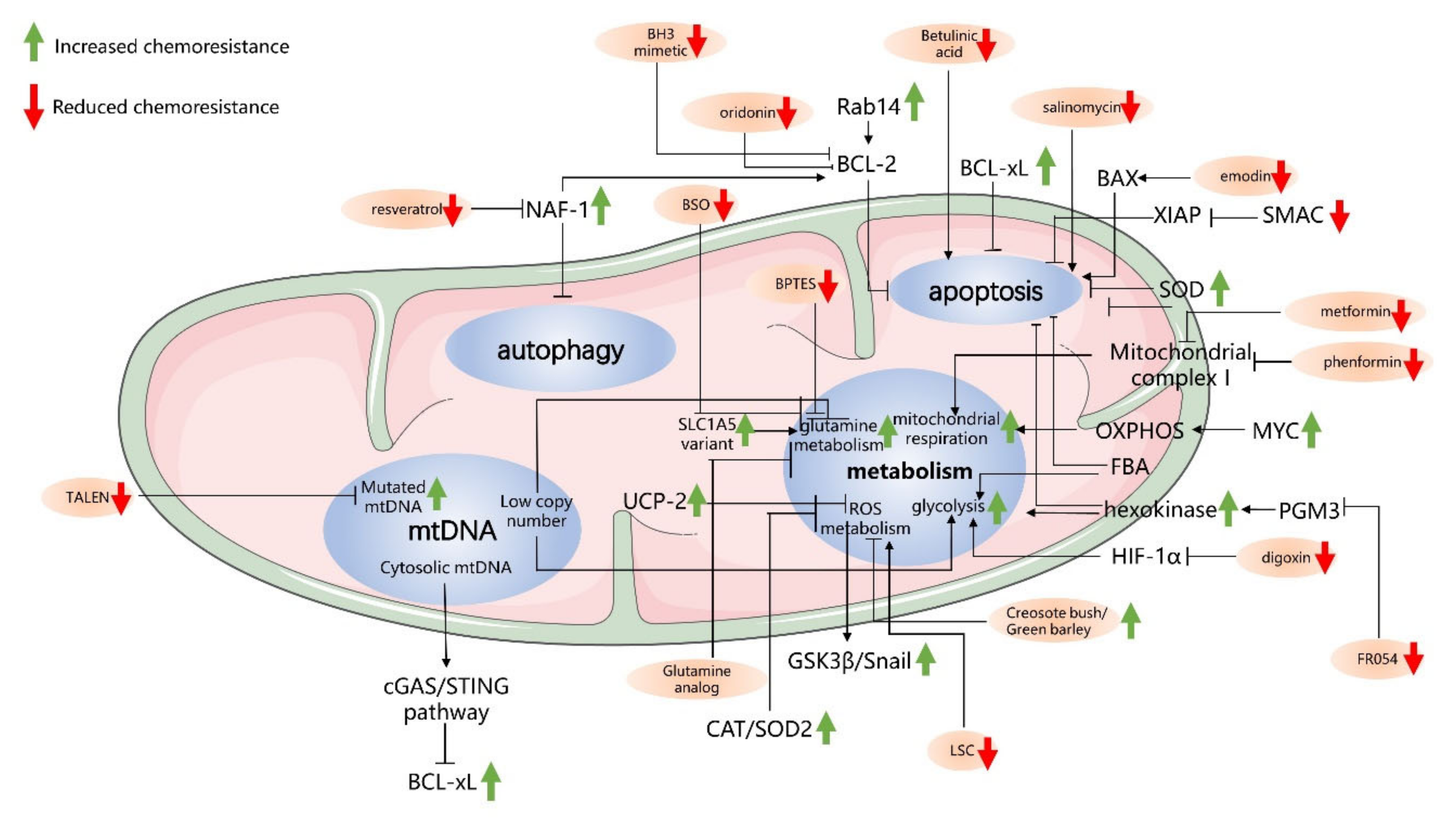
3. Mitochondria-Driven Mechanisms of Drug Resistance in Pancreatic Cancer Cells
3.1. Antiapoptosis-Related Mechanisms in Mitochondria
3.2. Autophagy-Related Mechanisms in Mitochondria and Chemoresistance
3.3. Cancer Metabolism and Chemoresistance
3.4. mtDNA and Chemoresistance in PC Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Process | Related Targets | Function on Chemoresistance | Reference |
|---|---|---|---|
| Apoptosis | BCL-xL | Inducing chemoresistance | [172] |
| RAB14 | Reducing chemosensitivity | [171] | |
| SMAC | Enhancing apoptosis and chemosensitivity | [175,220] | |
| Autophagy and mitophagy | NAF-1 | Regulating autophagy and inducing chemoresistance | [179,182] |
| Mitophagy | Either promoting glycolysis and PC progression or causing the loss of mitochondrial function and chemosensitivity | [188,191] | |
| Metabolism | Mitochondrial Complex I | Contributing to chemoresistance | [195,196] |
| Glycolytic enzymes including HK and FBA | Inducing chemoresistance | [17,192,208] | |
| ROS | Mediating the activation of AKT/GSK3β/Snail signaling and contributing to gemcitabine resistance | [201] | |
| Glutaminase | Promoting tumor proliferation and chemoresistance | [204] | |
| mtDNA-related | mtDNA mutations | Leading to the destruction of the electron transfer chain and OXPHOS inefficiency and enhancing glucose uptake and chemoresistance | [213,219] |
| Low mtDNA copy number | Enhancing the switch from mitochondrial respiration to the Warburg effect | [213] | |
| Released mtDNA | Activating the cGAS/STING-mediated cytosolic DNA-sensing pathway that leads to IFN-β transcription and IFN-β secretion | [216] |
4. Targeting Mitochondria to Overcome Chemoresistance
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pandiri, A.R. Overview of Exocrine Pancreatic Pathobiology. Toxicol. Pathol. 2014, 42, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Paoli, C.; Carrer, A. Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers 2020, 12, 2606. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, S.; Niu, K.; Wang, J.; Dai, J.; Ganguly, A.; Gao, C.; Tian, Y.; Lin, Z.; Yang, X.; Zhang, X.; et al. LINC00671 suppresses cell proliferation and metastasis in pancreatic cancer by inhibiting AKT and ERK signaling pathway. Cancer Gene Ther. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Vargas, T.R.; Apetoh, L. Danger signals: Chemotherapy enhancers? Immunol. Rev. 2017, 280, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, C.S.; Traub, B.; Hackert, T.; Klaiber, U.; Strobel, O.; Büchler, M.W.; Neoptolemos, J.P. Adjuvant and neoadjuvant chemotherapy in pancreatic ductal adenocarcinoma. J. Pancreatol. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Fesler, A.; Ju, J. Development of microRNA-based therapy for pancreatic cancer. J. Pancreatol. 2019, 2, 147–151. [Google Scholar] [CrossRef]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Binenbaum, Y.; Na’Ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Yang, G.; Qiu, J.; Luan, J.; Zhang, Y.; You, L.; Feng, M.; Zhao, F.; Liu, Y.; Cao, Z.; et al. Novel discoveries targeting gemcitabine-based chemoresistance and new therapies in pancreatic cancer: How far are we from the destination? Cancer Med. 2019, 8, 6403–6413. [Google Scholar] [CrossRef] [Green Version]
- Rauchwerger, D.R.; Firby, P.S.; Hedley, D.W.; Moore, M.J. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res. 2000, 60, 6075–6079. [Google Scholar] [PubMed]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The challenge of drug resistance in pancreatic ductal adenocarcinoma: A current overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [PubMed]
- Du, J.; Gu, J.; Li, J. Mechanisms of drug resistance of pancreatic ductal adenocarcinoma at different levels. Biosci. Rep. 2020, 40, BSR20200401. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. 2017, 114, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.B.; Yang, Y.; Zhao, Y.-P.; Zhang, T.-P.; Liao, Q.; Shu, H. Recent studies of 5-fluorouracil resistance in pancreatic cancer. World J. Gastroenterol. 2014, 20, 15682–15690. [Google Scholar] [CrossRef]
- Bauduin, H.; Colin, M.; Dumont, J. Energy sources for protein synthesis and enzymatic secretion in rat pancreas in vitro. Biochim. Biophys. Acta 1969, 174, 722–733. [Google Scholar] [CrossRef]
- Voronina, S.G.; Barrow, S.L.; Simpson, A.W.; Gerasimenko, O.V.; Xavier, G.D.S.; Rutter, G.A.; Petersen, O.H.; Tepikin, A.V. Dynamic Changes in Cytosolic and Mitochondrial ATP Levels in Pancreatic Acinar Cells. Gastroenterology 2010, 138, 1976–1987. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Ashby, M.C.; Erdemli, G.; Petersen, O.H.; Tepikin, A.V. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001, 20, 1863–1874. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.H. Specific mitochondrial functions in separate sub-cellular domains of pancreatic acinar cells. Pflügers Archiv 2012, 464, 77–87. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.P.; Renard-Rooney, D.C.; Hajnóczky, G.; Robb-Gaspers, L.D.; Lin, C.; Rooney, T.A. Subcellular Organization of Calcium Signalling in Hepatocytes and the Intact Liver. Ciba Found. Symp. 2007, 188, 18–49. [Google Scholar] [CrossRef]
- Criddle, D.N.; Gerasimenko, J.V.; Baumgartner, H.K.; Jaffar, M.; Voronina, S.; Sutton, R.; Petersen, O.H.; Gerasimenko, O.V. Calcium signalling and pancreatic cell death: Apoptosis or necrosis? Cell Death Differ. 2007, 14, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial Dysfunction, Through Impaired Autophagy, Leads to Endoplasmic Reticulum Stress, Deregulated Lipid Metabolism, and Pancreatitis in Animal Models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.A.; Cash, N.J.; Ouyang, Y.; Morton, J.C.; Chvanov, M.; Latawiec, D.; Awais, M.; Tepikin, A.V.; Sutton, R.; Criddle, D.N. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J. Biol. Chem. 2018, 293, 8032–8047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maléth, J.; Rakonczay, Z.; Venglovecz, V.; Dolman, N.J.; Hegyi, P. Central role of mitochondrial injury in the pathogenesis of acute pancreatitis. Acta Physiol. 2012, 207, 226–235. [Google Scholar] [CrossRef]
- Voronina, S.G.; Barrow, S.L.; Gerasimenko, O.V.; Petersen, O.H.; Tepikin, A.V. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: Comparison of different modes of evaluating DeltaPsim. J. Biol. Chem. 2004, 279, 27327–27338. [Google Scholar] [CrossRef] [Green Version]
- Cassano, G.B.; Hansson, E. Uptake of [14C]glutamine in the tissues of the mouse studied by whole-body autoradiography. J. Neurochem. 1965, 12, 851–855. [Google Scholar] [CrossRef]
- Rooman, I.; Lutz, C.; Pinho, A.V.; Huggel, K.; Reding, T.; Lahoutte, T.; Verrey, F.; Graf, R.; Camargo, S.M. Amino acid transporters expression in acinar cells is changed during acute pancreatitis. Pancreatology 2013, 13, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Araya, S.; Kuster, E.; Gluch, D.; Mariotta, L.; Lutz, C.; Reding, T.V.; Graf, R.; Verrey, F.; Camargo, S.M. Exocrine pancreas glutamate secretion help to sustain enterocyte nutritional needs under protein restriction. Am. J. Physiol. Liver Physiol. 2018, 314, G517–G536. [Google Scholar] [CrossRef]
- Maleth, J.; Venglovecz, V.; Razga, Z.; Tiszlavicz, L.; Rakonczay, Z.; Hegyi, P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 2010, 60, 136–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criddle, D.N. Keeping mitochondria happy—Benefits of a pore choice in acute pancreatitis. J. Physiol. 2019, 597, 5741–5742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maléth, J.; Hegyi, P. Calcium signaling in pancreatic ductal epithelial cells: An old friend and a nasty enemy. Cell Calcium 2014, 55, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Roux, C.; Riganti, C.; Borgogno, S.F.; Curto, R.; Curcio, C.; Catanzaro, V.; Digilio, G.; Padovan, S.; Puccinelli, M.P.; Isabello, M.; et al. Endogenous glutamine decrease is associated with pancreatic cancer progression. Oncotarget 2017, 8, 95361–95376. [Google Scholar] [CrossRef]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic beta-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H. Transcribing β-cell mitochondria in health and disease. Mol. Metab. 2017, 6, 1040–1051. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; Van De Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Dos Santos, M.S.; Exposito, R.T.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Ji, B. Ras Activity in Acinar Cells Links Chronic Pancreatitis and Pancreatic Cancer. Clin. Gastroenterol. Hepatol. 2009, 7, S40–S43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, M.; Maisonneuve, P.; Lowenfels, A.B. K-Ras Mutations and Benign Pancreatic Disease. Int. J. Pancreatol. 2000, 27, 093–104. [Google Scholar] [CrossRef]
- Berthelemy, P.; Bouisson, M.; Escourrou, J.; Vaysse, N.; Rumeau, J.L.; Pradayrol, L. Identification of K-ras Mutations in Pancreatic Juice in the Early Diagnosis of Pancreatic Cancer. Ann. Intern. Med. 1995, 123, 188–191. [Google Scholar] [CrossRef]
- Nakaizumi, A.; Uehara, H.; Takenaka, A.; Uedo, N.; Sakai, N.; Yano, H.; Ohigashi, H.; Ishikawa, O.; Ishiguro, S.; Sugano, K.; et al. Diagnosis of pancreatic cancer by cytology and measurement of oncogene and tumor markers in pure pancreatic juice aspirated by endoscopy. Hepatogastroenterology 1999, 46, 31–37. [Google Scholar] [PubMed]
- Shi, C.; Pan, F.C.; Kim, J.N.; Washington, M.K.; Padmanabhan, C.; Meyer, C.T.; Kopp, J.L.; Sander, M.; Gannon, M.; Beauchamp, D.R.; et al. Differential Cell Susceptibilities to Kras(G12D) in the Setting of Obstructive Chronic Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 579–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Löhr, M.; Klöppel, G.; Maisonneuve, P.; Lowenfels, A.B.; Lüttges, J. Frequency of K-ras Mutations in Pancreatic Intraductal Neoplasias Associated with Pancreatic Ductal Adenocarcinoma and Chronic Pancreatitis: A Meta-Analysis. Neoplasia 2005, 7, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Deramaudt, T.; Rustgi, A.K. Mutant KRAS in the initiation of pancreatic cancer. Biochim. Biophys. Acta 2005, 1756, 97–101. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, P.; Petersen, O.H. The Exocrine Pancreas: The Acinar-Ductal Tango in Physiology and Pathophysiology. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 1–30. [Google Scholar] [CrossRef]
- Mukherjee, R.; Criddle, D.; Gukvoskaya, A.; Pandol, S.; Petersen, O.; Sutton, R. Mitochondrial injury in pancreatitis. Cell Calcium 2008, 44, 14–23. [Google Scholar] [CrossRef]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.R.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwahashi, K.; Hikita, H.; Makino, Y.; Shigekawa, M.; Ikezawa, K.; Yoshioka, T.; Kodama, T.; Sakamori, R.; Tatsumi, T.; Takehara, T. Autophagy impairment in pancreatic acinar cells causes zymogen granule accumulation and pancreatitis. Biochem. Biophys. Res. Commun. 2018, 503, 2576–2582. [Google Scholar] [CrossRef]
- Alberghina, L.; Gaglio, D.; Gelfi, C.; Moresco, R.M.; Mauri, G.; Bertolazzi, P.; Messa, C.; Gilardi, M.C.; Chiaradonna, F.; Vanoni, M. Cancer cell growth and survival as a system-level property sustained by enhanced glycolysis and mitochondrial metabolic remodeling. Front. Physiol. 2012, 3, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [Green Version]
- Kodydkova, J.; Vavrova, L.; Stankova, B.; Macasek, J.; Krechler, T.; Zak, A. Antioxidant Status and Oxidative Stress Markers in Pancreatic Cancer and Chronic Pancreatitis. Pancreas 2013, 42, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. KRas, ROS and the initiation of pancreatic cancer. Small GTPases 2016, 8, 38–42. [Google Scholar] [CrossRef]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nat. Cell Biol. 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, G.; Kamada, P.; Chari, S.T. Prevalence of Diabetes Mellitus in Pancreatic Cancer Compared to Common Cancers. Pancreas 2013, 42, 198–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, G.; Rauenzahn, S.; Maitra, A. In vitro models of pancreatic cancer for translational oncology research. Expert Opin. Drug Discov. 2009, 4, 429–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.M.; Tien, S.-C.; Hsieh, P.-K.; Jeng, Y.-M.; Chang, M.-C.; Chang, Y.-T.; Chen, Y.-J.; Lee, E.Y.-T.P.; Lee, W.-H. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab. 2019, 29, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nat. Cell Biol. 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Daemen, A.; Peterson, D.W.; Sahu, N.; Mccord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.G.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Li, B.; Liu, J.; Fu, Y.; Luo, Y. Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardiello, F.; Gang, Y.; Palorini, R.; Li, Q.; Giampà, M.; Zhao, F.; You, L.; La Ferla, B.; De Vitto, H.; Guan, W.; et al. Hexosamine pathway inhibition overcomes pancreatic cancer resistance to gemcitabine through unfolded protein response and EGFR-Akt pathway modulation. Oncogene 2020, 39, 4103–4117. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose Induces Transketolase Flux to Promote Pancreatic Cancer Growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87. [Google Scholar] [CrossRef] [Green Version]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef]
- Yu, L.; Teoh, S.T.; Ensink, E.; Ogrodzinski, M.P.; Yang, C.; Vazquez, A.I.; Lunt, S.Y. Cysteine catabolism and the serine biosynthesis pathway support pyruvate production during pyruvate kinase knockdown in pancreatic cancer cells. Cancer Metab. 2019, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mattaini, K.R.; Sullivan, M.R.; Heiden, M.G.V. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- De Vitto, H.; Pérez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumor Biol. 2015, 37, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.M.-Y.; Ferreira, R.M.M.; Almagro, J.; Evan, T.; Legrave, N.; Thin, M.Z.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P.; et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Souba, W.W. Glutamine and Cancer. Ann. Surg. 1993, 218, 715–728. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef]
- Yang, S.; Hwang, S.; Kim, M.; Bin Seo, S.; Lee, J.-H.; Jeong, S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018, 9, 55. [Google Scholar] [CrossRef]
- Kam, P.C.A.; Ferch, N.I. Apoptosis: Mechanisms and clinical implications. Anaesthesia 2000, 55, 1081–1093. [Google Scholar] [CrossRef]
- Cory, S.; Huang, D.C.S.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Cory, S. The BCL-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Gerhard, M.C.; Schmid, R.M.; Häcker, G. Analysis of the cytochrome c-dependent apoptosis apparatus in cells from human pancreatic carcinoma. Br. J. Cancer 2002, 86, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cao, Z.; Yan, H.; Wood, W.C. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: Implication for cancer specific therapy. Cancer Res. 2003, 63, 6815–6824. [Google Scholar]
- Westphal, S.; Kalthoff, H. Apoptosis: Targets in Pancreatic Cancer. Mol. Cancer 2003, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Campani, D.; Esposito, I.; Boggi, U.; Cecchetti, D.; Menicagli, M.; De Negri, F.; Colizzi, L.; Del Chiaro, M.; Mosca, F.; Fornaciari, G.; et al. Bcl-2 expression in pancreas development and pancreatic cancer progression. J. Pathol. 2001, 194, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wang, B.; Gu, S.; Li, X.; Sun, S. Expression of Beclin 1 and Bcl-2 in pancreatic neoplasms and its effect on pancreatic ductal adenocarcinoma prognosis. Oncol. Lett. 2017, 14, 7849–7861. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Lu, Z.; Andrén-Sandberg, A.; Berberat, P.; Zimmermann, A.; Adler, G.; Schmid, R.; Büchler, M.W. Moderate activation of the apoptosis inhibitor BCL-xL worsens the prognosis in pancreatic cancer. Ann. Surg. 1998, 228, 780–787. [Google Scholar] [CrossRef]
- Ikezawa, K.; Hikita, H.; Shigekawa, M.; Iwahashi, K.; Eguchi, H.; Sakamori, R.; Tatsumi, T.; Takehara, T. Increased BCL-xL Expression in Pancreatic Neoplasia Promotes Carcinogenesis by Inhibiting Senescence and Apoptosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 185–200. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Liu, S.; Kleeff, J.; Friess, H.; Büchler, M.W. Acquired resistance of pancreatic cancer cells towards 5-Fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology 2002, 62, 354–362. [Google Scholar] [CrossRef]
- Friess, H.; Lu, Z.; Graber, H.U.; Zimmermann, A.; Adler, G.; Korc, M.; Schmid, R.M.; Büchler, M.W. bax, but notbcl-2, influences the prognosis of human pancreatic cancer. Gut 1998, 43, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graber, H. Bak expression and cell death occur in peritumorous tissue but not in pancreatic cancer cells. J. Gastrointest. Surg. 1999, 3, 74–81. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.-G. Somatic Mitochondrial DNA Mutations in Mammalian Aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Wong, L.-J.C. Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion 2007, 7, 45–52. [Google Scholar] [CrossRef]
- Rahman, S.; Poulton, J.; Marchington, D.; Suomalainen, A. Decrease of 3243 A→G mtDNA Mutation from Blood in MELAS Syndrome: A Longitudinal Study. Am. J. Hum. Genet. 2001, 68, 238–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Singh, K.K. Mitochondrial DNA Mutations in Aging, Disease and Cancer; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Dianov, G.L.; Souza-Pinto, N.; Nyaga, S.G.; Thybo, T.; Stevnsner, T.; Bohr, V.A. Base excision repair in nuclear and mitochondrial DNA. Base Excision Repair 2001, 68, 285–297. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-C.; Yin, P.-H.; Lin, J.-C.; Wu, C.-C.; Chen, C.-Y.; Wu, C.-W.; Chi, C.-W.; Tam, T.-N.; Wei, Y.-H. Mitochondrial Genome Instability and mtDNA Depletion in Human Cancers. Ann. N. Y. Acad. Sci. 2005, 1042, 109–122. [Google Scholar] [CrossRef]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navaglia, F.; Basso, D.; Fogar, P.; Sperti, C.; Greco, E.; Stranges, A.; Falda, A.; Pizzi, S.; Parenti, A.; Pedrazzoli, S.; et al. Mitochondrial DNA D-Loop in Pancreatic Cancer: Somatic Mutations Are Epiphenomena While the Germline 16519 T Variant Worsens Metabolism and Outcome. Am. J. Clin. Pathol. 2006, 126, 593–601. [Google Scholar] [CrossRef]
- Maassen, J.A.; Janssen, G.M.C.; Hart, L.M. ’T Molecular mechanisms of mitochondrial diabetes (MIDD). Ann. Med. 2005, 37, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-K.; Chen, S.-D.; Wang, P.-W.; Wei, Y.-H.; Lee, C.-F.; Chen, T.-L.; Chuang, Y.-C.; Tan, T.-Y.; Chang, K.-C.; Liou, C.-W. Increased Oxidative Damage with Altered Antioxidative Status in Type 2 Diabetic Patients Harboring the 16189 T to C Variant of Mitochondrial DNA. Ann. N. Y. Acad. Sci. 2005, 1042, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Weinstein, S.J.; Virtamo, J.; Lan, Q.; Liu, C.-S.; Cheng, W.-L.; Rothman, N.; Albanes, D.; Stolzenberg-Solomon, R.Z. Mitochondrial DNA Copy Number and Pancreatic Cancer in the Alpha-Tocopherol Beta-Carotene Cancer Prevention Study. Cancer Prev. Res. 2011, 4, 1912–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.M.; Vrieling, A.; Lubin, J.H.; Kraft, P.; Mendelsohn, J.B.; Hartge, P.; Canzian, F.; Steplowski, E.; Arslan, A.A.; Gross, M.; et al. Cigarette Smoking and Pancreatic Cancer: A Pooled Analysis From the Pancreatic Cancer Cohort Consortium. Am. J. Epidemiol. 2009, 170, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-C.; Lu, C.-Y.; Fahn, H.-J.; Wei, Y.-H. Aging- and smoking-associated alteration in the relative content of mitochondrial DNA in human lung. FEBS Lett. 1998, 441, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.T.; Bracci, P.M.; Holly, E.A.; Chu, C.; Poon, A.; Wan, E.; White, K.; Kwok, P.-Y.; Pawlikowska, L.; Tranah, G.J. Mitochondrial DNA Sequence Variation and Risk of Pancreatic Cancer. Cancer Res. 2012, 72, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.B.; Song, J.J.; Hempen, P.M.; Parmigiani, G.; Hruban, R.H.; Kern, S.E. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001, 61, 1299–1304. [Google Scholar] [PubMed]
- Sui, G.; Zhou, S.; Wang, J.; Canto, M.; Lee, E.E.; Eshleman, J.R.; A Montgomery, E.; Sidransky, D.; Califano, J.A.; Maitra, A. Mitochondrial DNA mutations in preneoplastic lesions of the gastrointestinal tract: A biomarker for the early detection of cancer. Mol. Cancer 2006, 5, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassauei, K.; Habbe, N.; Mullendore, M.E.; Karikari, C.A.; Maitra, A.; Feldmann, G. Mitochondrial DNA Mutations in Pancreatic Cancer. Int. J. Pancreatol. 2006, 37, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Hardie, R.-A.; Initiative, A.P.C.G.; Van Dam, E.; Cowley, M.; Han, T.-L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Marín-Hernández, A.; Goyri-Aguirre, M.; Anaya-Ruiz, M.; Reyes-Leyva, J.; Cortés-Hernández, P. Mitochondrial dynamics and cancer. Tumor Biol. 2017, 39, 1010428317698391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J. Cell Biol. 1968, 37, 345–369. [Google Scholar] [CrossRef] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Purnell, P.R.; Fox, H.S. Autophagy-mediated turnover of Dynamin-related Protein 1. BMC Neurosci. 2013, 14, 86. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Frank, S. Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life 2008, 60, 448–455. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; De Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaradonna, F.; Gaglio, D.; Vanoni, M.; Alberghina, L. Expression of transforming K-Ras oncogene affects mitochondrial function and morphology in mouse fibroblasts. Biochim. Biophys. Acta 2006, 1757, 1338–1356. [Google Scholar] [CrossRef] [Green Version]
- Nagdas, S.; Kashatus, J.A.; Nascimento, A.; Hussain, S.S.; Trainor, R.E.; Pollock, S.R.; Adair, S.J.; Michaels, A.D.; Sesaki, H.; Stelow, E.B.; et al. Drp1 Promotes KRas-Driven Metabolic Changes to Drive Pancreatic Tumor Growth. Cell Rep. 2019, 28, 1845–1859. [Google Scholar] [CrossRef] [Green Version]
- Palorini, R.; De Rasmo, D.; Gaviraghi, M.; Danna, L.S.; Signorile, A.; Cirulli, C.; Chiaradonna, F.; Alberghina, L.; Papa, S. Oncogenic K-ras expression is associated with derangement of the cAMP/PKA pathway and forskolin-reversible alterations of mitochondrial dynamics and respiration. Oncogene 2012, 32, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Palorini, R.; Votta, G.; Pirola, Y.; De Vitto, H.; De Palma, S.; Airoldi, C.; Vasso, M.; Ricciardiello, F.; Lombardi, P.P.; Cirulli, C.; et al. Protein Kinase A Activation Promotes Cancer Cell Resistance to Glucose Starvation and Anoikis. PLoS Genet. 2016, 12, e1005931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Yang, Y.; Bai, L.; Li, F.; Li, E. DRP1 upregulation promotes pancreatic cancer growth and metastasis through increased aerobic glycolysis. J. Gastroenterol. Hepatol. 2019, 35, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Chattaragada, M.S.; Riganti, C.; Sassoe, M.; Principe, M.; Santamorena, M.M.; Roux, C.; Curcio, C.; Evangelista, A.; Allavena, P.; Salvia, R.; et al. FAM49B, a novel regulator of mitochondrial function and integrity that suppresses tumor metastasis. Oncogene 2018, 37, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; Lucas, F.A.S.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Meng, Q.; Lu, D.; Liu, X.; Wang, Y.; Hao, J. Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/mTOR Signaling Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. MCT4 Defines a Glycolytic Subtype of Pancreatic Cancer with Poor Prognosis and Unique Metabolic Dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anania, S.; Peiffer, R.; Rademaker, G.; Hego, A.; Thiry, M.; Deldicque, L.; Francaux, M.; Maloujahmoum, N.; Agirman, F.; Bellahcène, A.; et al. Myoferlin Is a Yet Unknown Interactor of the Mitochondrial Dynamics’ Machinery in Pancreas Cancer Cells. Cancers 2020, 12, 1643. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, G.; Costanza, B.; Anania, S.; Agirman, F.; Maloujahmoum, N.; Di Valentin, E.; Goval, J.J.; Bellahcène, A.; Castronovo, V.; Peulen, O.; et al. Myoferlin Contributes to the Metastatic Phenotype of Pancreatic Cancer Cells by Enhancing Their Migratory Capacity through the Control of Oxidative Phosphorylation. Cancers 2019, 11, 853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademaker, G.; Hennequière, V.; Brohée, L.; Nokin, M.-J.; Lovinfosse, P.; Durieux, F.; Gofflot, S.; Bellier, J.; Costanza, B.; Herfs, M.; et al. Myoferlin controls mitochondrial structure and activity in pancreatic ductal adenocarcinoma, and affects tumor aggressiveness. Oncogene 2018, 37, 4398–4412. [Google Scholar] [CrossRef] [Green Version]
- Turtoi, A.; Blomme, A.; Bellahcène, A.; Gilles, C.; Hennequière, V.; Peixoto, P.; Bianchi, E.; Noel, A.; De Pauw, E.; Lifrange, E.; et al. Myoferlin Is a Key Regulator of EGFR Activity in Breast Cancer. Cancer Res. 2013, 73, 5438–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.S.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Kheir, T.B.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.M.; Jajeh, J.; Guinan, P.; Rubenstein, M. In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res. 2009, 29, 4579–4588. [Google Scholar] [PubMed]
- Madhok, B.M.; Yeluri, S.; Perry, S.L.; Hughes, T.A.; Jayne, D.G. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br. J. Cancer 2010, 102, 1746–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2009, 120, 253–260. [Google Scholar] [CrossRef]
- Tataranni, T.; Agriesti, F.; Pacelli, C.; Ruggieri, V.; Laurenzana, I.; Mazzoccoli, C.; Della Sala, G.; Panebianco, C.; Pazienza, V.; Capitanio, N.; et al. Dichloroacetate Affects Mitochondrial Function and Stemness-Associated Properties in Pancreatic Cancer Cell Lines. Cells 2019, 8, 478. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nat. Cell Biol. 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Fu, Z.; Chen, R.; Zhao, X.; Zhou, Y.; Zeng, B.; Yu, M.; Zhou, Q.; Lin, Q.; Gao, W.; et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget 2015, 6, 31151–31163. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2014, 6, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Brandi, J.; Dando, I.; Pozza, E.D.; Biondani, G.; Jenkins, R.; Elliott, V.; Park, K.; Fanelli, G.; Zolla, L.; Costello, E.; et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J. Proteom. 2017, 150, 310–322. [Google Scholar] [CrossRef]
- Brandi, J.; Pozza, E.D.; Dando, I.; Biondani, G.; Robotti, E.; Jenkins, R.; Elliott, V.; Park, K.; Marengo, E.; Costello, E.; et al. Secretome protein signature of human pancreatic cancer stem-like cells. J. Proteom. 2016, 136, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Isayev, O.; Rausch, V.; Bauer, N.; Liu, L.; Fan, P.; Zhang, Y.; Gladkich, J.; Nwaeburu, C.C.; Mattern, J.; Mollenhauer, M.; et al. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget 2014, 5, 5177–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quint, K.; Tonigold, M.; Di Fazio, P.; Montalbano, R.; Lingelbach, S.; Rückert, F.; Alinger, B.; Ocker, M.; Neureiter, D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epitheli-al-mesenchymal transition. Int. J. Oncol. 2012, 41, 2093–2102. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Qin, R.; Wei, C.; Wang, M.; Shi, C.; Tian, R.; Peng, C. Pancreatic cancer cells resistant to chemoradiotherapy rich in “stem-cell-like” tumor cells. Dig. Dis. Sci. 2011, 56, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Duan, Q.; Zhang, Z.; Li, H.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell. Mol. Med. 2017, 21, 2055–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; Lucas, F.A.S.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Schniewind, B.; Christgen, M.; Kurdow, R.; Haye, S.; Kremer, B.; Kalthoff, H.; Ungefroren, H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int. J. Cancer 2004, 109, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Ge, C. Rab14 overexpression regulates gemcitabine sensitivity through regulation of Bcl-2 and mitochondrial function in pancreatic cancer. Virchows Arch. 2019, 474, 59–69. [Google Scholar] [CrossRef]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-Binding Proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef]
- Gundry, C.; Marco, S.; Rainero, E.; Miller, B.; Dornier, E.; Mitchell, L.; Caswell, P.T.; Campbell, A.D.; Hogeweg, A.; Sansom, O.J.; et al. Phosphorylation of Rab-coupling protein by LMTK3 controls Rab14-dependent EphA2 trafficking to promote cell:cell repulsion. Nat. Commun. 2017, 8, 14646. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c–Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Hashim, Y.M.; Vangveravong, S.; Sankpal, N.V.; Binder, P.S.; Liu, J.; Goedegebuure, S.P.; Mach, R.H.; Spitzer, D.; Hawkins, W.G. The Targeted SMAC Mimetic SW IV-134 is a strong enhancer of standard chemotherapy in pancreatic cancer. J. Exp. Clin. Cancer Res. 2017, 36, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Li, Y.; Jian, Z.; Xia, K.; Li, X.; Lv, X.; Pei, H.; Chen, Z.; Li, J. XIAP Is Related to the Chemoresistance and Inhibited Its Expression by RNA Interference Sensitize Pancreatic Carcinoma Cells to Chemotherapeutics. Pancreas 2006, 32, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yan, B.; Chen, K.; Jiang, Z.; Zhou, C.; Cao, J.; Qian, W.; Li, J.; Sun, L.; Ma, J.; et al. Resveratrol-Induced Downregulation of NAF-1 Enhances the Sensitivity of Pancreatic Cancer Cells to Gemcitabine via the ROS/Nrf2 Signaling Pathways. Oxid. Med. Cell. Longev. 2018, 2018, 1–16. [Google Scholar] [CrossRef]
- Holt, S.H.; Darash-Yahana, M.; Sohn, Y.S.; Song, L.; Karmi, O.; Tamir, S.; Michaeli, D.; Luo, Y.; Paddock, M.L.; Jennings, P.A.; et al. Activation of apoptosis in NAF-1-deficient human epithelial breast cancer cells. J. Cell Sci. 2016, 129, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, T.; Cheng, L.; Xiao, Y.; Qian, W.; Li, J.; Wu, Z.; Wang, Z.; Xu, Q.; Duan, W.; Wong, L.; et al. NAF-1 Inhibition by Resveratrol Suppresses Cancer Stem Cell-Like Properties and the Invasion of Pancreatic Cancer. Front. Oncol. 2020, 10, 1038. [Google Scholar] [CrossRef]
- Iosub-Amir, A.; Bai, F.; Sohn, Y.-S.; Song, L.; Tamir, S.; Marjault, H.-B.; Mayer, G.; Karmi, O.; Jennings, P.A.; Mittler, R.; et al. The anti-apoptotic proteins NAF-1 and iASPP interact to drive apoptosis in cancer cells. Chem. Sci. 2019, 10, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Tamir, S.; Rotem-Bamberger, S.; Katz, C.; Morcos, F.; Hailey, K.L.; Zuris, J.A.; Wang, C.; Conlan, A.R.; Lipper, C.H.; Paddock, M.L.; et al. Integrated strategy reveals the protein interface between cancer targets Bcl-2 and NAF-1. Proc. Natl. Acad. Sci. USA 2014, 111, 5177–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2009, 29, 606–618. [Google Scholar] [CrossRef]
- Yan, C.; Li, T.-S. Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res. 2018, 38, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Luo, L.; Guo, C.-Y.; Goto, S.; Urata, Y.; Shao, J.-H.; Li, T.-S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Naik, P.P.; Panigrahi, D.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Sethi, G.; Bhutia, S.K. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: Its implication in cancer therapeutics. Cell. Mol. Life Sci. 2019, 76, 1641–1652. [Google Scholar] [CrossRef]
- Humpton, T.J.; Alagesan, B.; DeNicola, G.M.; Lu, D.; Yordanov, G.N.; Leonhardt, C.S.; Yao, M.A.; Alagesan, P.; Zaatari, M.N.; Park, Y.; et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov. 2019, 9, 1268–1287. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, Y.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, Å.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Peng, Y.; Zhu, Y.; Xu, D.; Zhu, F.; Xu, W.; Chen, Q.; Zhu, X.; Liu, T.; Hou, C.; et al. Glycolysis promotes the progression of pancreatic cancer and reduces cancer cell sensitivity to gemcitabine. Biomed. Pharmacother. 2020, 121, 109521. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Dauer, P.; Gupta, V.; McGinn, O.; Arora, N.; Majumdar, K.; Iii, C.U.; Dalluge, J.; Dudeja, V.; Saluja, A.; et al. Microenvironment mediated alterations to metabolic pathways confer increased chemo-resistance in CD133+ tumor initiating cells. Oncotarget 2016, 7, 56324–56337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J. Mitochondrial Complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Masoud, R.; Lac, S.; Garcia, J.; Reyes-Castellanos, G.; Iovanna, J.; Carrier, A. Targeting mitochondrial energy metabolism in PDAC is a promising strategy to overcome resistance to chemotherapy. Pancreatology 2018, 18, S154. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Morán-Santibañez, K.; Vasquez, A.H.; Varela-Ramirez, A.; Henderson, V.; Sweeney, J.; Odero-Marah, V.; Fenelon, K.; Skouta, R. Larrea tridentata Extract Mitigates Oxidative Stress-Induced Cytotoxicity in Human Neuroblastoma SH-SY5Y Cells. Antioxidants 2019, 8, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Medina, B.E.; Lerma, D.; Hwang, M.; Ross, J.A.; Skouta, R.; Aguilera, R.J.; Kirken, R.A.; Varela-Ramirez, A.; Robles-Escajeda, E. Green barley mitigates cytotoxicity in human lymphocytes undergoing aggressive oxidative stress, via activation of both the Lyn/PI3K/Akt and MAPK/ERK pathways. Sci. Rep. 2019, 9, 6005. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, Y. Acquisition of resistance of pancreatic cancer cells to 2-methoxyestradiol is associated with the upregulation of manganese superoxide dismutase. Mol. Cancer Res. 2012, 10, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Yu, X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene 2018, 37, 5843–5857. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.-K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; A Lai, L.; Sullivan, Y.; Wong, M.; Wang, L.; Riddell, J.; Jung, L.; Pillarisetty, V.G.; Brentnall, T.A.; Pan, S. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci. Rep. 2017, 7, 7950. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2016, 8, 56081–56094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.; Fan, Z.; Cheng, H.; Huang, Q.; Yang, C.; Jin, K.; Luo, G.; Yu, X.; Liu, C. Hexokinase 2 dimerization and interaction with voltage-dependent anion channel promoted resistance to cell apoptosis induced by gemcitabine in pancreatic cancer. Cancer Med. 2019, 8, 5903–5915. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kang, H.J.; Lee, S.Y.; Chung, S.J.; Kang, S.; Chi, S.W.; Cho, S.; Lee, S.C.; Lee, C.-K.; Park, B.C.; et al. Glyceraldehyde-3-phosphate, a glycolytic intermediate, plays a key role in controlling cell fate via inhibition of caspase activity. Mol. Cells 2009, 28, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, T.C.; Gomez, M.L.; Germain, D. Mitohormesis, UPRmt, and the Complexity of Mitochondrial DNA Landscapes in Cancer. Cancer Res. 2019, 79, 6057–6066. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; John, J.C.S. The role of the mtDNA set point in differentiation, development and tumorigenesis. Biochem. J. 2016, 473, 2955–2971. [Google Scholar] [CrossRef]
- Giampazolias, E.; Zunino, B.; Dhayade, S.; Bock, F.; Cloix, C.; Cao, K.; Roca, A.; Lopez, J.; Ichim, G.; Proïcs, E.; et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat. Cell Biol. 2017, 19, 1116–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Lohard, S.; Bourgeois, N.; Maillet, L.; Gautier, F.; Fétiveau, A.; Lasla, H.; Nguyen, F.; Vuillier, C.; Dumont, A.; Moreau-Aubry, A.; et al. STING-dependent paracriny shapes apoptotic priming of breast tumors in response to anti-mitotic treatment. Nat. Commun. 2020, 11, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Mou, J.-J.; Peng, J.; Shi, Y.-Y.; Li, N.; Wang, Y.; Ke, Y.; Zhou, Y.-F.; Zhou, F.-X. Mitochondrial DNA content reduction induces aerobic glycolysis and reversible resistance to drug-induced apoptosis in SW480 colorectal cancer cells. Biomed. Pharmacother. 2018, 103, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, S.; Miyato, Y.; Shidara, Y.; Asoh, S.; Tokunaga, A.; Tajiri, T.; Ohta, S. Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 2009, 100, 1680–1687. [Google Scholar] [CrossRef]
- Fan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA Determines Chemoresistance by Reprogramming Metabolism and Regulating Mitochondrial Transcription. Cancer Res. 2019, 79, 1069–1084. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]
- Quinn, B.A.; Dash, R.; Sarkar, S.; Azab, B.; Bhoopathi, P.; Das, S.K.; Emdad, L.; Wei, J.; Pellecchia, M.; Sarkar, D.; et al. Pancreatic Cancer Combination Therapy Using a BH3 Mimetic and a Synthetic Tetracycline. Cancer Res. 2015, 75, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liu, H.; Xue, R.; Tang, W.; Zhang, S. BH3 Mimetic ABT-199 Enhances the Sensitivity of Gemcitabine in Pancreatic Cancer in vitro and in vivo. Dig. Dis. Sci. 2018, 63, 3367–3375. [Google Scholar] [CrossRef] [PubMed]
- Pandita, A.; Kumar, B.; Manvati, S.; Vaishnavi, S.; Singh, S.K.; Bamezai, R.N.K. Synergistic Combination of Gemcitabine and Dietary Molecule Induces Apoptosis in Pancreatic Cancer Cells and Down Regulates PKM2 Expression. PLoS ONE 2014, 9, e107154. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-L.; Bu, H.-Q.; Jin, H.-M.; Zhao, J.-F.; Li, Y.; Huang, H. Enhancement of the effects of gemcitabine against pancreatic cancer by oridonin via the mitochondrial caspase-dependent signaling pathway. Mol. Med. Rep. 2014, 10, 3027–3034. [Google Scholar] [CrossRef]
- Fuchs, D.; Daniel, V.; Sadeghi, M.; Opelz, G.; Naujokat, C. Salinomycin overcomes ABC transporter-mediated multidrug and apoptosis resistance in human leukemia stem cell-like KG-1a cells. Biochem. Biophys. Res. Commun. 2010, 394, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, D.; Heinold, A.; Opelz, G.; Daniel, V.; Naujokat, C. Salinomycin induces apoptosis and overcomes apoptosis resistance in human cancer cells. Biochem. Biophys. Res. Commun. 2009, 390, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-N.; Liang, Y.; Zhou, L.-J.; Chen, S.-P.; Chen, G.; Zhang, T.-P.; Kang, T.; Zhao, Y.-P. Combination of salinomycin and gemcitabine eliminates pancreatic cancer cells. Cancer Lett. 2011, 313, 137–144. [Google Scholar] [CrossRef]
- Liu, D.-L.; Bu, H.; Li, H.; Chen, H.; Guo, H.-C.; Wang, Z.-H.; Tong, H.-F.; Ni, Z.-L.; Liu, H.-B.; Lin, S.-Z. Emodin reverses gemcitabine resistance in pancreatic cancer cells via the mitochondrial apoptosis pathway in vitro. Int. J. Oncol. 2011, 40, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candido, S.; Abrams, S.L.; Steelman, L.; Lertpiriyapong, K.; Martelli, A.M.; Cocco, L.; Ratti, S.; Follo, M.Y.; Murata, R.M.; Rosalen, P.L.; et al. Metformin influences drug sensitivity in pancreatic cancer cells. Adv. Biol. Regul. 2018, 68, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Liang, C.; Sun, L.; Zhou, C.; Gao, L.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Anbil, S.; Bulin, A.-L.; Obaid, G.; Mai, Z.; Baglo, Y.; Rizvi, I.; Hasan, T. Modulation of redox metabolism negates cancer-associated fibroblasts-induced treatment resistance in a heterotypic 3D culture platform of pancreatic cancer. Biomaterials 2019, 222, 119421. [Google Scholar] [CrossRef]
- Hosseini, M.; Rezvani, H.R.; Aroua, N.; Bosc, C.; Farge, T.; Saland, E.; Guyonnet-Dupérat, V.; Zaghdoudi, S.; Jarrou, L.; Larrue, C.; et al. Targeting Myeloperoxidase Disrupts Mitochondrial Redox Balance and Overcomes Cytarabine Resistance in Human Acute Myeloid Leukemia. Cancer Res. 2019, 79, 5191–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Gao, Z.; Liu, X.; Agarwal, P.; Zhao, S.; Conroy, D.W.; Ji, G.; Yu, J.; Jaroniec, C.P.; Liu, Z.; et al. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat. Commun. 2018, 9, 562. [Google Scholar] [CrossRef]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The Mitochondrial Uncoupling Protein-2 Promotes Chemoresistance in Cancer Cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Shi, L.; Lin, W.; Lu, B.; Zhao, Y. UCP2 promotes proliferation and chemoresistance through regulating the NF-κB/β-catenin axis and mitochondrial ROS in gallbladder cancer. Biochem. Pharmacol. 2020, 172, 113745. [Google Scholar] [CrossRef] [PubMed]
- Pustylnikov, S.; Costabile, F.; Beghi, S.; Facciabene, A. Targeting mitochondria in cancer: Current concepts and immunotherapy approaches. Transl. Res. 2018, 202, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mitochondrial genome changes and neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Ricciardiello, F.; Yang, G.; Qiu, J.; Huang, H.; Xiao, J.; Cao, Z.; Zhao, F.; Liu, Y.; Luo, W.; et al. The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells. Cells 2021, 10, 497. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030497
Fu Y, Ricciardiello F, Yang G, Qiu J, Huang H, Xiao J, Cao Z, Zhao F, Liu Y, Luo W, et al. The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells. Cells. 2021; 10(3):497. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030497
Chicago/Turabian StyleFu, Yibo, Francesca Ricciardiello, Gang Yang, Jiangdong Qiu, Hua Huang, Jianchun Xiao, Zhe Cao, Fangyu Zhao, Yueze Liu, Wenhao Luo, and et al. 2021. "The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells" Cells 10, no. 3: 497. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030497