
Increased Alveolar Heparan Sulphate and Reduced Pulmonary Surfactant Amount and Function in the Mucopolysaccharidosis IIIA Mouse

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Tissue and Lavage (BALF) Collection
2.3. Tissue Homogenisation
2.4. Sample Preparation and Analysis—Heparan Sulphate
2.5. Sample Preparation and Analysis—Gangliosides, Bis(Monoacylglycero)Phosphate (BMP), and Cholesterol
2.6. Tissue and Lavage Collection for Total Phospholipid Pool Analysis
2.7. Sample Preparation and Analysis—Total Phospholipids
- Positive and negative ESI scans were performed for each sample using an injection volume of 50 µL and an analysis time of 13 min, measuring the total number of ionised molecules in the samples. This was a control for the presence of any contaminants and overall signal intensity and were not used for ion peak assignments.
- Diagnostic MS/MS precursor scans were performed with the second injection (100 µL) and analysis time of 22.40 min to quantify PC. Scans were based on the detection of the major collision-induced dissociation (CID) phosphorylcholine fragment ion, detected at m/z 184.
- The third injection (100 µL, analysis 22.40 min) included neutral loss (NL) scans for the DPPC molecule and diagnostic scans for the other major phospholipid classes, P153 for acidic phospholipids, P241 for phosphatidylinositol (PI), NL141 for phosphatidylethanolamine (PE) and NL87 for phosphatidylserine (PS).
- The final injection (100 µL, analysis 22.40 min) included precursor scans to determine the distributions of fatty acyl moieties. Each of the lipids indicated Supplementary Materials—Table S1) were detected by the indicated type of scan to provide a comprehensive analysis of unlabelled phospholipids.
2.8. Captive Bubble Surfactometry (CBS)
2.9. Gene Expression Using Quantitative Real-Time PCR
2.10. Protein Expression Using Western Blot Analysis
2.11. Statistics
3. Results
3.1. Heparan Sulphate and Lipid Analyses by Liquid Chromatography Tandem Mass Spectrometry
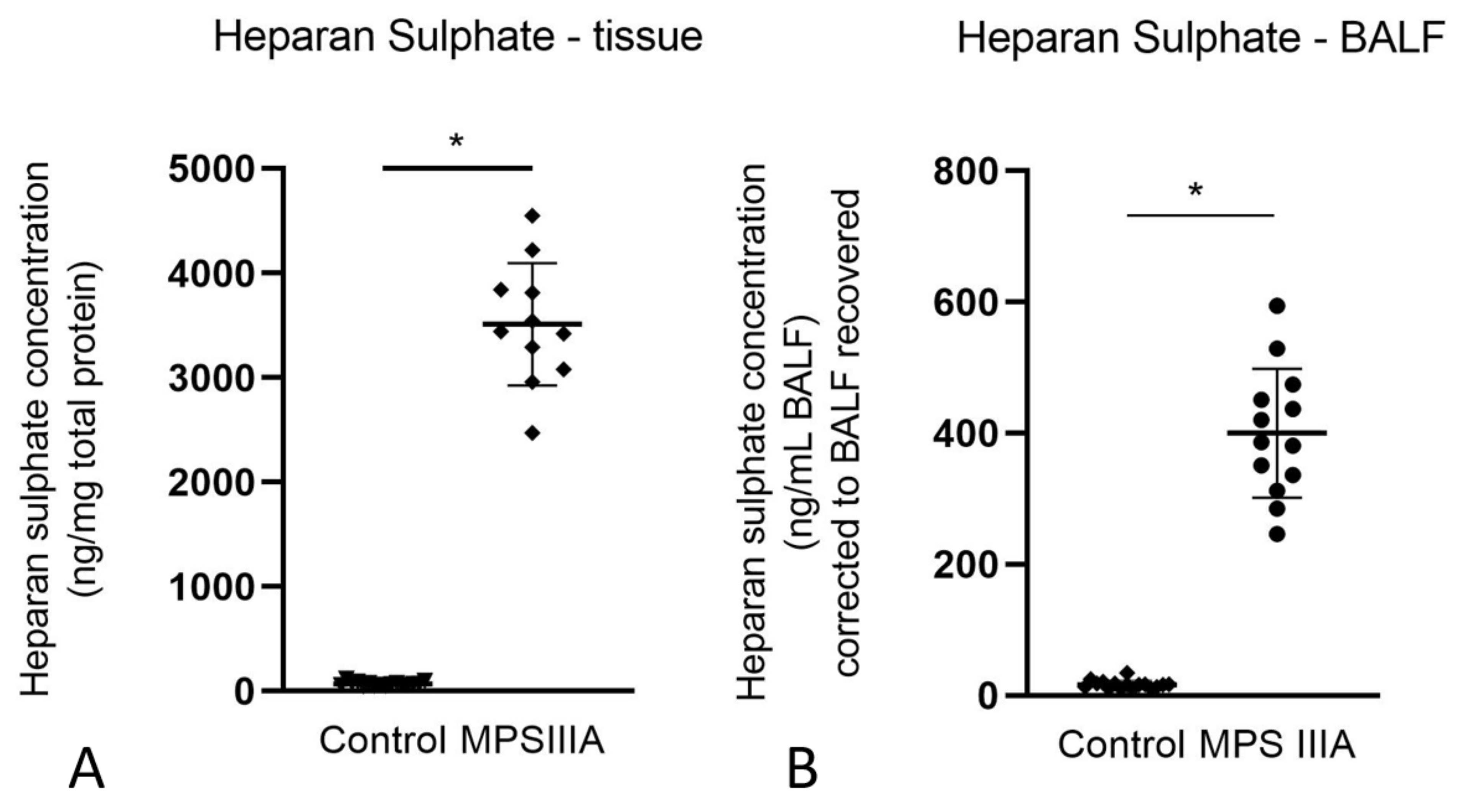
3.1.1. Presence of Heparan Sulphate (HS) in Lung Tissue and Bronchoalveolar Lavage Fluid (BALF)
3.1.2. Alterations in Ganglioside Concentration in Lung Tissue
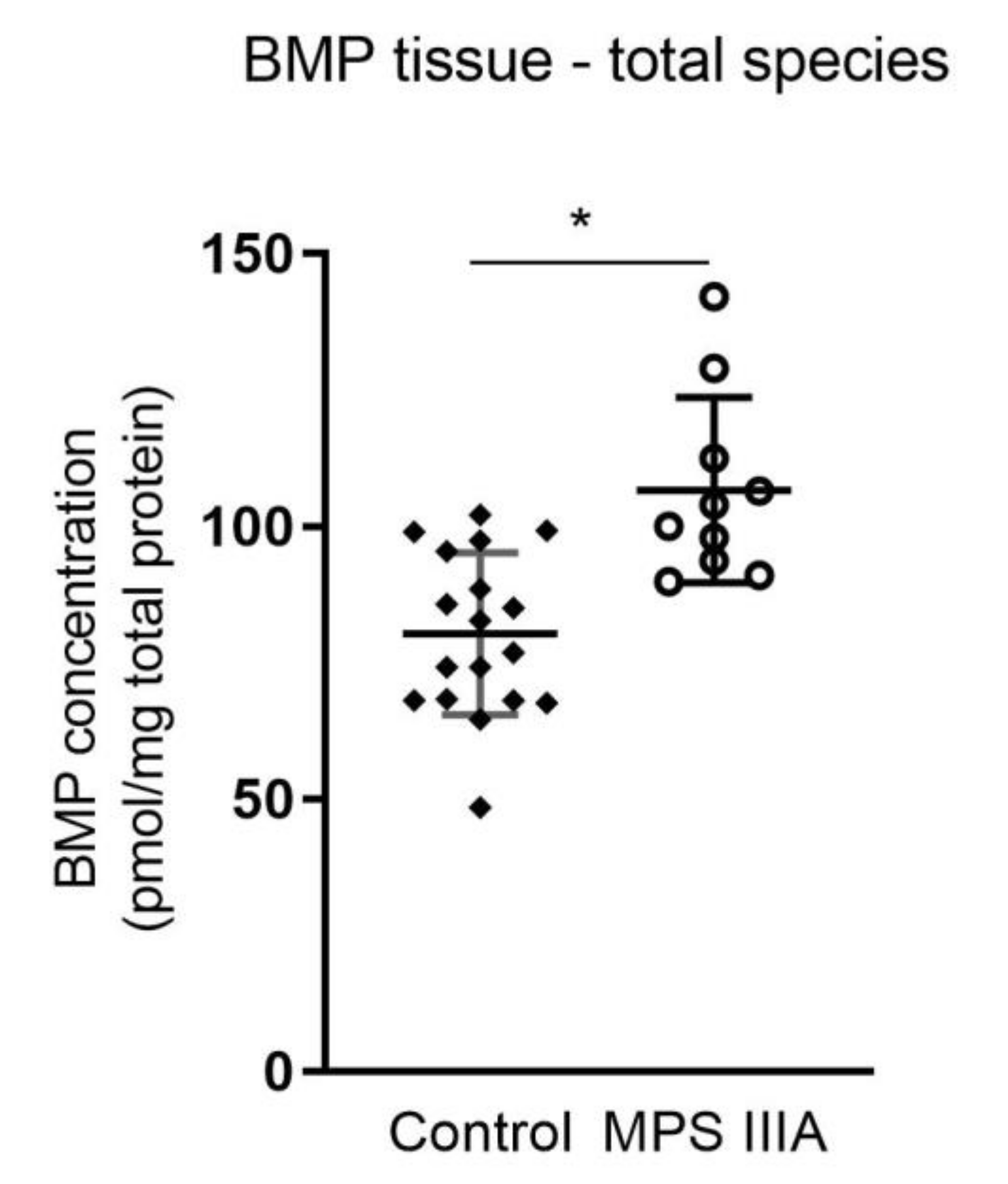
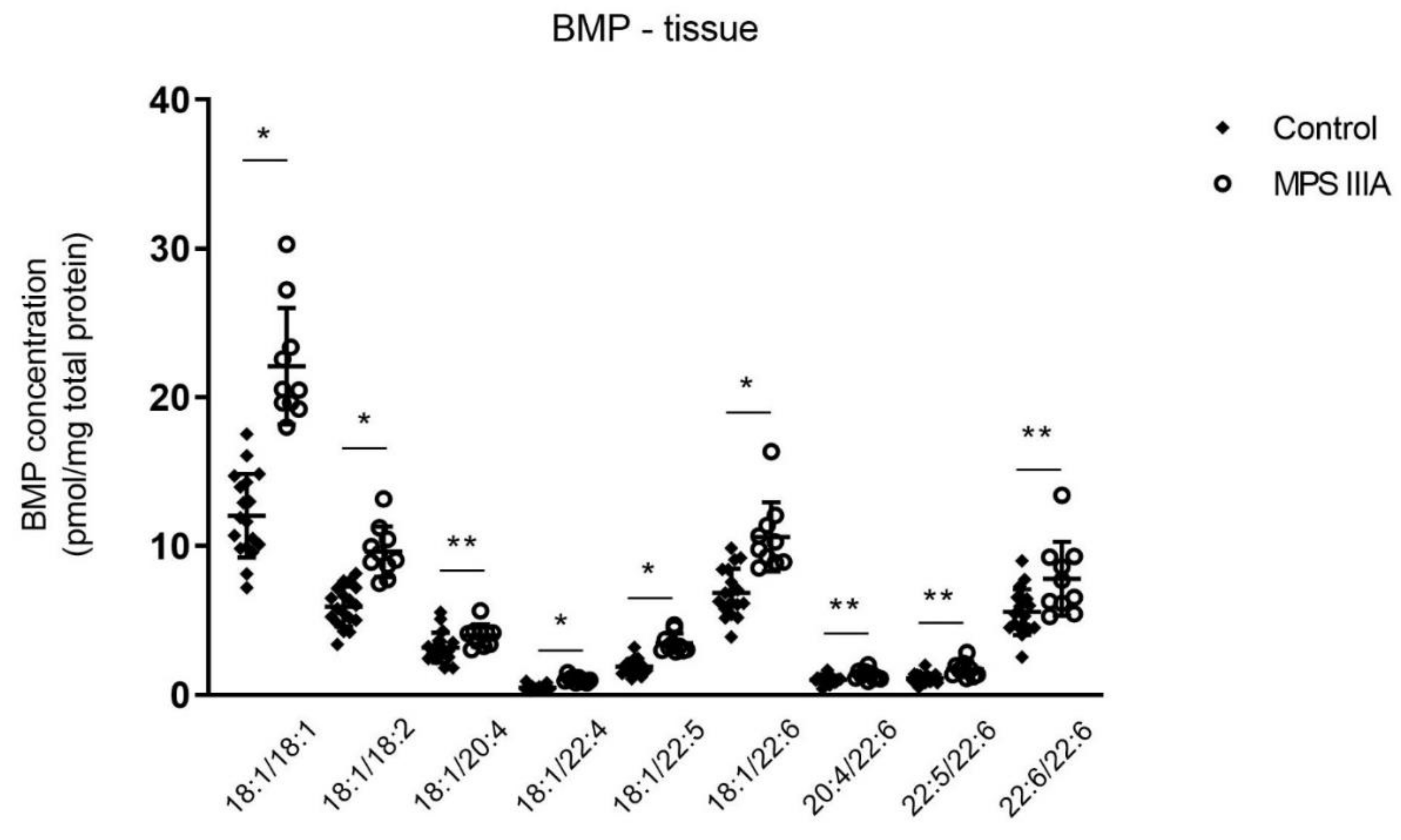
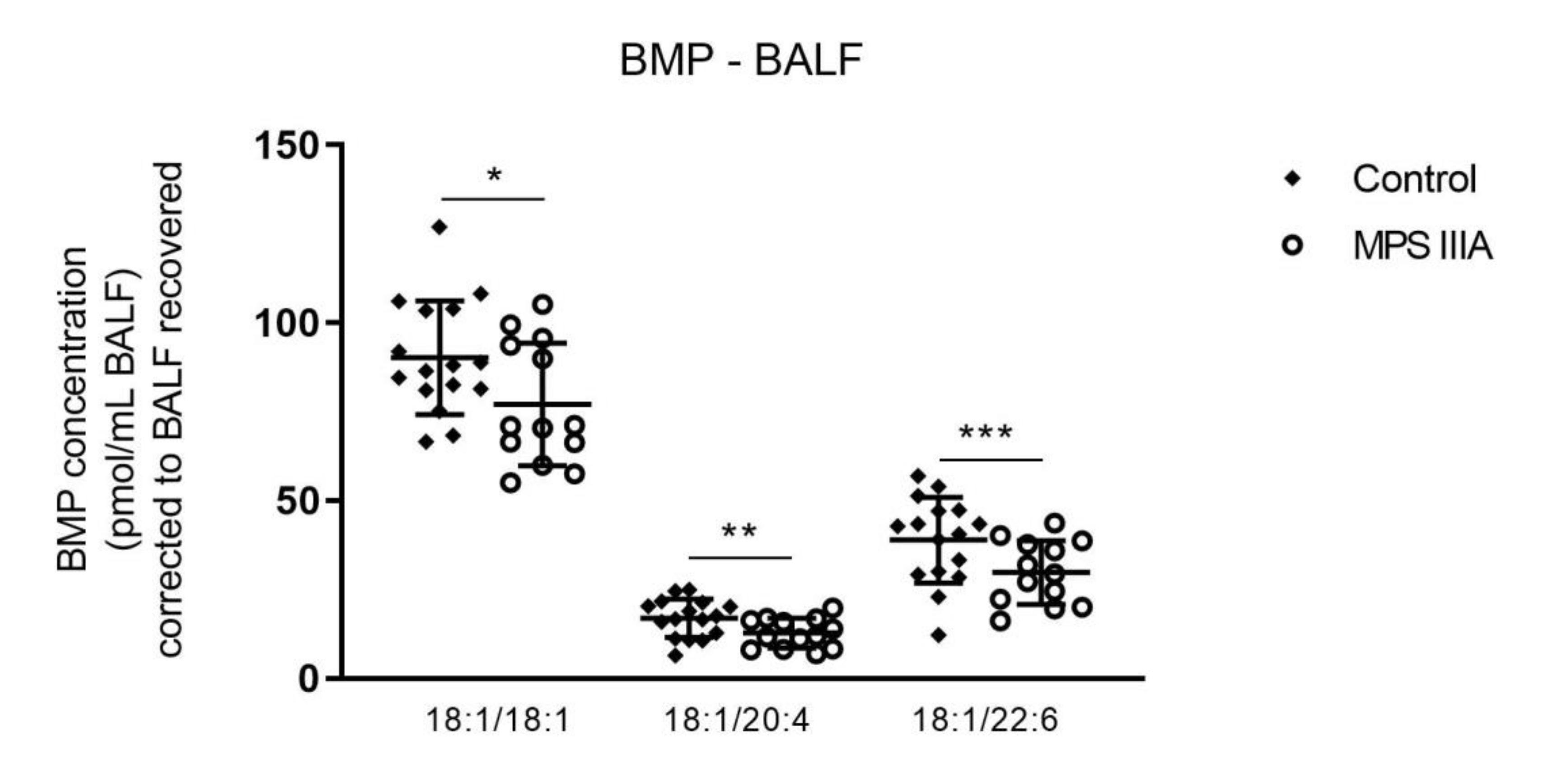
3.1.3. Alterations in Bis(Monoacylglycerol)Phosphate (BMP) Concentration in Lung Tissue and BALF
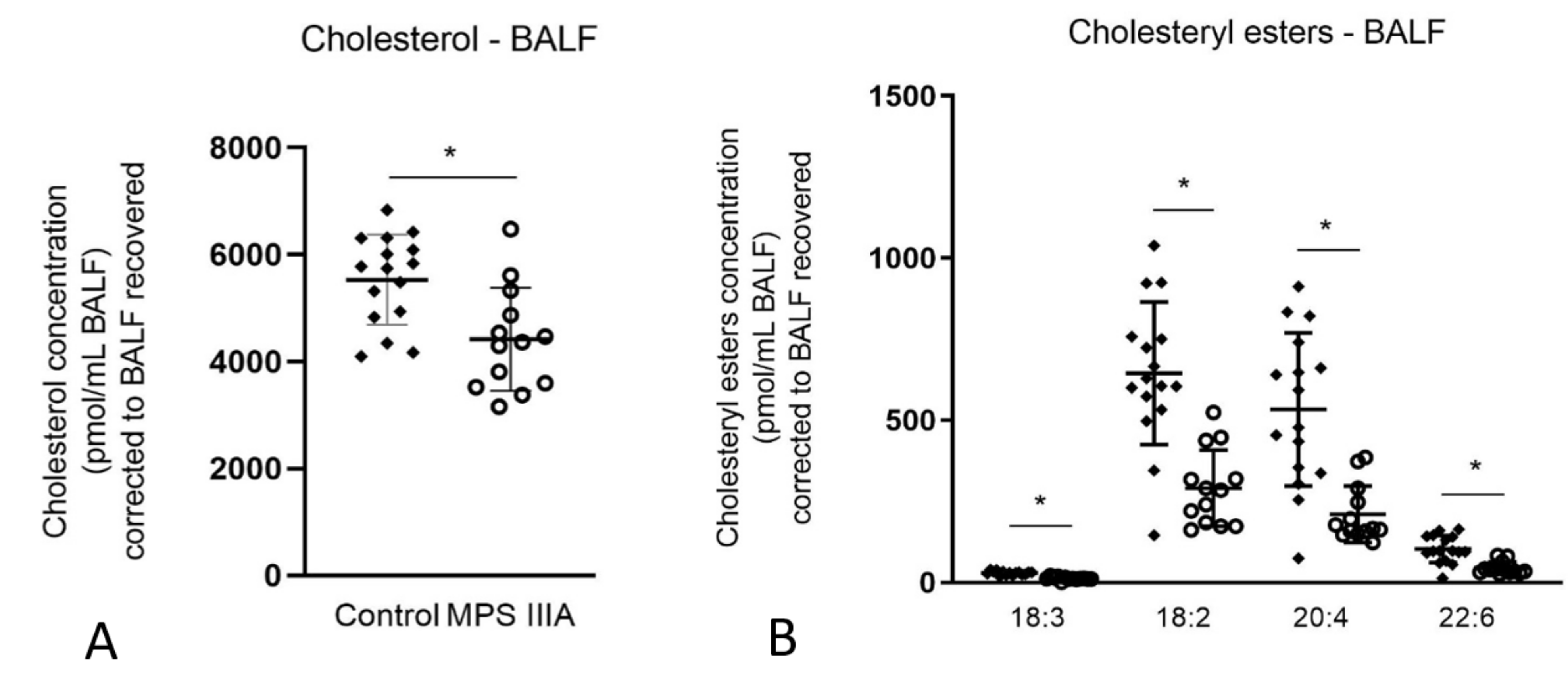
3.1.4. Alterations in Cholesterol Concentration in Lung Tissue and BALF
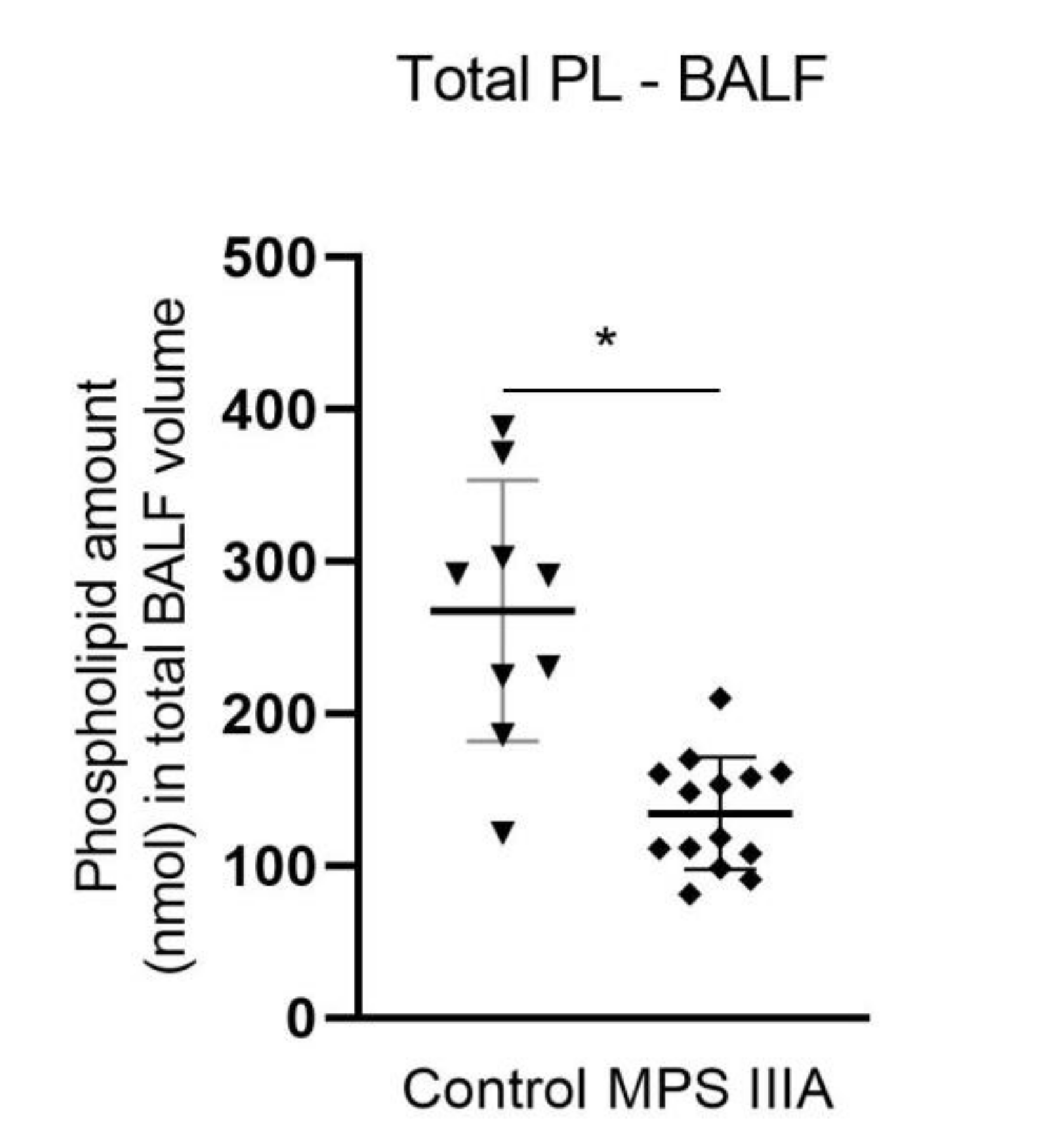
3.2. Phospholipid Composition and Alterations in Total Alveolar Phospholipid Pool Size
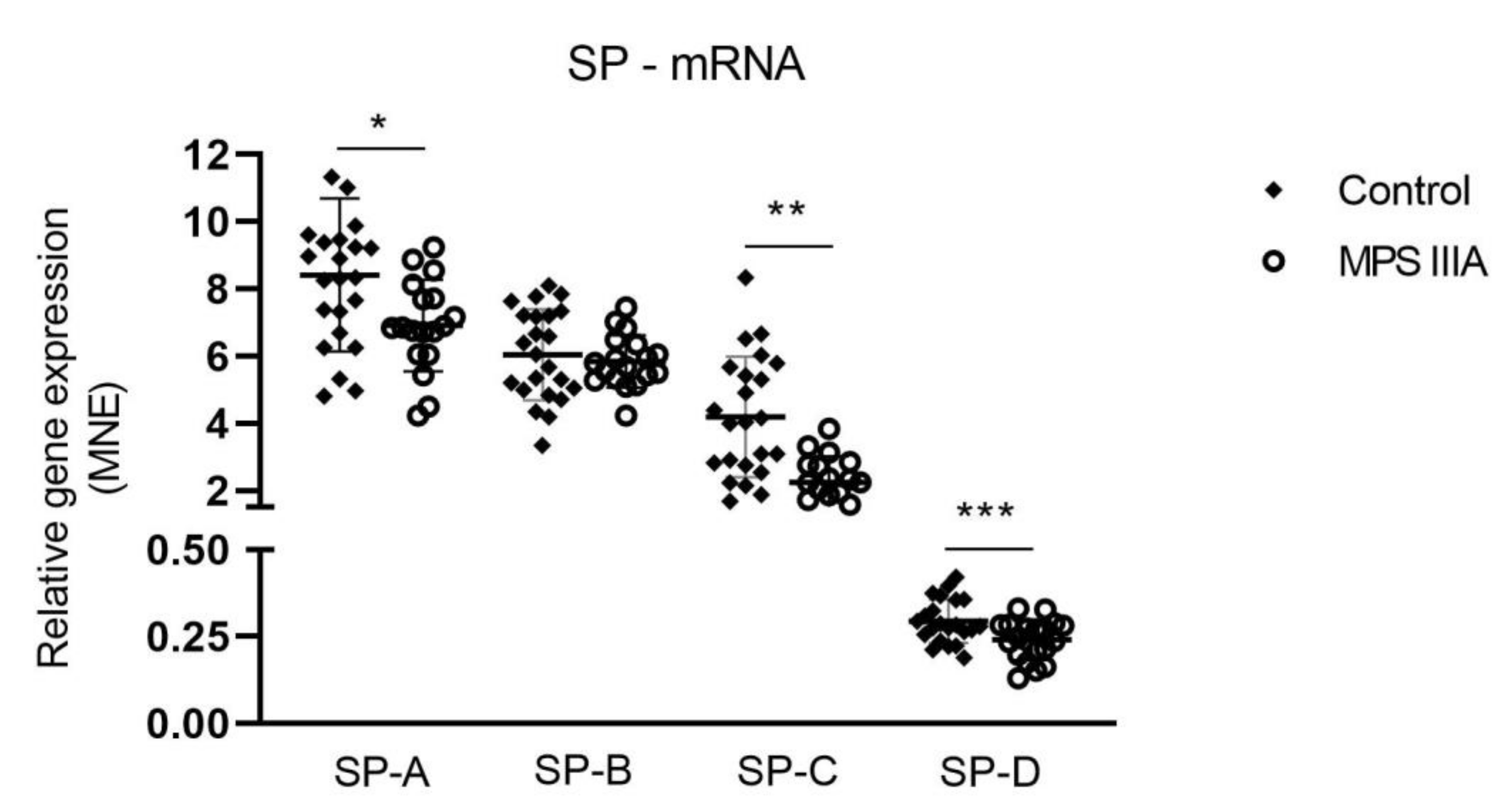
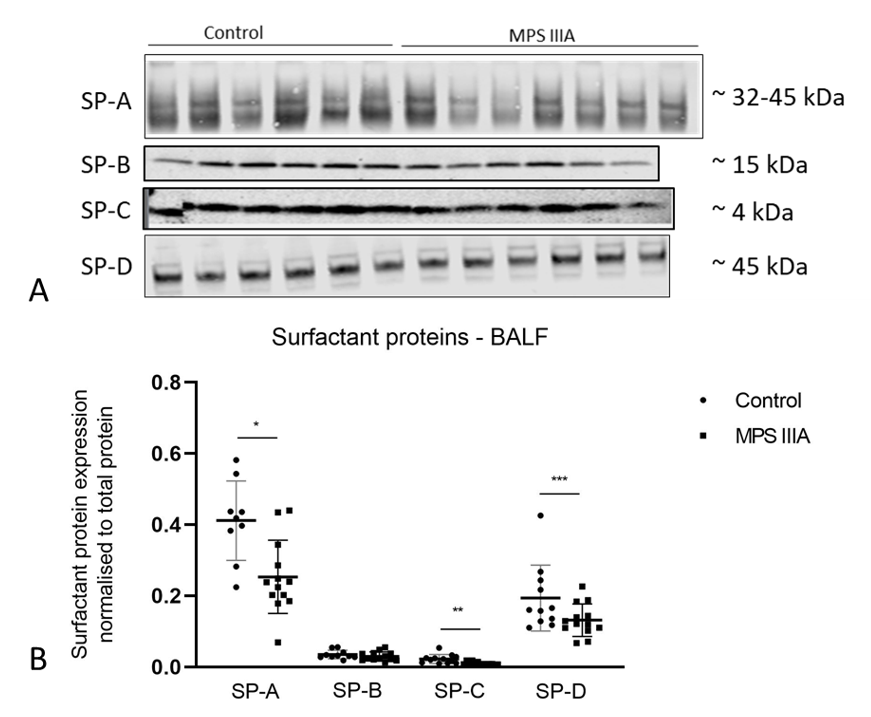
3.3. Surfactant Protein Gene Expression and Protein Analysis
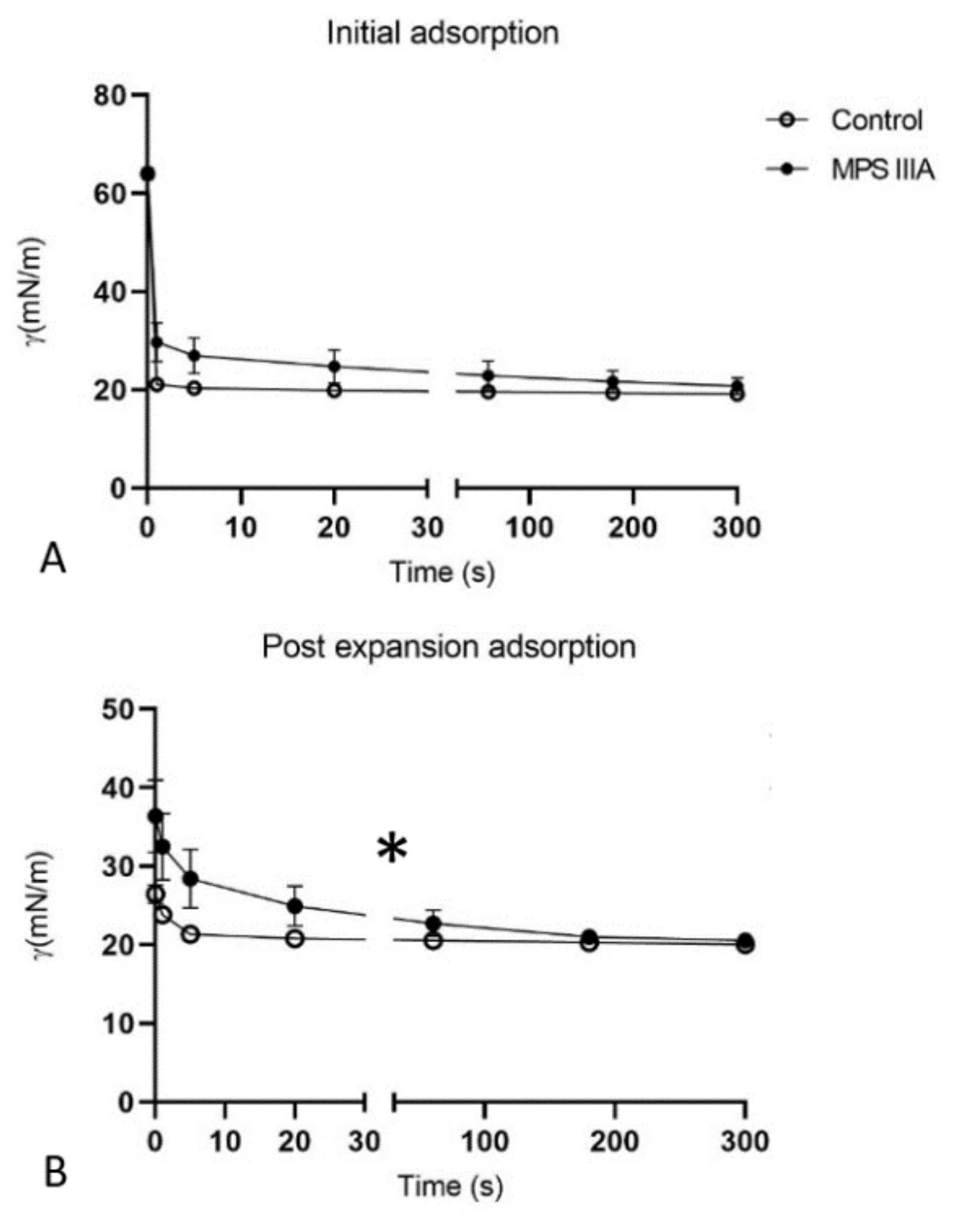
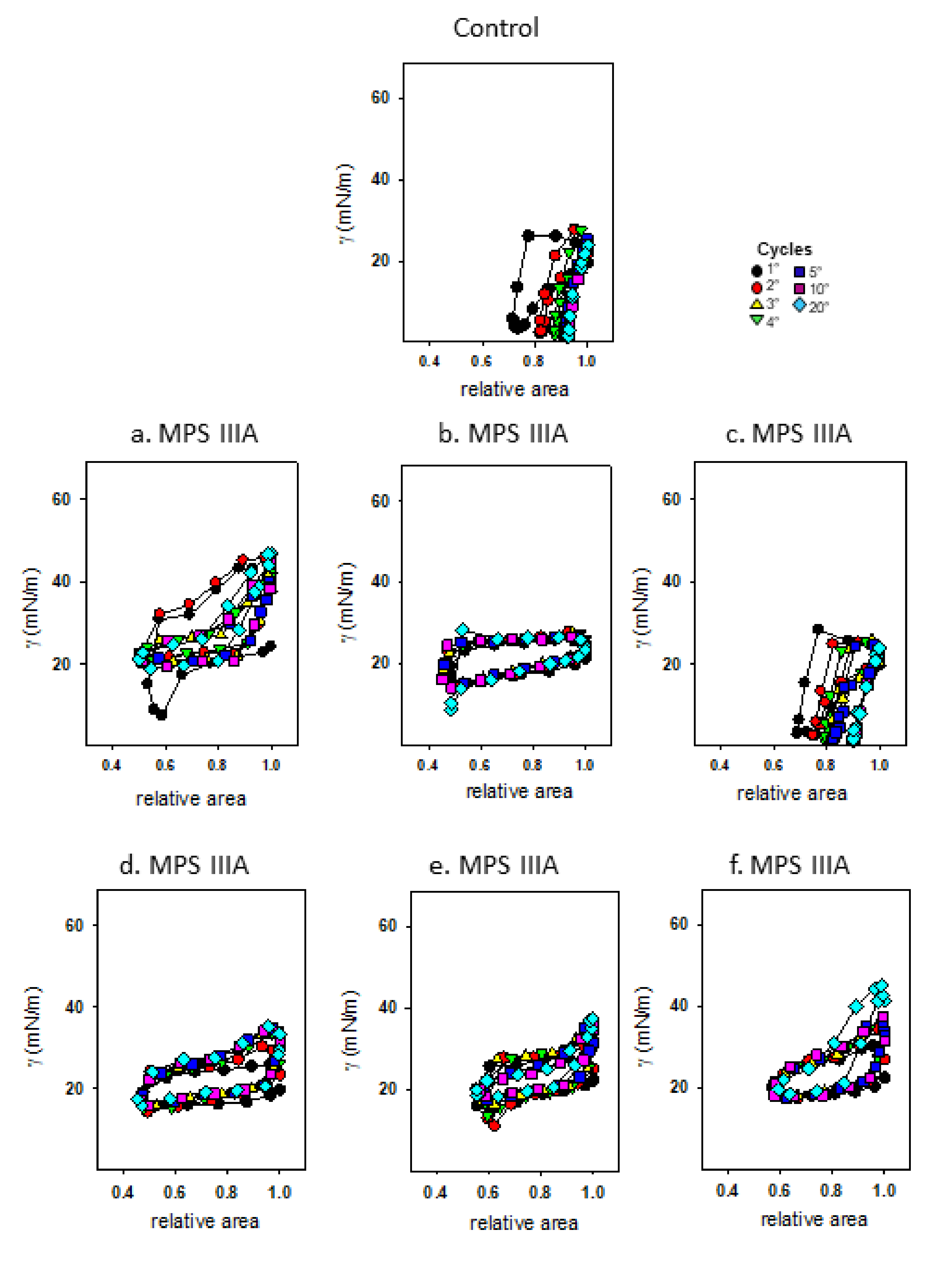
3.4. Captive Bubble Surfactometry
4. Discussion
4.1. Heparan Sulphate Accumulation in Lung Tissue and the Alveolar Compartment
4.2. The Role of Pulmonary Surfactant
4.3. Alterations in Gangliosides, BMP and Cholesterol Composition
4.4. Total Alveolar Phospholipid Pool
4.5. Surfactant Proteins A-D
4.6. Surfactant Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cox, T.; Cachon-Gonzalez, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2012, 226, 241–254. [Google Scholar] [CrossRef]
- Valstar, M.J.; Marchal, J.P.; Grootenhuis, M.; Colland, V.; Wijburg, F.A. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet J. Rare Dis. 2011, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Valstar, M.; Ruijter, G.; Van Diggelen, O.; Poorthuis, B.; Wijburg, F. Sanfilippo syndrome: A mini-review. J. Inherit. Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef]
- Neufeld, E.; Meunzer, J. The metabolic and molecular bases of inherited disease (Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., and Vogelstein, B. eds. 8th edn. Biochemistry 2001, 67, 611–612. [Google Scholar]
- Chiang, J.; Raiman, J.; Cutz, E.; Solomon, M.; Dell, S. Tachypnea of infancy as the first sign of Sanfilippo syndrome. Pediatrics 2014, 134, e884–e888. [Google Scholar] [CrossRef] [Green Version]
- Cleary, M.A.; Wraith, J.E. Management of mucopolysaccharidosis type III. Arch. Dis. Child. 1993, 69, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; Guo, N.; DeAngelis, V.; Tomatsu, S.; Schuchman, E.H. Evaluation of pentosan polysulfate in mucopolysaccharidosis type IIIA mice. Mol. Genet. Metab. 2017, 120, S123. [Google Scholar] [CrossRef]
- Leighton, S.; Papsin, B.; Vellodi, A.; Dinwiddie, R.; Lane, R. Disordered breathing during sleep in patients with mucopolysaccharidoses. Int. J. Pediatric Otorhinolaryngol. 2001, 58, 127–138. [Google Scholar] [CrossRef]
- Lavery, C.; Hendriksz, C.J.; Jones, S.A. Mortality in patients with Sanfilippo syndrome. Orphanet J. Rare Dis. 2017, 12, 168. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Kossow, K.; Gal, A.; Mühlhausen, C.; Ullrich, K.; Braulke, T.; Muschol, N. Scoring evaluation of the natural course of Mucopolysaccharidosis Type IIIA (Sanfilippo Syndrome Type A). Pediatrics 2007, 120, e1255–e1261. [Google Scholar] [CrossRef] [Green Version]
- Mühlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory manifestations in mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef]
- Bush, D.; Sremba, L.; Lomax, K.; Lipsett, J.; Ketteridge, D.; Bratkovic, D.; Eachautegul-Colon, Y.; Welsfeld-Adams, J.; Galambos, C.; Lummus, S.; et al. Neonatal onset interstitial lung disease as a primary presenting manifestation of mucopolysaccharidosis type I. J. Inherit. Metab. Dis. Rep. 2018, 43, 71–77. [Google Scholar]
- Kurolap, A.; del Toro, M.; Spiegel, R.; Gutstein, A.; Shafir, G.; Cohen, I.J.; Barrabés, J.A.; Feldman, H.B. Gaucher disease type 3c: New patients with unique presentations and review of the literature. Mol. Genet. Metab. 2019, 127, 138–146. [Google Scholar] [CrossRef]
- Buccoliero, R.; Ginzburg, L.; Futerman, A.H. Elevation of lung surfactant phosphatidylcholine in mouse models of Sandhoff and of Niemann-Pick A disease. J. Inherit. Metab. Dis. 2004, 27, 641–648. [Google Scholar] [CrossRef]
- Buccoliero, R.; Palmeri, S.; Ciarleglio, G.; Collodoro, A.; De Santi, M.M.; Federico, A. Increased lung surfactant phosphatidylcholine in patients affected by lysosomal storage diseases. J. Inherit. Metab. Dis. 2007, 30, 983. [Google Scholar] [CrossRef]
- Griese, M.; Brasch, F.; Aldana, V.; Cabrera, M.; Goelnitz, U.; Ikonen, E.; Karam, B.; Liebisch, G.; Linder, M.; Lohse, P. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin. Genet. 2010, 77, 119–130. [Google Scholar] [CrossRef]
- Roszell, B.R.; Tao, J.-Q.; Yu, K.J.; Huang, S.; Bates, S.R. Characterization of the Niemann-Pick C pathway in alveolar type II cells and lamellar bodies of the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L919–L932. [Google Scholar] [CrossRef] [Green Version]
- Crawley, A.C.; Gliddon, B.L.; Auclair, D.; Brodie, S.L.; Hirte, C.; King, B.M.; Fuller, M.; Hemsley, K.M.; Hopwood, J.J. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006, 1104, 1–17. [Google Scholar] [CrossRef]
- Bhaumik, M.; Muller, V.J.; Rozaklis, T.; Johnson, L.; Dobrenis, K.; Bhattacharyya, R.; Wurzelmann, S.; Finamore, P.; Hopwood, J.J.; Walkley, S.U.; et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology 1999, 9, 1389–1396. [Google Scholar] [CrossRef]
- He, Q.Q.; Trim, P.J.; Lau, A.A.; King, B.M.; Hopwood, J.J.; Hemsley, K.M.; Snel, M.F.; Ferro, V. Synthetic disaccharide standards enable quantitative analysis of stored heparan sulfate in MPS IIIA murine brain regions. ACS Chem. Neurosci. 2019, 10, 3847–3858. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane-Stanley, G. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Job, E.R.; Saelens, X.; Roose, K. Bronchoalveolar lavage of murine lungs to analyze inflammatory cell infiltration. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Postle, A.D.; Henderson, N.G.; Koster, G.; Clark, H.W.; Hunt, A.N. Analysis of lung surfactant phosphatidylcholine metabolism in transgenic mice using stable isotopes. Chem. Phys. Lipids 2011, 164, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Goss, V.; Hunt, A.N.; Postle, A.D. Regulation of lung surfactant phospholipid synthesis and metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 448–458. [Google Scholar] [CrossRef]
- Schürch, S.; Green, F.H.; Bachofen, H. Formation and structure of surface films: Captive bubble surfactometry. Biochim. Biophys. Acta 1998, 1408, 180–202. [Google Scholar] [CrossRef] [Green Version]
- Nanjee, M.N.; Gebre, A.K.; Miller, N.E. Enzymatic fluorometric procedure for phospholipid quantification with an automated microtiter plate fluorometer. Clin. Chem. 1991, 37, 868–874. [Google Scholar] [CrossRef]
- Naito, H.K.; David, J.A. Laboratory considerations: Determination of cholesterol, triglyceride, phospholipid, and other lipids in blood and tissues. Lab. Res. Methods Biol. Med. 1984, 10, 1–76. [Google Scholar]
- Schoel, W.M.; Schürch, S.; Goerke, J. The captive bubble method for the evaluation of pulmonary surfactant: Surface tension, area, and volume calculations. Biochim. Biophys. Acta (BBA) Gen. Subj. 1994, 1200, 281–290. [Google Scholar] [CrossRef]
- Soo, P.S.; Hiscock, J.; Botting, K.J.; Roberts, C.T.; Davey, A.K.; Morrison, J.L. Maternal undernutrition reduces P-glycoprotein in guinea pig placenta and developing brain in late gestation. Reprod. Toxicol. 2012, 33, 374–381. [Google Scholar] [CrossRef]
- McGillick, E.V.; Orgeig, S.; McMillen, I.C.; Morrison, J.L. The fetal sheep lung does not respond to cortisol infusion during the late canalicular phase of development. Physiol. Rep. 2013, 1, 1–15. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Darby, J.R.T.; McMillen, I.C.; Morrison, J.L. Maternal undernutrition in late gestation increases IGF2 signalling molecules and collagen deposition in the right ventricle of the fetal sheep heart. J. Physiol. 2018, 596, 2345–2358. [Google Scholar] [CrossRef] [Green Version]
- Pillai-Kastoori, L.; Schutz-Geschwender, A.R.; Harford, J.A. A systematic approach to quantitative Western blot analysis. Anal. Biochem. 2020, 593, 113608. [Google Scholar] [CrossRef]
- Vieira, F.; Kung, J.W.; Bhatti, F. Structure, genetics and function of the pulmonary associated surfactant proteins A and D: The extra-pulmonary role of these C type lectins. Ann. Anat. Anat. Anz. 2017, 211, 184–201. [Google Scholar] [CrossRef]
- Davis, D.A.S.; Parish, C.R. Heparan sulfate: A ubiquitous glycosaminoglycan with multiple roles in immunity. Front. Immunol. 2013, 4, 470. [Google Scholar]
- Ochs, M.; Hegermann, J.; Lopez-Rodriguez, E.; Timm, S.; Nouailles, G.; Matuszak, J.; Simmons, S.; Witzenrath, M.; Kuebler, W.M. On Top of the Alveolar Epithelium: Surfactant and the Glycocalyx. Int. J. Mol. Sci. 2020, 21, 3075. [Google Scholar] [CrossRef]
- Stringer, S.E.; Gallagher, J.T. Heparan sulphate. Int. J. Biochem. Cell Biol. 1997, 29, 709–714. [Google Scholar] [CrossRef]
- Gallagher, J.T.; Lyon, M.; Steward, W.P. Structure and function of heparan sulphate proteoglycans. Biochem. J. 1986, 236, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, U.; Kjellén, L. Pathophysiology of heparan sulphate: Many diseases, few drugs. J. Intern. Med. 2013, 273, 555–571. [Google Scholar] [CrossRef] [Green Version]
- Smits, N.C.; Shworak, N.W.; Dekhuijzen, P.N.; van Kuppevelt, T.H. Heparan sulfates in the lung: Structure, diversity, and role in pulmonary emphysema. Anat. Rec. 2010, 293, 955–967. [Google Scholar] [CrossRef]
- Haeger, S.M.; Yang, Y.; Schmidt, E.P. Heparan sulfate in the developing, healthy, and injured lung. Am. J. Respir. Cell Mol. Biol. 2016, 55, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Li, J.P.; Kusche-Gullberg, M. Heparan sulfate: Biosynthesis, structure, and function. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Amsterdam, The Netherlands, 2016; Volume 325, pp. 215–273. [Google Scholar]
- Yang, Y.; Schmidt, E.P. The endothelial glycocalyx. Tissue Barriers 2013, 1, e23494. [Google Scholar] [CrossRef] [Green Version]
- Haeger, S.M.; Liu, X.; Han, X.; McNeil, J.B.; Oshima, K.; McMurtry, S.A.; Yang, Y.; Ouyang, Y.; Zhang, F.; Nozik-Grayck, E.; et al. Epithelial heparan sulfate contributes to alveolar barrier function and is shed during lung injury. Am. J. Respir. Cell Mol. Biol. 2018, 59, 363–374. [Google Scholar] [CrossRef]
- LaRivière, W.B.; Schmidt, E.P. Chapter Two—The pulmonary endothelial glycocalyx in ARDS: A critical role for heparan sulfate. In Current Topics in Membranes; Belvitch, P., Dudek, S., Eds.; Academic Press: Amsterdam, The Netherlands, 2018; Volume 82, pp. 33–52. [Google Scholar]
- Maccari, F.; Sorrentino, N.C.; Mantovani, V.; Galeotti, F.; Fraldi, A.; Volpi, N. Glycosaminoglycan levels and structure in a mucopolysaccharidosis IIIA mice and the effect of a highly secreted sulfamidase engineered to cross the blood-brain barrier. Metab. Brain Dis. 2017, 32, 203–210. [Google Scholar] [CrossRef]
- Cañadas, O.; Keough, K.M.W.; Casals, C. Bacterial lipopolysaccharide promotes destabilization of lung surfactant-like films. Biophys. J. 2011, 100, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Parra, E.; Pérez-Gil, J. Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem. Phys. Lipids 2015, 185, 153–175. [Google Scholar] [CrossRef]
- Krafft, M.P. Overcoming inactivation of the lung surfactant by serum proteins: A potential role for fluorocarbons? Soft Matter 2015, 11, 5982–5994. [Google Scholar] [CrossRef] [Green Version]
- Devendra, G.; Spragg, R.G. Lung surfactant in subacute pulmonary disease. Respir. Res. 2002, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Amigoni, A.; Pettenazzo, A.; Stritoni, V.; Circelli, M. Surfactants in acute respiratory distress syndrome in infants and children: Past, present and future. Clin. Drug Investig. 2017, 37, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.E.; Yamashita, C.M.; Veldhuizen, R.A. Lung remodeling associated with recovery from acute lung injury. Cell Tissue Res. 2017, 367, 495–509. [Google Scholar] [CrossRef]
- Archer, L.D.; Langford-Smith, K.J.; Bigger, B.W.; Fildes, J.E. Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity. J. Inherit. Metab. Dis. 2014, 37, 1–12. [Google Scholar] [CrossRef]
- Bigger, B.; Boutin, H.; Wilkinson, F.; Holley, R.; Brough, D.; Pinteaux, E.; Parker, H. Mucopolysaccharidosis type IIIA storage substrate drives an innate immune neuro-inflammatory response. Mol. Genet. Metab. 2016, 120, S27–S28. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, E.; Pérez-Gil, J. Structure-function relationships in pulmonary surfactant membranes: From biophysics to therapy. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1568–1585. [Google Scholar] [CrossRef] [Green Version]
- Reid, K.B.M. Functional roles of the lung surfactant proteins SP-A and SP-D in innate immunity. Immunobiology 1998, 199, 200–207. [Google Scholar] [CrossRef]
- Al-Saiedy, M.; Tarokh, A.; Nelson, S.; Hossini, K.; Green, F.; Ling, C.-C.; Prenner, E.J.; Amrein, M. The role of multilayers in preventing the premature buckling of the pulmonary surfactant. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1372–1380. [Google Scholar] [CrossRef]
- Svennerholm, L. The gangliosides. J. Lipid Res. 1964, 5, 145–155. [Google Scholar] [CrossRef]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef]
- Dautel, S.E.; Kyle, J.E.; Clair, G.; Sontag, R.L.; Weitz, K.K.; Shukla, A.K.; Nguyen, S.N.; Kim, Y.-M.; Zink, E.M.; Luders, T.; et al. Lipidomics reveals dramatic lipid compositional changes in the maturing postnatal lung. Sci. Rep. 2017, 7, 40555. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Tsukuda, Y.; Iwasaki, N.; Seito, N.; Kanayama, M.; Fujitani, N.; Shinohara, Y.; Kasahara, Y.; Onodera, T.; Suzuki, K.; Asano, T.; et al. Ganglioside GM3 Has an Essential Role in the Pathogenesis and Progression of Rheumatoid Arthritis. PLoS ONE 2012, 7, e40136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jou, I.; Lee, J.H.; Park, S.Y.; Yoon, H.J.; Joe, E.-H.; Park, E.J. Gangliosides trigger inflammatory responses via TLR4 in brain glia. Am. J. Pathol. 2006, 168, 1619–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somogyi, A.; Petcherski, A.; Beckert, B.; Huebecker, M.; Priestman, D.A.; Banning, A.; Cotman, S.L.; Platt, F.M.; Ruonala, M.O.; Tikkanen, R. Altered expression of ganglioside metabolizing enzymes results in GM3 ganglioside accumulation in cerebellar cells of a mouse model of juvenile neuronal ceroid lipofuscinosis. Int. J. Mol. Sci. 2018, 19, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langeveld, M.; Ghauharali, K.J.; Sauerwein, H.P.; Ackermans, M.T.; Groener, J.E.; Hollak, C.E.; Aerts, J.M.; Serlie, M.J. Type I Gaucher disease, a glycosphingolipid storage disorder, is associated with insulin resistance. J. Clin. Endocrinol. Metab. 2008, 93, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Ghauharali-van der Vlugt, K.; Langeveld, M.; Poppema, A.; Kuiper, S.; Hollak, C.E.; Aerts, J.M.; Groener, J.E. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin. Chim. Acta 2008, 389, 109–113. [Google Scholar] [CrossRef]
- Hullin-Matsuda, F.; Luquain-Costaz, C.; Bouvier, J.; Delton-Vandenbroucke, I. Bis (monoacylglycero) phosphate, a peculiar phospholipid to control the fate of cholesterol: Implications in pathology. Prostaglandinsleukotrienes Essent. Fat. Acids 2009, 81, 313–324. [Google Scholar] [CrossRef]
- McCauliff, L.A.; Langan, A.; Li, R.; Ilnytska, O.; Bose, D.; Waghalter, M.; Lai, K.; Kahn, P.C.; Storch, J. Intracellular cholesterol trafficking is dependent upon NPC2 interaction with lysobisphosphatidic acid. eLife 2019, 8, e50832. [Google Scholar] [CrossRef]
- Frederick, T.E.; Chebukati, J.N.; Mair, C.E.; Goff, P.C.; Fanucci, G.E. Bis (monoacylglycero) phosphate forms stable small lamellar vesicle structures: Insights into vesicular body formation in endosomes. Biophys. J. 2009, 96, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Wardhan, R.; Mudgal, P. Introduction to Biomembranes. In Textbook of Membrane Biology; Springer: Singapore, 2017; pp. 1–28. [Google Scholar] [CrossRef]
- Gallala, H.D.; Sandhoff, K. Biological function of the cellular lipid BMP—BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem. Res. 2011, 36, 1594–1600. [Google Scholar] [CrossRef]
- Jabs, S.; Quitsch, A.; Käkelä, R.; Koch, B.; Tyynelä, J.; Brade, H.; Glatzel, M.; Walkley, S.; Saftig, P.; Vanier, M.T.; et al. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 2008, 106, 1415–1425. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Roldan, N.; Pérez-Gil, J.; Morrow, M.R.; García-Álvarez, B. Divide & Conquer: SurfactantpProtein SP-C and cholesterol modulate phase segregation in lung surfactant. Biophys. J. 2017, 113, 847–859. [Google Scholar] [CrossRef]
- Orgeig, S.; Daniels, C.B. The comparative biology of pulmonary surfactant: Past, present and future. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 129, 75–89. [Google Scholar] [CrossRef]
- Orgeig, S.; Daniels, C.B. The roles of cholesterol in pulmonary surfactant: Insights from comparative and evolutionary studies. Comp. Biochem. Physiol. Part Amolecular Integr. Physiol. 2010, 129, 75–89. [Google Scholar] [CrossRef]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Investig. 2002, 110, 597–603. [Google Scholar] [CrossRef]
- Veldhuizen, R.; Nag, K.; Orgeig, S.; Possmayer, F. The role of lipids in pulmonary surfactant. Biochim. Biophys. Acta 1998, 1408, 90–108. [Google Scholar] [CrossRef]
- Wüstneck, R.; Perez-Gil, J.; Wüstneck, N.; Cruz, A.; Fainerman, V.B.; Pison, U. Interfacial properties of pulmonary surfactant layers. Adv. Colloid Interface Sci. 2005, 117, 33–58. [Google Scholar] [CrossRef]
- Vockeroth, D.; Gunasekara, L.; Amrein, M.; Possmayer, F.; Lewis, J.F.; Veldhuizen, R.A.W. Role of cholesterol in the biophysical dysfunction of surfactant in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L117–L125. [Google Scholar] [CrossRef] [Green Version]
- Serrano, A.G.; Pérez-Gil, J. Protein–lipid interactions and surface activity in the pulmonary surfactant system. Chem. Phys. Lipids 2006, 141, 105–118. [Google Scholar] [CrossRef]
- Perez-Gil, J.; Weaver, T. Pulmonary surfactant pathophysiology: Current models and open questions. Physiology 2010, 25, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Haller, T.; Ortmayr, J.; Friedrich, F.; Völkl, H.; Dietl, P. Dynamics of surfactant release in alveolar type II cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1579–1584. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Hull, W.M.; Yoshida, M.; Wert, S.E.; Whitsett, J.A. SP-D and GM-CSF regulate surfactant homeostasis via distinct mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L697–L703. [Google Scholar] [CrossRef] [Green Version]
- Schürch, S.; Qanbar, R.; Bachofen, H.; Possmayer, F. The surface-associated surfactant reservoir in the alveolar lining. Neonatology 1995, 67 (Suppl. 1), 61–76. [Google Scholar] [CrossRef]
- Baumgart, F.; Ospina, O.L.; Mingarro, I.; Rodríguez-Crespo, I.; Pérez-Gil, J. Palmitoylation of pulmonary surfactant protein SP-C is critical for its functional cooperation with SP-B to sustain compression/expansion dynamics in cholesterol-containing surfactant films. Biophys. J. 2010, 99, 3234–3243. [Google Scholar] [CrossRef] [Green Version]
- Alcorn, J.L. Chapter 4—Pulmonary surfactant trafficking and homeostasis. In Lung Epithelial Biology in the Pathogenesis of Pulmonary Disease; Sidhaye, V.K., Koval, M., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 59–75. [Google Scholar] [CrossRef]
- Korfhagen, T.R.; Sheftelyevich, V.; Burhans, M.S.; Bruno, M.D.; Ross, G.F.; Wert, S.E.; Stahlman, M.T.; Jobe, A.H.; Ikegami, M.; Whitsett, J.A. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J. Biol. Chem. 1998, 273, 28438–28443. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Na, C.-L.; Korfhagen, T.R.; Whitsett, J.A. Surfactant protein D influences surfactant ultrastructure and uptake by alveolar type II cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2005, 288, L552–L561. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, M.; Grant, S.; Korfhagen, T.; Scheule, R.K.; Whitsett, J.A. Surfactant protein-D regulates the postnatal maturation of pulmonary surfactant lipid pool sizes. J. Appl. Physiol. 2009, 106, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Calle, M.; Olmeda, B.; Dietl, P.; Frick, M.; Pérez-Gil, J. Pulmonary surfactant protein SP-B promotes exocytosis of lamellar bodies in alveolar type II cells. FASEB J. 2018, 32, 4600–4611. [Google Scholar] [CrossRef] [Green Version]
- Botas, C.; Poulain, F.; Akiyama, J.; Brown, C.; Allen, L.; Goerke, J.; Clements, J.; Carlson, E.; Gillespie, A.M.; Epstein, C.; et al. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc. Natl. Acad. Sci. USA 1998, 95, 11869–11874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, T.E. Synthesis, processing and secretion of surfactant proteins B and C. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1998, 1408, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Voorhout, W.F.; Veenendaal, T.; Haagsman, H.P.; Weaver, T.E.; Whitsett, J.A.; van Golde, L.M.; Geuze, H.J. Intracellular processing of pulmonary surfactant protein B in an endosomal/lysosomal compartment. Am. J. Physiol. Lung Cell. Mol. Physiol. 1992, 263, L479–L486. [Google Scholar] [CrossRef] [PubMed]
- Nogee, L.M.; Garnier, G.; Dietz, H.C.; Singer, L.; Murphy, A.M.; deMello, D.E.; Colten, H.R. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J. Clin. Investig. 1994, 93, 1860–1863. [Google Scholar] [CrossRef] [PubMed]
- Danhaive, O.; Chapin, C.; Horneman, H.; Cogo, P.E.; Ballard, P.L. Surface film formation in vitro by infant and therapeutic surfactants: Role of surfactant protein B. Pediatric Res. 2015, 77, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardino de la Serna, J.; Perez-Gil, J.; Simonsen, A.C.; Bagatolli, L.A. Cholesterol Rules: Direct observation of the coexistend of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J. Biol. Chem. 2004, 279, 40715–40722. [Google Scholar] [CrossRef] [Green Version]
- Ruwisch, J.; Sehlmeyer, K.; Roldan, N.; Garcia-Alvarez, B.; Perez-Gil, J.; Weaver, T.E.; Ochs, M.; Knudsen, L.; Lopez-Rodriguez, E. Air space distension precedes spontaneous fibrotic remodeling and impaired cholesterol metabolism in the absence of surfactant protein C. Am. J. Respir. Cell Mol. Biol. 2020, 62, 466–478. [Google Scholar] [CrossRef]
- Glasser, S.W.; Burhans, M.S.; Korfhagen, T.R.; Na, C.-L.; Sly, P.D.; Ross, G.F.; Ikegami, M.; Whitsett, J.A. Altered stability of pulmonary surfactant in SP-C-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 6366–6371. [Google Scholar] [CrossRef] [Green Version]
- Kazzi, B.; Lederer, D.; Arteaga-Solis, E.; Saqi, A.; Chung, W.K. Recurrent diffuse lung disease due to surfactant protein C deficiency. Respir. Med. Case Rep. 2018, 25, 91–95. [Google Scholar] [CrossRef]
- Glasser, S.W.; Senft, A.P.; Whitsett, J.A.; Maxfield, M.D.; Ross, G.F.; Richardson, T.R.; Prows, D.R.; Xu, Y.; Korfhagen, T.R. Macrophage Dysfunction and Susceptibility to Pulmonary Pseudomonas aeruginosa Infection in Surfactant Protein C-Deficient Mice. J. Immunol. 2008, 181, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef]
- Nogee, L. Alterations in SP-B and SP-C expression in neonatal lung disease. Annu. Rev. Physiol. 2004, 66, 601–623. [Google Scholar] [CrossRef]
- Delestrain, C.; Blanchon, S.; Abbal, J.; Nattes, E.; Aissat, A.; Delattre, V.; Simon, S.; Tarze, A.; Bremont, F.; Epaud, R.; et al. A novel non-BRICHOS surfactant protein C mutation causing infantile interstitial lung disease is associated with reduced mature SP-C level. Eur. Respir. J. 2018, 52, PA633. [Google Scholar]
- Griese, M.; Lorenz, E.; Hengst, M.; Schams, A.; Wesselak, T.; Rauch, D.; Wittmann, T.; Kirchberger, V.; Escribano, A.; Schaible, T.; et al. Surfactant proteins in pediatric interstitial lung disease. Pediatric Res. 2016, 79, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Dai, D.; Liu, J.; Zhang, C.; Jin, T.; Shi, Y.; Jiang, G.; Mei, M.; Wang, L.; Qian, L. Clinical and genetic spectrum of interstitial lung disease in Chinese children associated with surfactant protein C mutations. Ital. J. Pediatrics 2019, 45, 117. [Google Scholar] [CrossRef]
- McCormack, F.X.; Whitsett, J.A. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J. Clin. Investig. 2002, 109, 707–712. [Google Scholar] [CrossRef]
- Kishore, U.; Greenhough, T.J.; Waters, P.; Shrive, A.K.; Ghai, R.; Kamran, M.F.; Bernal, A.L.; Reid, K.B.; Madan, T.; Chakraborty, T. Surfactant proteins SP-A and SP-D: Structure, function and receptors. Mol. Immunol. 2006, 43, 1293–1315. [Google Scholar] [CrossRef]
- Pastva, A.M.; Wright, J.R.; Williams, K.L. Immunomodulatory roles of surfactant proteins A and D: Implications in lung disease. Proc. Am. Thorac. Soc. 2007, 4, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Schürch, S.; Possmayer, F.; Cheng, S.; Cockshutt, A.M. Pulmonary SP-A enhances adsorption and appears to induce surface sorting of lipid extract surfactant. Am. J. Physiol. Lung Cell. Mol. Physiol. 1992, 263, L210–L218. [Google Scholar] [CrossRef]
- Schürch, S.; Schurch, D.; Curstedt, T.; Robertson, B. Surface activity of lipid extract surfactant in relation to film area compression and collapse. J. Appl. Physiol. 1994, 77, 974–986. [Google Scholar] [CrossRef]
- Clark, H.; Palaniyar, N.; Strong, P.; Edmondson, J.; Hawgood, S.; Reid, K.B.M. Surfactant Protein D Reduces Alveolar Macrophage Apoptosis In Vivo. J. Immunol. 2002, 169, 2892–2899. [Google Scholar] [CrossRef] [Green Version]
- Autilio, C.; Pérez-Gil, J. Understanding the principle biophysics concepts of pulmonary surfactant in health and disease. Arch. Dis. Child. Fetal Neonatal Ed. 2019, 104, F443–F451. [Google Scholar] [CrossRef]
- Nag, K.; Rodriguez-Capote, K.; Panda, A.K.; Frederick, L.; Hearn, S.A.; Petersen, N.O.; Schürch, S.; Possmayer, F. Disparate effects of two phosphatidylcholine binding proteins, C-reactive protein and surfactant protein A, on pulmonary surfactant structure and function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1145–L1153. [Google Scholar] [CrossRef] [PubMed]
- Holm, B.A.; Wang, Z.; Notter, R.H. Multiple Mechanisms of Lung Surfactant Inhibition. Pediatric Res. 1999, 46, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomaznik, M.; Nova, Z.; Calkovska, A. Pulmonary surfactant and bacterial lipopolysaccharide: The interaction and its functional consequences. Physiol. Res. 2017, 66. [Google Scholar] [CrossRef] [PubMed]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewith, H.; Pandit, J.J. Lung ventilation and the physiology of breathing. Surgery 2020, 38, 233–239. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Solvent A (%) | Solvent B (%) | Flow (µL/min) |
|---|---|---|---|
| 0.01 | 99 | 1 | 0.350 |
| 2.00 | 95 | 5 | 0.350 |
| 2.01 | 80 | 20 | 0.350 |
| 5.00 | 75 | 25 | 0.350 |
| 5.01 | 1 | 99 | 0.350 |
| 7.50 | 99 | 1 | 0.350 |
| 8.50 | 99 | 1 | 0.350 |
| Standard Name | nmol Added to Each Sample at Extraction |
|---|---|
| Dimyristoyl-phosphatidylcholine (DMPC) | 10 |
| Dimyristoyl phosphatidylethanolamine (DMPE) | 4 |
| Dimyristoyl phosphatidylserine (DMPS) | 2 |
| Dimyristoyl phosphatidylglycerol (DMPG) | 2 |
| Dimyristoyl phosphatidic acid (DMPA) | 1 |
| Lysophosphatidylcholine (LPC17:0) | 1 |
| Sphingomyelin (SM16:0) | 1 |
| Ceramide 12:0 | 2 |
| Triacylglycerol (TAG) | 10 |
| MS | Scan | MS/MS Mode | Range (m/z) |
|---|---|---|---|
| ES+ | a | Full positive | 2–1300 |
| ES- | a | Full Negative | 2–1300 |
| ES- | a | Partial Negative | 400–900 |
| Gene | Primer Sequence (5′ → 3′) | Accession Numbers |
|---|---|---|
| ACTB | FWD-AGCTGTGCTATGTTGCTC REV-CACTTCATGATGGAATTGAATGTAG | BC138614.1 |
| HPRT1 | FWD-GCTGGATTACATTAAAGCACTGAAT REV-AAAGTTTGCATTGTTTTACCAGTGT | NM_013556 |
| YWHAZ | FWD-TTGAGCAGAAGACGGAAGGT REV-GAAGCATTGGGGATCAAGAA | NM_011740 |
| PGK1 | FWD-GTCGTGATGAGGGTGGACTT REV-TTTGATGCTTGGAACAGCAG | NM_008828 |
| RpPo1 | FWD-CAACCCTGAAGTGCTTGACAT REV-AGGCAGATGGATCAGCCA | NM_007475.5 |
| PPIA | FWD-ACCAAACACAAACGGTTCCC REV-TGCCTTCTTTCACCTTCCCAAA | NM_008907.1 |
| SP-A | FWD-TTTCCACCAATGGGCAGTCA REV-AGAAGCCCCATCCAGGTAGT | NM_023134 |
| SP-B | FWD-GCTACTGCTGCTTCCTACCC REV-TGGCACAGGTCATTAGCTCC | NM_147779 |
| SP-C | FWD-GGAGCACCGGAAACTCAGAA REV-GGAGCCGCTGGTAGTCATAC | NM_011359 |
| SP-D | FWD-AGCCCAACAACAATGGTGGA REV-CACAGATAACAAGGCGCTGC | NM_009160 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paget, T.L.; Parkinson-Lawrence, E.J.; Trim, P.J.; Autilio, C.; Panchal, M.H.; Koster, G.; Echaide, M.; Snel, M.F.; Postle, A.D.; Morrison, J.L.; et al. Increased Alveolar Heparan Sulphate and Reduced Pulmonary Surfactant Amount and Function in the Mucopolysaccharidosis IIIA Mouse. Cells 2021, 10, 849. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10040849
Paget TL, Parkinson-Lawrence EJ, Trim PJ, Autilio C, Panchal MH, Koster G, Echaide M, Snel MF, Postle AD, Morrison JL, et al. Increased Alveolar Heparan Sulphate and Reduced Pulmonary Surfactant Amount and Function in the Mucopolysaccharidosis IIIA Mouse. Cells. 2021; 10(4):849. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10040849
Chicago/Turabian StylePaget, Tamara L., Emma J. Parkinson-Lawrence, Paul J. Trim, Chiara Autilio, Madhuriben H. Panchal, Grielof Koster, Mercedes Echaide, Marten F. Snel, Anthony D. Postle, Janna L. Morrison, and et al. 2021. "Increased Alveolar Heparan Sulphate and Reduced Pulmonary Surfactant Amount and Function in the Mucopolysaccharidosis IIIA Mouse" Cells 10, no. 4: 849. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10040849