
The Effects of Warfarin and Direct Oral Anticoagulants on Systemic Vascular Calcification: A Review

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Warfarin
2.1. History
2.2. Mechanism of Action and Properties
3. Direct Oral Anticoagulants
3.1. History
3.2. Mechanism of Action and Properties
4. Advantages and Disadvantages of Warfarin and Direct Oral Anticoagulants
5. Vascular Calcification
5.1. Pathophysiology of Vascular Calcification
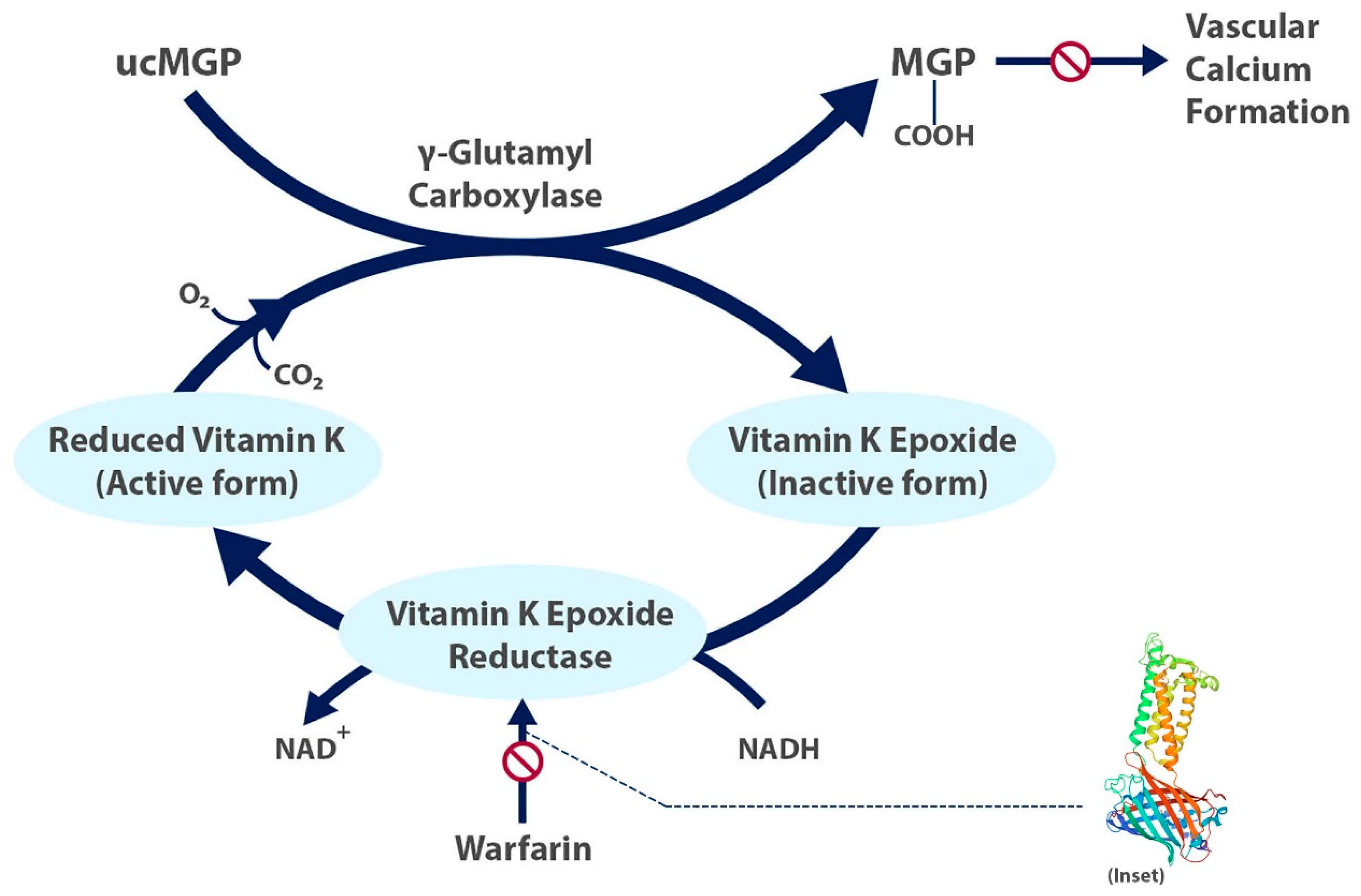
5.2. The Role of Vitamin K
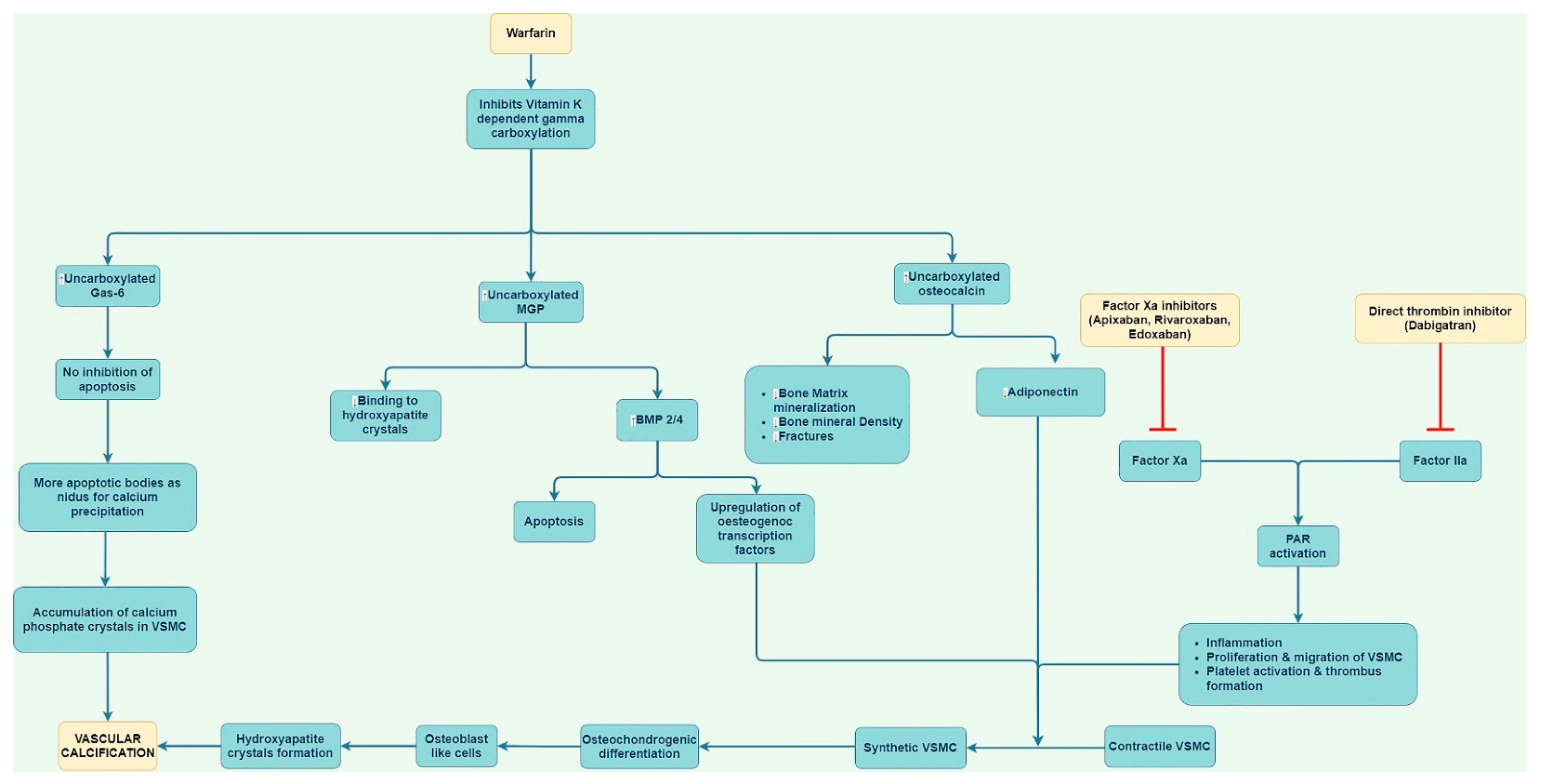
6. Mechanisms by Which Warfarin May Cause Calcification
6.1. Impact of Matrix Gla Protein
6.2. Other Mechanisms
7. Mechanisms by Which Direct Oral Anticoagulants May Prevent Vascular Calcification
8. Studies Linking Warfarin to Calcification
9. Effects of Warfarin vs. DOACs on Vascular Calcification
10. Effects of Warfarin and Direct Oral Anticoagulants on Valvular Calcification
11. Considerations in Patients with Kidney Disease
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Perreault, S.; de Denus, S.; White-Guay, B.; Côté, R.; Schnitzer, M.E.; Dubé, M.P.; Dorais, M.; Tardif, J.C. Oral anticoagulant prescription trends, profile use, and determinants of adherence in patients with atrial fibrillation. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 40–54. [Google Scholar] [CrossRef]
- Yu, Z.; Yu, L.; Shan, C. Trends of ambulatory oral anticoagulant prescription in five major cities of China, 2012–2017. BMC Health Serv. Res. 2020, 20, 1–6. [Google Scholar] [CrossRef]
- Poterucha, T.J.; Goldhaber, S.Z. Warfarin and vascular calcification. Am. J. Med. 2016, 129, 635.e1–635.e4. [Google Scholar] [CrossRef] [Green Version]
- Lim, G.B. Warfarin: From rat poison to clinical use. Nat. Rev. Cardiol. 2017. [Google Scholar] [CrossRef]
- Merck Manual Veterinary Manual. Sweet Clover Poisoning in Animals. 2021. Available online: https://www.merckvetmanual.com/toxicology/sweet-clover-poisoning/sweet-clover-poisoning-in-animals (accessed on 21 March 2021).
- Schofield, F.W. Damaged sweet clover: The cause of a new disease in cattle simulating hemorrhagic septicemia and blackleg. J. Am. Vet. Med. Assoc. 1924, 64, 553–575. [Google Scholar]
- Link, K.P. The discovery of dicumarol and its sequels. Circulation 1959, 19, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzung, B.G. Basic & Clinical Pharmacology, 13th ed.; McGraw-Hill: New York, NY, USA, 2017. [Google Scholar]
- Silberstein, L.E.; Anastasi, J. Hematology: Basic Principles and Practice E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Herman, D.; Locatelli, I.; Grabnar, I.; Peternel, P.; Stegnar, M.; Mrhar, A.; Breskvar, K.; Dolzan, V. Influence of CYP2C9 polymorphisms, demographic factors and concomitant drug therapy on warfarin metabolism and maintenance dose. Pharm. J. 2005, 5, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, S.; Shen, G.; Sukumar, N.; Krezel, A.M.; Li, W. Structural basis of antagonizing the vitamin K catalytic cycle for anticoagulation. Science 2021, 371. [Google Scholar] [CrossRef]
- Haycraft, J.B. IV On the action of a secretion obtained from the medicinal leech on the coagulation of the blood. Proc. R. Soc. Lond. 1883, 36, 478–487. [Google Scholar]
- Tuszynski, G.; Gasic, T.; Gasic, G. Isolation and characterization of antistasin. An inhibitor of metastasis and coagulation. J. Biol. Chem. 1987, 262, 9718–9723. [Google Scholar] [CrossRef]
- Cabral, K.P.; Ansell, J.E. The role of factor Xa inhibitors in venous thromboembolism treatment. Vasc. Health Risk Manag. 2015, 11, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokorney, S.D.; Simon, D.N.; Thomas, L.; Fonarow, G.C.; Kowey, P.R.; Chang, P.; Singer, D.E.; Ansell, J.; Blanco, R.G.; Gersh, B.; et al. Patients’ time in therapeutic range on warfarin among US patients with atrial fibrillation: Results from ORBIT-AF registry. Am. Heart J. 2015, 170, 141–148.e141. [Google Scholar] [CrossRef] [Green Version]
- Calabrò, P.; Gragnano, F.; Cesaro, A.; Marsico, F.; Pariggiano, I.; Patti, G.; Moscarella, E.; Cavallari, I.; Sardu, C.; Parato, V.M.; et al. Non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation and atrial thrombosis: An appraisal of current evidence. Arch. Cardiovasc. Dis. 2020, 113, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Bos, D.; Leening, M.J.; Kavousi, M.; Hofman, A.; Franco, O.H.; van de Lugt, A.; Vernooij, M.W.; Ikram, M.A. Comparison of atherosclerotic calcification in major vessel beds on the risk of all-cause and cause-specific mortality: The Rotterdam study. Circ. Cardiovasc. Imaging 2015, 8, e003843. [Google Scholar] [CrossRef] [Green Version]
- Iribarren, C.; Sidney, S.; Sternfeld, B.; Browner, W.S. Calcification of the aortic arch: Risk factors and association with coronary heart disease, stroke, and peripheral vascular disease. JAMA 2000, 283, 2810–2815. [Google Scholar] [CrossRef] [Green Version]
- Kohsaka, S.; Jin, Z.; Rundek, T.; Boden-Albala, B.; Homma, S.; Sacco, R.L.; Di Tullio, M.R. Impact of mitral annular calcification on cardiovascular events in a multiethnic community: The Northern Manhattan Study. JACC Cardiovasc. Imaging 2008, 1, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.B.; Falk, E.; Li, D.; Nasir, K.; Blaha, M.J.; Sandfort, V.; Rodriguez, C.J.; Ouyang, P.; Budoff, M. Statin trials, cardiovascular events, and coronary artery calcification: Implications for a trial-based approach to statin therapy in MESA. JACC Cardiovasc. Imaging 2018, 11, 221–230. [Google Scholar] [CrossRef]
- Andrews, J.; Psaltis, P.J.; Bayturan, O.; Shao, M.; Stegman, B.; Elshazly, M.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E.; Nicholls, S.J. Warfarin use is associated with progressive coronary arterial calcification: Insights from serial intravascular ultrasound. JACC Cardiovasc. Imaging 2018, 11, 1315–1323. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular calcification—New insights into its mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [Green Version]
- Magdič, J.; Cmor, N.; Kaube, M.; Hojs Fabjan, T.; Hauer, L.; Sellner, J.; Pikija, S. Intracranial Vertebrobasilar Calcification in Patients with Ischemic Stroke Is a Predictor of Recurrent Stroke, Vascular Disease, and Death: A Case-Control Study. Int. J. Environ. Res. Public Health 2020, 17, 2013. [Google Scholar] [CrossRef] [Green Version]
- McMullen, E.R.; Harms, P.W.; Lowe, L.; Fullen, D.R.; Chan, M.P. Clinicopathologic features and calcium deposition patterns in calciphylaxis. Am. J. Surg. Pathol. 2019, 43, 1273–1281. [Google Scholar] [CrossRef]
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 2012, 14, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toussaint, N.D.; Kerr, P.G. Vascular calcification and arterial stiffness in chronic kidney disease: Implications and management. Nephrology 2007, 12, 500–509. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.; Aster, J. Robbins Basic Pathology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in atherosclerotic plaque vulnerability: Friend or foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Geleijnse, J.M.; Vermeer, C.; Grobbee, D.E.; Schurgers, L.J.; Knapen, M.H.; van der Meer, I.M.; Hofman, A.; Witteman, J.C. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: The Rotterdam Study. J. Nutr. 2004, 134, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M. Matrix Gla-Protein (MGP) not only inhibits calcification in large arteries but also may be renoprotective: Connecting the dots. EBioMedicine 2016, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- O’Young, J.; Liao, Y.; Xiao, Y.; Jalkanen, J.; Lajoie, G.; Karttunen, M.; Goldberg, H.A.; Hunter, G.K. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 2011, 133, 18406–18412. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Chatrou, M.L.; Winckers, K.; Hackeng, T.M.; Reutelingsperger, C.P.; Schurgers, L.J. Vascular calcification: The price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev. 2012, 26, 155–166. [Google Scholar] [CrossRef]
- Proudfoot, D.; Shanahan, C.M. Molecular mechanisms mediating vascular calcification: Role of matrix Gla protein. Nephrology 2006, 11, 455–461. [Google Scholar] [CrossRef]
- Rennenberg, R.J.; Schurgers, L.J.; Kroon, A.A.; Stehouwer, C.D. Arterial calcifications. J. Cell. Mol. Med. 2010, 14, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Siltari, A.; Vapaatalo, H. Vascular calcification, vitamin K and warfarin therapy–possible or plausible connection? Basic Clin. Pharmacol. Toxicol. 2018, 122, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.-P.; van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Barrett, H.; O’Keeffe, M.; Kavanagh, E.; Walsh, M.; O’Connor, E.M. Is matrix Gla protein associated with vascular calcification? A systematic review. Nutrients 2018, 10, 415. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Teunissen, K.J.; Knapen, M.H.; Kwaijtaal, M.; van Diest, R.; Appels, A.; Reutelingsperger, C.P.; Cleutjens, J.P.; Vermeer, C. Novel conformation-specific antibodies against matrix γ-carboxyglutamic acid (Gla) protein: Undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1629–1633. [Google Scholar] [CrossRef] [Green Version]
- Marietta, M.; Coluccio, V.; Boriani, G.; Luppi, M. Effects of Anti-vitamin k oral anticoagulants on bone and cardiovascular health. Eur. J. Intern. Med. 2020, 79, 1–11. [Google Scholar] [CrossRef]
- Luo, X.H.; Zhao, L.L.; Yuan, L.Q.; Wang, M.; Xie, H.; Liao, E.Y. Development of arterial calcification in adiponectin-deficient mice: Adiponectin regulates arterial calcification. J. Bone Mineral. Res. 2009, 24, 1461–1468. [Google Scholar] [CrossRef]
- Marques, J.V.O.; Nalevaiko, J.Z.; Oliveira, M.F.; Raetsch, A.W.P.; Marques, G.L.; Petterle, R.R.; Moreira, C.A.; Borba, V.Z.C. Trabecular bone score (TBS) and bone mineral density in patients with long-term therapy with warfarin. Arch. Osteoporos. 2020, 15, 1–7. [Google Scholar] [CrossRef]
- Xiao, H.; Chen, J.; Duan, L.; Li, S. Role of emerging vitamin K-dependent proteins: Growth arrest-specific protein 6, Gla-rich protein and periostin. Int. J. Mol. Med. 2021, 47, 1–2. [Google Scholar] [PubMed]
- Esmon, C.T. Targeting factor Xa and thrombin: Impact on coagulation and beyond. Thromb. Haemost. 2014, 111, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, R.H.; Schurgers, L.J. New insights into the pros and cons of the clinical use of vitamin K antagonists (VKAs) versus direct oral anticoagulants (DOACs). Nutrients 2015, 7, 9538–9557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petzold, T.; Thienel, M.; Dannenberg, L.; Mourikis, P.; Helten, C.; Ayhan, A.; M’Pembele, R.; Achilles, A.; Trojovky, K.; Konsek, D. Rivaroxaban reduces arterial thrombosis by inhibition of FXa-driven platelet activation via protease activated receptor-1. Circ. Res. 2020, 126, 486–500. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Moustardas, P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Schafer, K.; Kostakis, A.; Liapis, C.D. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice. Cardiovasc. Drugs Ther. 2012, 26, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A. Apixaban versus warfarin in patients with atrial fibrillation. New Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef]
- Millenaar, D.; Bachmann, P.; Böhm, M.; Custodis, F.; Schirmer, S.H. Effects of edoxaban and warfarin on vascular remodeling: Atherosclerotic plaque progression and collateral artery growth. Vasc. Pharmacol. 2020, 127, 106661. [Google Scholar] [CrossRef]
- Namba, S.; Yamaoka-Tojo, M.; Kakizaki, R.; Nemoto, T.; Fujiyoshi, K.; Hashikata, T.; Kitasato, L.; Hashimoto, T.; Kameda, R.; Meguro, K. Effects on bone metabolism markers and arterial stiffness by switching to rivaroxaban from warfarin in patients with atrial fibrillation. Heart Vessel. 2017, 32, 977–982. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Joosen, I.A.; Laufer, E.M.; Chatrou, M.L.; Herfs, M.; Winkens, M.H.; Westenfeld, R.; Veulemans, V.; Krueger, T.; Shanahan, C.M. Vitamin K-antagonists accelerate atherosclerotic calcification and induce a vulnerable plaque phenotype. PLoS ONE 2012, 7, e43229. [Google Scholar]
- Bui, Q.M.; Daniels, L.B. A review of the role of breast arterial calcification for cardiovascular risk stratification in women. Circulation 2019, 139, 1094–1101. [Google Scholar] [CrossRef]
- Turner, M.E.; Holden, R.M. Risks of Vitamin K Antagonism: Mammograms as a Powerful Tool to Assess Calcification Progression. Am. Heart. Assoc. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tantisattamo, E.; Han, K.H.; O’Neill, W.C. Increased vascular calcification in patients receiving warfarin. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- COLE, W.G. Structure of growth plate and bone matrix. In Pediatric Bone; Elsevier: Amsterdam, The Netherlands, 2003; pp. 1–41. [Google Scholar]
- Han, K.H.; O’Neill, W.C. Increased peripheral arterial calcification in patients receiving warfarin. J. Am. Heart Assoc. 2016, 5, e002665. [Google Scholar] [CrossRef] [Green Version]
- Peeters, F.E.; Dudink, E.A.; Kimenai, D.M.; Weijs, B.; Altintas, S.; Heckman, L.I.; Mihl, C.; Schurgers, L.J.; Wildberger, J.E.; Meex, S.J. Vitamin K antagonists, non–vitamin K antagonist oral anticoagulants, and vascular calcification in patients with atrial fibrillation. TH Open Companion J. Thromb. Haemost. 2018, 2, e391–e398. [Google Scholar] [CrossRef] [PubMed]
- Plank, F.; Beyer, C.; Friedrich, G.; Stühlinger, M.; Hintringer, F.; Dichtl, W.; Wildauer, M.; Feuchtner, G. Influence of vitamin K antagonists and direct oral anticoagulation on coronary artery disease: A CTA analysis. Int. J. Cardiol. 2018, 260, 11–15. [Google Scholar] [CrossRef]
- Win, T.T.; Nakanishi, R.; Osawa, K.; Li, D.; Susaria, S.S.; Jayawardena, E.; Hamal, S.; Kim, M.; Broersen, A.; Kitslaar, P.H.; et al. Apixaban versus warfarin in evaluation of progression of atherosclerotic and calcified plaques (prospective randomized trial). Am. Heart J. 2019, 212, 129–133. [Google Scholar] [CrossRef]
- Hasific, S.; Øvrehus, K.A.; Gerke, O.; Hallas, J.; Busk, M.; Lambrechtsen, J.; Urbonaviciene, G.; Sand, N.P.R.; Nielsen, J.S.; Diederichsen, L. Extent of arterial calcification by conventional vitamin K antagonist treatment. PLoS ONE 2020, 15, e0241450. [Google Scholar] [CrossRef]
- Lee, J.; Nakanishi, R.; Li, D.; Shaikh, K.; Shekar, C.; Osawa, K.; Nezarat, N.; Jayawardena, E.; Blanco, M.; Chen, M. Randomized trial of rivaroxaban versus warfarin in the evaluation of progression of coronary atherosclerosis. Am. Heart J. 2018, 206, 127–130. [Google Scholar] [CrossRef]
- Annweiler, G.; Labriffe, M.; Ménager, P.; Ferland, G.; Brangier, A.; Annweiler, C. Intracranial calcifications under vitamin K antagonists or direct oral anticoagulants: Results from the French VIKING study in older adults. Maturitas 2020, 132, 35–39. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Caluwé, R.; Pyfferoen, L.; De Bacquer, D.; De Boeck, K.; Delanote, J.; De Surgeloose, D.; Van Hoenacker, P.; Van Vlem, B.; Verbeke, F. Multicenter randomized controlled trial of vitamin K antagonist replacement by rivaroxaban with or without vitamin K2 in hemodialysis patients with atrial fibrillation: The Valkyrie Study. J. Am. Soc. Nephrol. 2020, 31, 186–196. [Google Scholar] [CrossRef]
- Zipes, D.P.; Libby, P.; Bonow, R.O.; Mann, D.L.; Tomaselli, G.F. Braunwald’s Heart Disease E-Book: A Textbook of Cardiovascular Medicine; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Tastet, L.; Pibarot, P.; Shen, M.; Clisson, M.; Côté, N.; Salaun, E.; Arsenault, M.; Bédard, É.; Capoulade, R.; Puri, R. Oral anticoagulation therapy and progression of calcific aortic valve stenosis. J. Am. Coll. Cardiol. 2019, 73, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, L.; Tripepi, G.; Ronco, C.; D’Arrigo, G.; Barbera, V.; Russo, D.; Di Iorio, B.R.; Uguccioni, M.; Paoletti, E.; Ravera, M. Cardiac valve calcification and use of anticoagulants: Preliminary observation of a potentially modifiable risk factor. Int. J. Cardiol. 2019, 278, 243–249. [Google Scholar] [CrossRef]
- Cozzolino, M.; Fusaro, M.; Ciceri, P.; Gasperoni, L.; Cianciolo, G. The role of vitamin K in vascular calcification. Adv. Chronic Kidney Dis. 2019, 26, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.; Schurgers, L.J.; Uiterwijk, H.H.; Beulens, J.W.; Dalmeijer, G.W.; Westerhuis, R.; Magdeleyns, E.J.; Herfs, M.; Vermeer, C.; Laverman, G.D. Vitamin K intake and status are low in hemodialysis patients. Kidney Int. 2012, 82, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhou, J.J.; Robertson, G.R.; Lee, V.W. Vitamin D in vascular calcification: A double-edged sword? Nutrients 2018, 10, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makani, A.; Saba, S.; Jain, S.K.; Bhonsale, A.; Sharbaugh, M.S.; Thoma, F.; Wang, Y.; Marroquin, O.C.; Lee, J.S.; Estes, N.M. Safety and efficacy of direct oral anticoagulants versus warfarin in patients with chronic kidney disease and atrial fibrillation. Am. J. Cardiol. 2020, 125, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Ou, S.-H.; Huang, C.-W.; Lee, P.-T.; Chou, K.-J.; Lin, P.-C.; Su, Y.-C. Efficacy and Safety of Direct Oral Anticoagulants vs Warfarin in Patients with Chronic Kidney Disease and Dialysis Patients: A Systematic Review and Meta-Analysis. Clin. Drug Investig. 2021, 1–11. [Google Scholar]
- Reilly, R.F.; Jain, N. Warfarin in nonvalvular atrial fibrillation—Time for a change? Proc. Semin. Dial. 2019, 32, 520–526. [Google Scholar] [CrossRef] [PubMed]
- King, B.J.; El-Azhary, R.A.; McEvoy, M.T.; Shields, R.C.; McBane, R.D.; McCarthy, J.T.; Davis, M.D. Direct oral anticoagulant medications in calciphylaxis. Int. J. Dermatol. 2017, 56, 1065–1070. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author | Study Design | Study Population | Vasculature Studied | Vascular Calcification Measurement Method | Principal Findings |
|---|---|---|---|---|---|
| Hasific et al., 2020 | Retrospective Cohort | 2892 patients who underwent cardiac CT for various indications | Coronary arteries | CT 1 | For each year of warfarin treatment, odds of being in higher CAC 2 category increased; DOAC 3 treatment duration was not associated with CAC category |
| Di Lullo et al., 2018 | Retrospective Cohort | 347 patients with CKD 4 stage 3b-4 | All cardiac valves | TTE 5 | Rivaroxaban compared to warfarin reduced both mitral and aortic valve calcifications independently of the degree of baseline calcification |
| Plank et al., 2018 | Retrospective Cohort | 303 patients with non-valvular AF 6 | Coronary arteries | CCTA 7 | Warfarin significantly associated with increased overall plaque and higher prevalence of high-risk plaque burden compared to DOACs and control group |
| Tastet et al., 2019 | Retrospective Cohort | 303 patients with at least mild aortic stenosis | Aortic valve | TTE and MDCT 8 | Median annualized increase in Vpeak 9 larger in warfarin group compared to DOAC and no anticoagulant therapy groups |
| Peeters et al., 2018 | Cross-Sectional | 236 patients with AF | Ascending aorta, descending aorta, aortic valve | CCTA | Warfarin significantly associated with AsAc 10 and DAC 11 and trend in AVC 12 compared with non-anticoagulation. These same associations were absent in DOAC use when compared to non-anticoagulation |
| De Vriese et al., 2020 | Prospective Randomized Trial | 132 hemodialysis patients | Coronary arteries, thoracic aortic plaques, and cardiac valves | CT | Patients on warfarin, rivaroxaban, or rivaroxaban plus vitamin K2 did not differ significantly in terms of coronary artery, thoracic aorta, and cardiac valve calcium scores. Initiation or continuation of warfarin further increased dp-ucMGP 13 |
| Lee et al., 2018 | Prospective Randomized Trial | 97 patients with non-valvular AF | Coronary arteries | CCTA | Warfarin significantly associated with progression of total plaque volume and calcified plaque volume when compared to rivaroxaban |
| Win et al., 2019 | Prospective Randomized Trial | 66 patients with non-valvular AF | Coronary arteries | CCTA | Significantly higher total, calcified, and low-attenuation plaque volume in warfarin group compared to apixaban |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elango, K.; Javaid, A.; Khetarpal, B.K.; Ramalingam, S.; Kolandaivel, K.P.; Gunasekaran, K.; Ahsan, C. The Effects of Warfarin and Direct Oral Anticoagulants on Systemic Vascular Calcification: A Review. Cells 2021, 10, 773. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10040773
Elango K, Javaid A, Khetarpal BK, Ramalingam S, Kolandaivel KP, Gunasekaran K, Ahsan C. The Effects of Warfarin and Direct Oral Anticoagulants on Systemic Vascular Calcification: A Review. Cells. 2021; 10(4):773. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10040773
Chicago/Turabian StyleElango, Kalaimani, Awad Javaid, Banveet K. Khetarpal, Sathishkumar Ramalingam, Krishna Prasad Kolandaivel, Kulothungan Gunasekaran, and Chowdhury Ahsan. 2021. "The Effects of Warfarin and Direct Oral Anticoagulants on Systemic Vascular Calcification: A Review" Cells 10, no. 4: 773. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10040773