
From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas


Neurology Clinic and National Center for Tumor Diseases, University Hospital Heidelberg, 69120 Heidelberg, Germany
*
Author to whom correspondence should be addressed.
Cells 2021, 10(5), 1225; https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051225
Submission received: 9 April 2021
/
Revised: 7 May 2021
/
Accepted: 7 May 2021
/
Published: 17 May 2021
(This article belongs to the Special Issue Molecular Basis of Brain Tumors)
Abstract
:In this review, we discuss the use of the alkylating agent temozolomide (TMZ) in the treatment of IDH-mutant gliomas. We describe the challenges associated with TMZ in clinical (drug resistance and tumor recurrence) and preclinical settings (variabilities associated with in vitro models) in treating IDH-mutant glioma. Lastly, we summarize the emerging therapeutic targets that can potentially be used in combination with TMZ.
1. Introduction
Gliomas are the most common primary malignant tumors in the central nervous system. Grade 2 and 3 gliomas are referred to as lower grade gliomas (LGG) and harbor mutations in the isocitrate dehydrogenase (IDH) gene [1]. IDH-mutant gliomas have a slower growth rate and longer survival than IDH wild type (IDH-wt) tumors [1,2]. IDH-mutant gliomas are classified into two subgroups based on the presence (astrocytoma) or absence (oligodendroglioma) of chromosome arms 1p/19q [3] and histological criteria [4]. Recently, the European Association of Neuro-Oncology (EANO) stratified IDH-mutant gliomas into three WHO grades: oligodendroglioma, WHO grade 2 or 3; astrocytoma, WHO grade 2 or 3; astrocytoma, WHO grade 4 [5]. Although slower growing (at a rate of ~4–5 mm per year [6]), the majority of IDH-mutant LGGs eventually undergo malignant progression due to activation of the PI3K/mTOR pathway as a result of PTEN loss [7,8] or enhanced PDGF signaling [9]. Detailed molecular diagnostic markers, and other common molecular and pathway alterations in IDH-mutant gliomas are summarized in Table 1.
The IDH gene encodes the enzyme isocitrate dehydrogenase, which converts isocitrate to α-ketoglutarate (α-KG). α-KG an intermediate of the tricarboxylic acid (TCA) cycle that contributes to the production of NADPH. NADPH is necessary to reduce oxidized glutathione to glutathione, which directly neutralizes free radicals and reactive oxygen species (ROS). Overall, 65% of total NADPH in glioblastoma (GBM) is driven by the enzymatic activity of IDH, which is reduced to 38% when IDH is mutated [10]. There are three IDH isoforms, IDH1, IDH2, and IDH3, which are encoded by different genes. IDH1 is localized in the cytosol and peroxisomes, while IDH2 and IDH3 are located in the mitochondria. Among them, IDH1 is most frequently mutated in gliomas and harbors a monoallelic missense mutation of arginine to histidine at position 132 (IDH1R132H) at the catalytic site of the enzyme. IDH mutation produces a neomorphic enzyme that converts α-KG to D-(R)-2-hydroxyglutarate (2-HG), leading to the accumulation of 2-HG in the tumor [11]. The oncometabolite 2-HG is a competitive inhibitor of α-KG-dependent enzymes, including DNA demethylases (family of TET enzymes) and histone demethylases (family of Jumonji enzymes) [12,13]. This inhibition modifies the epigenetic status of histones and DNA, resulting in a plethora of cellular changes, including DNA hypermethylation [14] and altered histone methylation [15] (Figure 1).
Treatment of LGGs includes surgery, radiation, and chemotherapy with either procarbazine/lomustine/vincristine (PCV) or temozolomide (TMZ). Here, we focus on the use of TMZ in IDH-mutant LGGs. First, we will present the effect of IDH mutation on cellular metabolism, epigenetic modifications, and the targeted therapies associated with these alterations. Second, we will discuss the use of TMZ in the treatment of IDH-mutant gliomas, including its toxicity, TMZ-associated molecular signature in tumor recurrence, and drug resistance, and discuss the synthetic lethality opportunities that emerge with TMZ treatment of IDH-mutant gliomas. Third, we will discuss the challenges of using TMZ to treat IDH-mutant gliomas in the preclinical setting, including non-consensus TMZ dosage and regimen, variable methods in measuring cell viability, and difficulties in culturing IDH-mutant glioma cell lines. To conclude the review, we will discuss targeted vaccine therapy that may facilitate the treatment of IDH-mutant gliomas.
2. Cellular Alterations upon IDH Mutation and Targeted Therapies
Redox balance. IDH is a central enzyme of the TCA cycle; therefore, many studies have sought to understand the unique metabolic profiles reprogrammed by mutant IDH1 to therapeutically exploit these metabolic vulnerabilities [16] (Figure 2). IDH activity contributes to the majority of NADPH, which is used to maintain redox balance by converting oxidized glutathione to reduced glutathione. Therefore, IDH mutation leads to an increased oxidative burden, making interrupted redox balance a good therapeutic target. Synthesis of glutathione is mediated by the transcription factor nuclear factor erythroid 2-related factor (NRF2). Suppression of NRF2 by the natural compound brusatol [17], triptolide [18] resulted in profound tumor suppression in IDH-mutant xenografts, accompanied by overwhelming oxidative stress.
Glutamate. Metabolomic analysis using glioma cell lines and surgical specimens indicated significantly reduced levels of glutamate in IDH-mutant gliomas [19,20,21,22], as glutamate used to produce α-KG is now used to synthesize 2-HG. IDH1 mutant tumor cells rely on glutaminase (GLS) activity to maintain α-KG homeostasis, making GLS inhibition a good therapeutic target [23,24]. However, preclinical studies indicated that the only GLS inhibitor CB-839 has only a moderate antiproliferative effect on IDH1-mutant cells, as cells compensate for reduced glutamate levels by upregulating asparagine synthetase (ASNS) [25], to generate glutamate via one of its amino acid precursors [26] (Figure 2).
Lipid synthesis. Altered lipid biosynthesis is another significant vulnerability of IDH-mutant gliomas. Biopsies from glioma patients showed lower phospholipid levels in IDH-mutant tumors compared with IDH-wt tumors. This effect was mediated by autophagic degradation of the endoplasmic reticulum (ER), termed ER-phagy [27]. Since ER is the site of phospholipid synthesis, late-stage autophagy inhibitors chloroquine (CQ) and bafilomycin A1 (BAF) restore phospholipid levels and trigger apoptosis in vitro and in vivo. This suggests that inhibition of ER-phagy may be a novel therapeutic option for IDH-mutant gliomas [27]. In a recent study, IDH1 mutation was found to increase monounsaturated fatty acids and their phospholipids [28]. The sphingolipid signaling pathway is also frequently activated in IDH-mutant gliomas, and inhibition of sphingosine kinase I (SphK1) with N,N-dimethylsphingosine (DNMS) specifically leads to cell death in IDH1-mutant gliomas [29]. All these studies provide therapeutic evidence for targeting lipid biosynthesis in IDH-mutant gliomas (Figure 2).
NAD. Another extreme metabolic vulnerability is the low NAD+ levels in IDH-mutant cells, making further depletion of NAD+ a good therapeutic target [30]. Biosynthetic and consumptive processes maintain the intracellular NAD+ pool. Inhibition of the NAD+ synthesis enzyme nicotinamide phosphoribosyl transferase (NAMPT) with FK866 and GMX1778 [30] or activation of the NAD+ consuming enzyme sirtuin (SIRT) with SIRT1-activating compounds, or a combination thereof, reduce cellular NAD+ levels and inhibit the growth of IDH1-mutant tumor cells [31]. This suggests that targeting NAD+ metabolites may be a promising treatment option for these tumors. IDH-mutant gliomas are also sensitive to biguanides such as metformin, a metabolic inhibitor, which alters whole-body and cellular energy metabolism [32]. This treatment is currently being investigated in a phase Ib/II clinical trial to assess the efficacy of CQ in IDH-mutant gliomas (NCT02496741) [33,34]. This trial will provide direct evidence for targeting the metabolic rewiring for cancer therapy (Figure 2).
Epigenetic modifications. IDH-mutant gliomas exhibit a cytosine-phosphate-guanine (CpG) island methylator phenotype (G-CIMP) [35] characterized by a genome-wide hypermethylation induced directly by mutant IDH1 [14] (Figure 1b). Several studies from us and others have shown that DNMT1 inhibitors, decitabine (DAC) [36,37] and azacitidine (AZA) [38], exerts an antiproliferative tumor effect in vitro and in vivo in IDH1-mutant gliomas.
3. Temozolomide Treatment in IDH-Mutant Gliomas
Standard-of-care treatment for gliomas includes maximal surgical resection, possibly followed by radiotherapy (RT) and chemotherapy with PCV or TMZ. Several randomized clinical trials [39] investigating dosing (EORTC 22844 [40]) and timing (EORTC 22845 [41]) for RT in LGGs show that RT alone provides no significant benefit for overall survival. Similarly, TMZ alone showed no significant difference in progression-free survival in patients with LGGs compared with the efficacy of RT (EORTC 22033-26033 [42]). However, RT combined with TMZ or PCV resulted in an overall survival benefit in patients [43]. IDH-mutant oligodendrogliomas benefit from the addition of PCV to RT (RTOG 9802 [44,45], RTOG 9402 [46], EORTC 26951 [47], and NOA-04 [48]), while RT plus TMZ treatment shows more benefit in astrocytomas in clinical (EORTC 26053-22054 (CATNON) [49], RTOG 0424 [50]), and retrospective [51] studies. The interim analysis of the CATNON trial indicate a trend toward benefit with concurrent TMZ in IDH-mutant tumors, but not in IDH-wt gliomas. Thus, EANO recommends RT + TMZ for the treatment of newly diagnosed astrocytomas. For oligodendrogliomas, EANO recommends RT + PCV for initial treatment, and TMZ is only recommended for recurrent tumors not being pre-treated with TMZ [5].
Currently, there are no mature data comparing TMZ and PCV or their combination with radiation for LGGs. The ongoing clinical trial ALLIANCE-N0577-CODEL comparing RT + TMZ with RT + PCV for anaplastic oligodendrogliomas with 1p/19q co-deletion could potentially provide a more definitive comparison between the two regimens [52]. Both PCV and TMZ have been associated with grade 3 and 4 hematologic toxicities. Clinicians largely suggest TMZ to patients instead of PCV (>85%) [53], considering the relative difficulty of administering intravenous vincristine and the greater toxicity of PCV [54], whereas TMZ is easy to administer and generally well tolerated [43,55]. The ongoing phase III EORTC-1635-BTG (Wait or Treat?) is a randomized phase III trial comparing early adjuvant treatment with radiotherapy and adjuvant temozolomide to active surveillance in patients with resected IDH-mutant astrocytoma. Here, we summarize the current challenges related to TMZ in gliomas with a particular focus on IDH-mutant tumors.
3.1. Mechanisms of TMZ Toxicity
TMZ is administered orally in capsules at a dose of 150–200 mg/m2 for 5 out of 28 days for 6–12 cycles [5]. TMZ is a lipophilic DNA alkylating prodrug, and the cytotoxicity of TMZ is mediated by the addition of methyl groups to DNA. TMZ is an imidazotetrazine derivative of dacarbazine. Under neutral pH and aqueous conditions, it spontaneously decarboxylates to generate 5-(3-methyltriazen-1-yl)-imidazole-4-carboxamide (MTIC), which is further degraded to 4-amino-5-imidazole-carboxamide (AIC), and a highly reactive methyldiazonium ion that acts as a DNA methylating species [56] (Figure 3a). About 60–80% of methyl groups are added at DNA guanine residues (N7-MeG), 10–20% of the methyl groups are added at adenine (N3-MeA), and 10% of methyl groups at guanine (O6-MeG) [57] (Figure 3b). Single damaged bases, N7-MeG and N3-MeA, are readily removed by the rapid and efficient base excision repair (BER) system before replication. Therefore, the key toxic insult of TMZ is attributed to the O6-meG lesions [58,59].
O6-meG is considered the most genotoxic base modification due to the subsequent nucleotide mispairing with thymine (T) instead of cytosine (C) during DNA replication (Figure 3c). During replication, DNA polymerase inserts T opposite O6-meG. The mismatch repair (MMR) system can detect and repair these mismatches through the MutS and MutL complexes. The MutS recognition complex, including MutSα (an MSH2/MSH6 heterodimer) and MutSb (MSH2/ MSH3 heterodimer), identifies base–base mismatches and binds the O6-meG: T mismatch. Upon binding to the mismatch, the MutS complex recruits MutL (MLH1/PMS2 dimer) to the site of DNA damage. Together, these proteins excise a stretch of single-stranded DNA (ssDNA) containing the mispaired T, creating a gap in the DNA, while leaving the O6-meG adduct on the template strand intact [60]. DNA polymerase fills the gap by reinserting T opposite O6-meG, triggering another round of MMR which leads to repeated attempts to repair the same base T. This futile MMR cycling and accumulation of ssDNA gaps lead to successively longer DNA reinsertion and excision, which generates double strand breaks (DSBs) in subsequent rounds of replication and induce cell cycle arrest in G2/M phase, apoptosis and autophagy [61]. Thus, it needs two cell divisions for the emergence of TMZ cytotoxicity [62] (Figure 3d).
However, O6-meG lesions can be directly removed by O6-methylguanine DNA methyltransferase (MGMT) through covalent transfer, a process that effectively repairs the alteration prior to replication (Figure 3e). MGMT promoter methylation is a predictive biomarker of TMZ response in GBM. In general, the repair of O6-meG depends on the number of MGMT molecules per cell and the rate of MGMT regeneration [63]. In summary, the cytotoxicity from TMZ depends on low MGMT levels [64] and an intact MMR pathway [65].
3.2. Maintenance of TMZ Sensitivity
Efforts have focused on maintaining TMZ sensitivity by reducing MGMT levels or attenuating the activity of the BER and HR pathways for the duration of TMZ treatment to prevent resistance.
MGMT. O6–benzylguanine (O6–BG) is a potent inhibitor of the repair protein O6–alkylguanine–DNA alkyltransferase (AGT) that effectively inhibits MGMT activity by suicide inactivation. O6-BG binds and inactivates AGT, and until new AGT protein is synthesized, the cells have increased sensitivity to TMZ [66,67,68], leading to several clinical trials combining O6-BG and TMZ [67,69,70,71,72,73]. A phase II study showed that one-day dosing of O6-BG plus TMZ restored TMZ sensitivity in patients with TMZ-resistant anaplastic IDH-mutant gliomas [73] (Table 2).
MMR. MMR is regulated by multiple signaling pathways and responds to many stimuli [74]. Mutations in MMR genes and loss of MMR function are frequently detected in tumor samples [75]. Nevertheless, there is little research on how to maintain MMR integrity or directly increase MMR activity. One study showed that EGFRvIII expression and MAPK activation lead to increased MMR and therefore TMZ sensitivity [76], with direct clinical relevance that anti-EGFRvIII and anti-MAPK strategies should be used with caution in combination with TMZ.
BER. Many efforts have been made to target the downstream ssDNA repair and HR system to maintain TMZ sensitivity. One target from the base-excision repair (BER) pathway is poly (ADP-ribose) polymerase 1 (PARP1), which facilitates DNA repair by binding to single-strand breaks and recruiting DNA repair proteins to the site of damage [77]. PARP inhibitors (PARPi), INO-1001 [78], NU1025 [79], ABT-888 (veliparib) [80,81] and pamiparib [82], restored sensitivity in TMZ resistant glioma cells and xenografts. Several PARPi in combination with TMZ in GBM have been registered for clinical trials [83,84,85]. However, there are only two phase I clinical trials testing the efficacy of pamiparib (BGB-290) in combination with TMZ in newly diagnosed or recurrent IDH1/2-mutant gliomas (NCT03914742 and NCT03749187), and its clinical efficacy in IDH-mutant gliomas has yet to be demonstrated [86] (Table 2).
HR. One target from the HR pathway is the homologous recombinase RAD51. RAD51 is involved in DNA strand exchange between homologous DNA sequences. Exogenously expressed mutant IDH1 increases RAD51-driven HR and leads to increased TMZ resistance, and RAD51 knockdown increases the sensitivity of glioma cells to TMZ [87]. Several drug screens have identified inhibitors of RAD51 [88], but there is only one active clinical trial directly targeting RAD51 using a small molecule inhibitor CYT-0851 (NCT03997968). Another HR target is PLK1 (Polo-like kinase 1), which phosphorylates BRCA1 [89] and RAD51 [90] to promote homologous recombination. The combination of TMZ with a PLK1 inhibitor, BI2536, significantly suppressed the growth of IDH1-mutant glioma tumors [91], induced G2/M arrest, and suppressed cell proliferation and sphere formation [92]. Due to the toxicity of the small molecule inhibitor, knockdown of PLK1 using a small interfering RNA (siRNA) was combined with TMZ for glioma treatment, which showed enhanced anti-tumor activity both in vitro and in vivo [93]. However, limited success has been reported in preclinical studies with PLK1 inhibitors. ATM (ataxia telangiectasia mutated), a protein kinase that is a central mediator of responses to DNA double-strand breaks in cells [94], can also be targeted therapeutically. KU-55933, an ATM inhibitor, enhances the cytotoxic effects of TMZ in IDH1-mutant glioma cell lines [95].
Tumor Microtubes. Recently, there has been increasing evidence that tumor microtubes (TM) are an important mechanism of therapy resistance in gliomas. Gliomas interconnect and communicate through a network of TMs [96]. TM-connected glioma cells can self-repair, and are resistant to radiotherapy [96] and TMZ [97]. Inhibition of gap junctions with INI-0602 sensitizes primary GBM cells to TMZ [98], which shows the importance of pharmacological inhibition of the TM network. Furthermore, disrupting TM-based networks with meclofenamate (MFA) [99] sensitized primary glioblastoma cells to TMZ. The fact that TMs are more abundant in astrocytomas than in oligodendrogliomas [96], might explain why astrocytoma patients have a better response to TMZ than oligodendroglioma patients. A phase I/II trial evaluating safety as well as feasibility of a combined MFA-TMZ approach in relapsed MGMT-methylated glioblastoma (“MecMeth” EudraCT2021-000708-39) is being initiated in Germany [99].
3.3. TMZ-Associated Hypermutation
The first report investigating the effect of TMZ in the treatment of LGGs [100] showed that although most tumors exhibited initial chemosensitivity, the majority of tumors resumed progressive growth within a year of TMZ treatment, with astrocytomas (20/33) exhibiting a higher regrowth rate than oligodendrogliomas (5/30), implying that astrocytomas acquire accelerated TMZ resistance than oligodendrogliomas. Another long-term follow-up study showed that most oligodendrogliomas resumed growth within 3 years after TMZ [101].
TMZ resistance can be acquired either by elevated MGMT levels [102,103] or by mutations in the MMR machinery [104,105,106,107,108] that prevent futile MMR cycles at unrepaired O6-meG lesions. In the absence of MGMT-mediated repair in conjunction with deficient MMR, long-term TMZ treatment causes cells to accumulate G:C>A:T transitions throughout the genome, resulting in a hypermutator phenotype in recurrent tumors [63,109] (Figure 3e). Long-term TMZ treatment could also inactivate MMR pathway genes leading to hypermutation [109].
TMZ-induced hypermutation is observed more frequently in IDH-mutant than in IDH-wt gliomas [110,111]. However, it is not clear which subtype is more prone to develop the hypermutator phenotype. Reports from paired primary and TMZ-treated recurrent tumors show that astrocytomas have a higher rate of hypermutation [111,112], while data from random patient samples show that TMZ-induced hypermutation is more prevalent in oligodendrogliomas [109,110]. TMZ-induced hypermutation has been associated with a worse prognosis [112]; however, a larger cohort from the Glioma Longitudinal Analysis (GLASS) consortium shows no differences in overall survival between hypermutators and non-hypermutators [111].
Increased tumor mutation burden correlates with an elevated neoantigen load, indicating the potential to induce a durable response to immunotherapy [113]. However, current data show no discernible differences in the extent of immunoediting between initial and TMZ-treated relapsed hyper-mutated gliomas [111], and neoantigens from the recurrent hypermutators have relatively poor immunogenic qualities which may result in a weak anti-tumor T-cell response and likely a poor response to immunotherapy [109]. A current clinical trial is evaluating the immune-activating antibody pembrolizumab (MK-3475) in recurrent malignant gliomas that exhibit the hypermutator phenotype (NCT02658279).
3.4. TMZ-Induced Cellular Adaptations and Combination Therapy in IDH-Mutant Glioma
In addition to the known MGMT activity and DNA repair pathways in conferring TMZ resistance, efforts have been made to understand genetic, epigenetic, or metabolic adaptations following TMZ treatment. This knowledge could lead to synthetic lethal targeted strategies, with combinations of targeted therapies to circumvent some resistance mechanisms to delay or prevent malignant progression and recurrence [114,115]. Previous research has mainly focused on TMZ resistance in IDH-wt GBM [116,117,118], and here we summarize the current literatures on TMZ resistance mechanisms and therapeutic options in IDH-mutant gliomas.
3.4.1. Genetic Mutations Associated with TMZ Treatment in IDH Mutant Glioma
Direct comparison of the genomic landscape of gliomas at initial diagnosis and recurrence has provided insight into the genomic alterations that may be associated with tumor recurrence after TMZ. Analysis of copy number alterations (CNAs) from primary IDH-mutant and IDH-wt gliomas of all grades revealed amplification of cyclins and cyclin-dependent kinase genes in IDH-mutant gliomas [119] (Table 1). A cohort of six pairs of initial untreated and TMZ treated recurrent IDH-mutant gliomas showed that recurrent tumors have driver mutations that activate retinoblastoma (Rb) and mammalian target of rapamycin (mTOR) pathways [112], which might drive malignant progression. The mTOR inhibitors such as rapamycin (RAPA) [120] have been reported to enhance TMZ-induced autophagic death of GBM cells and inhibition of the Akt-mTOR signaling pathway with amlexanox enhances TMZ-induced anti-tumor effects in preclinical GBM models [121]. An orally bioavailable dual PI3K/mTOR inhibitor, XL765 (voxtalisib), produced additive toxicity when combined with TMZ in genetically diverse GBM xenografts [122]. A phase I clinical trial (NCT00704080) demonstrated a favorable safety profile and a moderate inhibition of the PI3K/mTOR pathway in all glioma subtypes [123]. Sequential treatment of TMZ followed by PX-866, a PI3K inhibitor, inhibited TMZ-induced autophagy survival and enhanced apoptosis in GBM cells [124]. These findings suggest that PI3K/mTOR/Rb signaling pathways can be targeted separately or together to prevent tumor progression after TMZ treatment.
However, a study by the GLASS consortium comparing 23 pairs of untreated primary and TMZ-treated recurrent IDH-mutant gliomas [111] did not identify specific driver mutations associated with TMZ resistance. Across all cohorts, the hotspot IDH1R132H mutation was not lost during progression and remained clonal in all progressed tumors [110], providing a good rationale for IDHR132H vaccines for targeted therapies.
CRISPR-based screening enables sensitive detection of drug-gene interactions directly in human cells. Although no genome-wide CRISPR-Cas9 screen has been performed in IDH-mutant glioma models, results from GBM patient-derived lines [125] and GBM adherent lines [126] indicated that mismatch repair (MMR) and HR pathways are involved in TMZ resistance. In addition to the MMR pathways, an interesting molecular alteration detected in the human GBM cell line is NRF2 activation, and inhibition of NRF2 enhanced the anti-tumor effect of TMZ in glioma cells [127]. Since NRF2 is important for maintaining the redox balance in IDH-mutant gliomas and increasing ROS has been shown to augment chemosensitivity in IDH-mutant glioma [128,129], it is plausible that NRF2 inhibitors in combination with TMZ may be promising for the treatment of IDH-mutant gliomas.
Pathway analysis from RNA-seq data obtained from preclinical GBM models showed that epithelial–mesenchymal transition, Wnt signaling, and immune response were the most significantly activated pathways in TMZ-resistant cell lines [130]. In addition, negative regulation of telomere maintenance via telomerase was enriched in TMZ-sensitive glioma cell lines. A synergistic effect of a combination treatment of TMZ and a telomerase inhibitor, BIBR1532, was observed in in vitro models of GBM [130]. Whether telomerase inhibitors in combination with TMZ have an anti-tumor effect in IDH-mutant gliomas requires further investigation.
3.4.2. Epigenetic Alterations upon TMZ Treatment
DNA methylation. Preclinical studies have shown that high TMZ concentration leads to a short-term increase in total 5-methylcytosine (hypermethylation), while repeated low TMZ doses lead to DNA hypomethylation [131]. This indirect effect on DNA methylation status may partly explain why 5-azacytidine (AZA) in combination with TMZ has a better anti-tumor effect in IDH1-mutant glioma patient-derived xenograft (PDX) models [132]. A phase I clinical trial of AZA combined with TMZ in patients with unresectable or metastatic soft tissue sarcoma or malignant mesothelioma shows that both drugs can be administered at their full dose without dose-limiting toxicities (NCT00629343) [133]. DAC, another DNA methyltransferase inhibitor, has been shown to potentiate TMZ treatment by enhancing the effects of DNA damage [134] and DNA mismatch repair [135] in GBM. A phase I/II clinical trial of the combination of DAC and TMZ in metastatic melanoma has shown that DAC can be safely added to extended-schedule TMZ and leads to improved response rates and progression-free survival (PFS) and overall survival (OS) rates in patients [136]. Due to the dose-dependent effect of TMZ on epigenetic modifications, further studies are needed to determine appropriate treatment regimens for the combination of TMZ and epigenetic therapy to achieve optimal clinical benefit.
Histone methylation. An inhibitor of histone methyltransferase (HMT) G9a, BIX01294, also exerted a synergistic effect with TMZ in GBM [137], possibly by enhancing the autophagy pathway. JIB-04, a novel inhibitor of Jumonji demethylases [138], synergized strongly with TMZ [139,140] in GBM in vitro and in vivo. Since IDH-mutant gliomas exhibit increased histone H3 lysine 9 (H3K9) methylation [141], the combination of G9a and TMZ may be a potential therapeutic target in IDH-mutant gliomas.
Histone acetylation. Several histone deacetylase (HDAC) inhibitors synergize with TMZ. For example, vorinostat [142] is well tolerated in combination with TMZ in GBM patients in a phase II trial (NCT00731731) [143] and are currently evaluated in combination with RT. Valproic acid (VPA) [144], another HDAC inhibitor with concurrent RT and TMZ are also well tolerated in GBM in a phase II study (NCT00302159) [145,146]. In a phase I clinical trial, a triple agent of dual epigenetic therapy, a combination of DAC, panobinostat (an HDAC inhibitor) and TMZ was well-tolerated, and its further efficacy is currently being evaluated in a phase II trial (NCT00925132) [147]. As epigenetic alterations may represent a global mechanism of resistance in cancer [148], preclinical experiments and clinical trials will clarify whether epigenetic therapy can act synergistically with TMZ in IDH-mutant gliomas.
Here, we summarize the current epigenetic drugs with TMZ combination therapy in Table 3.
3.4.3. Metabolic Changes after TMZ Treatment
Glutamate. Previous studies have indicated that long-term TMZ treatment leads to changes in amino acid metabolism in preclinical models of oligodendroglioma [149]. Other reports identified that increased glutamate/glutamine/GLX (the sum of glutamate and glutamine) levels could be an early indication of response to TMZ treatment in IDH1-mutant gliomas [150,151]. IDH-mutant tumors have lower glutamate levels; thus, combination therapy of GLS inhibitor and TMZ may provide a greater benefit in IDH-mutant gliomas. Loss of xCT/SLC7A11, the glutamate exchanger that plays a role in ferroptosis, leads to increased vulnerability to TMZ [152,153]. These studies suggest that the effect of TMZ can be potentiated by ferroptosis inducing agents such as erastin and sorafenib. Another study showed that the addition of the glutaminase inhibitor CB-839 to TMZ significantly reduced aspartate and glutamate levels in an IDH-mutant patient-derived glioma xenograft model [154]. A phase I clinical study is currently evaluating the combination of CB-839, RT, and TMZ in IDH-mutated diffuse or anaplastic astrocytomas (NCT03528642) [155].
Phospholipid. The late-stage autophagy inhibitors chloroquine (CQ) and bafilomycin A1 (BAF) restore phospholipid levels and inhibit clonogenicity of IDH-mutant glioma cells. CQ enhances the cytotoxic effects of TMZ in GBM [156], and its clinical impact is being investigated in a phase I trial (NCT02378532). It is possible that the combination of CQ and TMZ disrupts the phospholipid balance and has greater synergistic effect in IDH-mutant gliomas.
NAD+. TMZ treatment leads to NAD+ consumption driven by PARP activation, as NAD+ is a known PARP cofactor. In IDH1-mutant cells with already low basal NAD+ levels, this surge in consumption leads to a further reduction in NAD+. Importantly, this metabolic imbalance introduces a window of hypervulnerability to NAD+ biosynthesis inhibitors [30]. Indeed, combined TMZ and NAMPT inhibition showed better efficacy in vivo than either agent alone [157]. Although the role of PARP in TMZ resistance is paradoxical as PARP needs to be inhibited to suppress its DNA repair function to maintain TMZ sensitivity but should be activated to drive NAD+ scarcity for its anti-tumor effect in IDH-mutant cells. The ongoing clinical trials of PARPi + TMZ in IDH-mutant glioma (NCT03914742, NCT03749187, NCT04394858, NCT01026493) [158] will give us a clear answer in the near future (Table 3). NAD+ is used for making poly (ADP-ribose) (PARylation) to recruit DNA repair factors [159]. PARylation is eventually degraded by PAR glycohydrolase (PARG), and NAD+ is regenerated. Therefore, combining TMZ with a PARG inhibitor COH34 leads to a scarcity of available NAD+, which is highly effective against IDH-mutant gliomas [160]. We expect PARG inhibitors with better toxicity profiles to be developed for preclinical and clinical trials in the near future.
Glucose. TMZ treatment has been shown to increase the expression of glucose transporters (GLUTs) [161,162], which triggers higher glycolytic activity and decreases the response to TMZ treatment, while inhibition of GLUT/SLC2A enhances the effect of TMZ [162]. Combination treatment with TMZ and paclitaxel (Taxol), a microtubule inhibitor, sensitized Taxol-resistant glioma cells via inhibition of glucose metabolism [163] and is currently in a phase II trial for the treatment of patients with metastatic melanoma (NCT01009515) [164]. Metformin, another metabolic inhibitor, alters both whole-body and cellular energy metabolism, and also shows a synergistic effect when combined with TMZ in GBM [165]. Trehalose, a natural disaccharide of glucose, combined with TMZ reduced clonogenicity and enhanced autophagic effects in melanoma cells [166]. Whether targeting glucose metabolism enhances the efficacy of TMZ in IDH-mutant gliomas requires further investigation.
A triple therapy combination with TMZ, CQ, and rapamycin decreased mitochondrial function and induced lysosome-dependent apoptotic cell death [167], suggesting that combinatorial targeting of metabolic and genetic alterations may be a good therapeutic option in cancer therapy in the future. Here, we summarize in Table 4 current vulnerable targets that could potentially be combined with TMZ in preclinical and clinical settings. Some of these have been tested only in GBM or other tumor entities but have the potential to be applicable to IDH-mutant gliomas as well.
4. Challenges of Using TMZ to Treat IDH-Mutant Glioma Cells in the Preclinical Setting
A recent review summarizes the four main limitations associated with the use of TMZ in preclinical models of GBM [168]: (1) the dosing and timing regimen of TMZ between clinical data and preclinical experimental design; (2) the dissolving agent, such as DMSO, and its low-dose toxicity; (3) cell lines that do not accurately represent the whole population; and (4) immunocompromised animal models with deficient immune systems. In this section, we focus on the use of TMZ in IDH-mutant glioma in vitro models, including TMZ administration, methods for determining cell viability, and difficulties in obtaining IDH-mutant glioma cell lines.
4.1. TMZ Treatment Dosage and Schedule
Human pharmacokinetic (PK) data determined exposure defined by the maximum plasma concentration (Cmax) and the integrated area under the plasma concentration-time curve (AUC), typically calculated from time zero to infinity, associated with the highest recommended dose of the drug [169]. With oral administration, 100% of the given TMZ dose enters the bloodstream. However, the site of action of TMZ requires its effective entry into the brain through the blood–brain barrier. There are no primary data showing how much of each metabolite eventually arrives at tumor cells in the human brain, with one report showing that the mean peak TMZ concentration in brain tissue was 0.6 ± 0.3 μg/mL, and the mean time to peak in the brain was 2.0 ± 0.8 h [170]. TMZ levels in the brain are only 20% of systemic drug levels, and it appears that the clinically relevant TMZ concentrations are around 5 µM [171]. Therefore, all in vitro studies should evaluate TMZ effects at low concentrations. There is no consensus on the concentrations and schedules used for TMZ in preclinical studies. To avoid the toxicity of DMSO [172,173] and maintain clinical relevance of TMZ dosage [174,175], we would recommend that TMZ dose should never be above 100 µM in vitro, and the dosing scheme should mimic that of a 5-day clinical schedule rather than a single high dose.
4.2. Cell Viability Assay In Vitro
Despite the wide range of cytotoxicity assays used in preclinical drug testing [176], the most commonly used assay methods in glioma research are the following: (1) dye exclusion assays using the trypan blue dye; (2) metabolism-based assays, including colorimetric assays that use MTT/MTS and luminometric assays that quantify ATP; (3) DNA synthesis proliferation assays using BrdU; (4) 3D soft agar growth assays.
The trypan blue dye exclusion assay [177] determines the number of viable cells based on the principle that live cells with intact membranes are impermeable to trypan blue. This method is used for suspension cells or trypsinized adherent cells, but it is associated with several pitfalls, including counting errors and reduced accuracy when viability is less than 80%. In addition, cell counting is usually performed with a hemocytometer, light microscope or automated cell counters, which makes simultaneous processing of a large number of samples difficult. On the other hand, MTT and MTS-based assays determine cell viability by the activity of mitochondrial enzymes such as succinate dehydrogenase [178]. Once MTT/MTS is reduced, the color changes from yellow to purple, which could be quantified by light absorbance at a specific wavelength, allowing the colorimetric measurement of cell viability using a spectrophotometer. Therefore, additional control experiments should be performed to reduce false-positive or false-negative results caused by background interference [179]. This method is applicable to both adherent and suspension cell lines and allows many assays to be performed simultaneously.
ATP-based assays are highly sensitive and quantify ATP content to determine the number of viable, metabolically active cells. In the presence of ATP, luciferin is oxidized to oxyluciferin, yielding a luminescent signal. Thus, the intensity of the luminescent signal is proportional to the concentration of ATP in active mitochondria. Because the IDH mutation alters multiple metabolic pathways, including NAD+ levels, redox balance, and oxidative phosphorylation, we should be extra cautious while interpreting the viability results from the metabolism-based assays, to ensure that we are only assessing the effect of drug treatment, and to rule out the effect of intrinsic metabolic alterations caused by 2-HG.
The BrdU (5-bromo-2′-deoxyuridine) cell proliferation assay monitors cell division by evaluating DNA synthesis through thymidine insertion. BrdU is a thymidine analog and can be incorporated into the newly synthesized DNA of replicating cells. BrdU levels can be detected by flow cytometry using a BrdU antibody conjugated with fluorochrome. Particular care must be taken with the incubation time for BrdU labeling as it is cell cycle dependent.
Soft agar colony formation assay measures the ability of single cells to form colonies in semi-solid soft agar medium in an anchorage-independent manner, which is closer to in vivo conditions. Depending on the cell lines and the number of cells seeded, the colony formation can take several weeks to months.
There is no perfect method to assess cell viability. While some assays can differentiate between dead and live cells, and others can just assess the relative performance of cells. To increase reliability of the results, researchers should use more than one assay method to better assess cell proliferation [180].
4.3. Cell Lines and PDX Models of IDH-Mutant Glioma
The IDH-mutant glioma cell lines used in preclinical research include four types: (1) genetically engineered lines overexpressing mutant IDH1 such as immortalized human astrocytes (IHA) [14,15,24,36,181], U87 [20,95,182,183], U251 [28,184,185], LN229 [95]; (2) patient-derived oligodendrogliomas: SF10417 [186], NCH612 [187], Hs683 [188], TS603 [28], BT088 and BT054 [189], BT237 and BT138 [190], BT054 [191], BT142 [192]; (3) patient-derived astrocytomas: SF10602 [186], NCH1681 and JHH273 [193], NCH551b and NCH620, and NCH645 and NCH3763 [194], MGG119 and MGG152 [30]; (4) non-glioma lines harboring endogenous IDH mutations, such as HT1080 [195].
Patient-derived IDH-mutant glioma cell lines are difficult to establish and propagate in vitro. One reason is the low incidence of IDH-mutant LGG patients at a given medical center, and another reason is that IDH-mutant tumor cells are difficult to culture [196] or to grow as xenografts [7,197] and can undergo loss of the IDH1 mutation in vitro. Therefore, regular cell line verification is required.
Due to rapid culturing and retention of the original genetic aberrations, some of the glioma cell lines have been used in several in vitro drug screens [194]. However, further evidence suggests that cell lines are poor representatives of the primary tumor isolated from patients in genetics, mRNA profiles, and protein expression [198]. Most experimental therapies with promising preclinical results using established cell lines have failed phase III clinical trials [199]. The development and propagation of mouse models and PDXs are more clinically representative but are time-consuming and costly [200].
Recently, 3D cultures of organoids have proven to be effective models. Organoids can be grown from patient-derived normal and tumor tissues [201] and eventually cultured in rotating bioreactors constituting various cell types, resembling defined brain regions [202]. Recently, Jacob et al. generated organoids for 96.4% IDH1-wt and 66.7% of IDH1-mutant gliomas [203]. Although the efficiency of generating organoids from IDH-mutant tumors remains to be optimized, IDH-mutant glioma organoids are likely to benefit personalized medicine and the development of individualized treatment for glioma patients [204].
5. Conclusions and Outlook
Here, we present the challenges and opportunities for treating IDH-mutant gliomas with the chemotherapy drug TMZ. We summarize the genetic, epigenetic, metabolic vulnerabilities that arise from IDH mutation and that could be exacerbated by treatment with TMZ. In the future, the fast-developing technologies, such as single-cell RNA-seq, or in vivo/in vitro shRNA and CRISPR screenings, will help us to identify more therapeutic targets in each specific molecular context, accelerating personalized medicine treatment in IDH-mutant gliomas. The recent success of the IDH1 mutation-specific vaccine in treating IDH1-mutated gliomas [205] may open new doors for the treatment of IDH-mutant gliomas. The integration of the immune system-assisted cancer cell elimination and chemotherapy-induced cancer cell death may be a new chapter for the treatment of patients with IDH-mutant gliomas.
Author Contributions
All authors contributed to the discussion of content and wrote, reviewed, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project-ID 404521405, SFB 1389—UNITE Glioblastoma, Work Package A04 to S.T.
Acknowledgments
The figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Cushing, H.W. A Classification of the Tumors of the Glioma Group on a Histo-Genetic Basis, with a Correlated Study of Prognosis; With 108 Illustrations; JB Lippincott Company: Philadelphia, PA, USA, 1926. [Google Scholar]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Pallud, J.; Blonski, M.; Mandonnet, E.; Audureau, E.; Fontaine, D.; Sanai, N.; Bauchet, L.; Peruzzi, P.; Frénay, M.; Colin, P.; et al. Velocity of tumor spontaneous expansion predicts long-term outcomes for diffuse low-grade gliomas. Neuro Oncol. 2013, 15, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakimoto, H.; Tanaka, S.; Curry, W.T.; Loebel, F.; Zhao, D.; Tateishi, K.; Chen, J.; Klofas, L.K.; Lelic, N.; Kim, J.C.; et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res. 2014, 20, 2898–2909. [Google Scholar] [CrossRef] [Green Version]
- Wiencke, J.K.; Zheng, S.; Jelluma, N.; Tihan, T.; Vandenberg, S.; Tamguney, T.; Baumber, R.; Parsons, R.; Lamborn, K.R.; Berger, M.S.; et al. Methylation of the PTEN promoter defines low-grade gliomas and secondary glioblastoma. Neuro Oncol. 2007, 9, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef]
- Yu, D.; Liu, Y.; Zhou, Y.; Ruiz-Rodado, V.; Larion, M.; Xu, G.; Yang, C. Triptolide suppresses IDH1-mutated malignancy via Nrf2-driven glutathione metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 9964–9972. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Tanaka, K.; Sasayama, T.; Irino, Y.; Sato, N.; Takeuchi, Y.; Kyotani, K.; Mukasa, A.; Mizukawa, K.; Sakata, J.; et al. Diagnostic value of glutamate with 2-hydroxyglutarate in magnetic resonance spectroscopy for IDH1 mutant glioma. Neuro Oncol. 2016, 18, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS ONE 2015, 10, e0118781. [Google Scholar] [CrossRef] [Green Version]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumour Biol. 2014, 35, 5911–5920. [Google Scholar] [CrossRef]
- Elkhaled, A.; Jalbert, L.E.; Phillips, J.J.; Yoshihara, H.A.I.; Parvataneni, R.; Srinivasan, R.; Bourne, G.; Berger, M.S.; Chang, S.M.; Cha, S.; et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci. Transl. Med. 2012, 4, 116ra115. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Rodado, V.; Lita, A.; Dowdy, T.; Celiku, O.; Saldana, A.C.; Wang, H.; Yang, C.Z.; Chari, R.; Li, A.; Zhang, W.; et al. Metabolic plasticity of IDH1-mutant glioma cell lines is responsible for low sensitivity to glutaminase inhibition. Cancer Metab. 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanath, P.; Radoul, M.; Izquierdo-Garcia, J.L.; Ong, W.Q.; Luchman, H.A.; Cairncross, J.G.; Huang, B.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. 2-Hydroxyglutarate-Mediated Autophagy of the Endoplasmic Reticulum Leads to an Unusual Downregulation of Phospholipid Biosynthesis in Mutant IDH1 Gliomas. Cancer Res. 2018, 78, 2290. [Google Scholar] [CrossRef] [Green Version]
- Lita, A.; Pliss, A.; Kuzmin, A.; Yamasaki, T.; Zhang, L.; Dowdy, T.; Burks, C.; de Val, N.; Celiku, O.; Ruiz-Rodado, V.; et al. IDH1 mutations induce organelle defects via dysregulated phospholipids. Nat. Commun. 2021, 12, 614. [Google Scholar] [CrossRef]
- Dowdy, T.; Zhang, L.; Celiku, O.; Movva, S.; Lita, A.; Ruiz-Rodado, V.; Gilbert, M.R.; Larion, M. Sphingolipid Pathway as a Source of Vulnerability in IDH1(mut) Glioma. Cancers 2020, 12, 2910. [Google Scholar] [CrossRef]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.J.; Fink, A.; Banagis, J.A.; Nagashima, H.; Subramanian, M.; Lee, C.K.; Melamed, L.; Tummala, S.S.; Tateishi, K.; Wakimoto, H.; et al. Sirtuin activation targets IDH-mutant tumors. Neuro-Oncol. 2021, 23, 53–62. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Khurshed, M.; Molenaar, R.J.; van Noorden, C.; Wilmink, J.W. Metabolic vulnerabilities in IDH1/2-mutated solid tumours lead to phase IB/II clinical trial using metformin and chloroquine. FASEB J. 2019, 33, 496.7. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, M.W.A.; van Linde, M.E.; Kouwenhoven, M.; Bramer, J.A.M.; Bovée, J.V.M.G.; Mathôt, R.A.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Sahm, F.; Steffl, B.; Arrillaga-Romany, I.; Cahill, D.; Monje, M.; Herold-Mende, C.; Wick, W.; Turcan, Ş. TERT and DNMT1 expression predict sensitivity to decitabine in gliomas. Neuro Oncol. 2021, 23, 76–87. [Google Scholar] [CrossRef]
- Borodovsky, A.; Salmasi, V.; Turcan, S.; Fabius, A.W.M.; Baia, G.S.; Eberhart, C.G.; Weingart, J.D.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.C.; Mehta, M.P. Low-Grade Glioma Radiotherapy Treatment and Trials. Neurosurg. Clin. N. Am. 2019, 30, 111–118. [Google Scholar] [CrossRef]
- Karim, A.B.M.F.; Maat, B.; Hatlevoll, R.; Menten, J.; Rutten, E.H.J.M.; Thomas, D.G.T.; Mascarenhas, F.; Horiot, J.C.; Parvinen, L.M.; van Reijn, M.; et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) study 22844. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 549–556. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Afra, D.; de Witte, O.; Ben Hassel, M.; Schraub, S.; Hoang-Xuan, K.; Malmström, P.O.; Collette, L.; Piérart, M.; Mirimanoff, R.; et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: The EORTC 22845 randomised trial. Lancet 2005, 366, 985–990. [Google Scholar] [CrossRef]
- Baumert, B.G.; Hegi, M.E.; van den Bent, M.J.; von Deimling, A.; Gorlia, T.; Hoang-Xuan, K.; Brandes, A.A.; Kantor, G.; Taphoorn, M.J.B.; Hassel, M.B.; et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): A randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016, 17, 1521–1532. [Google Scholar] [CrossRef] [Green Version]
- McDuff, S.G.R.; Dietrich, J.; Atkins, K.M.; Oh, K.S.; Loeffler, J.S.; Shih, H.A. Radiation and chemotherapy for high-risk lower grade gliomas: Choosing between temozolomide and PCV. Cancer Med. 2020, 9, 3–11. [Google Scholar] [CrossRef]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- Bell, E.H.; Won, M.; Fleming, J.; Becker, A.; McElroy, J.; Shaw, E.G.; Mehta, M.P.; Brachman, D.G.; Gertler, S.; Murtha, A.D.; et al. Comprehensive Prognostic and Predictive Molecular Subgroup Analysis within the High-risk Treatment Arms of NRG Oncology/RTOG 9802: A Phase III Trial of RT versus RT + PCV in High-risk Low-grade Gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, S78–S79. [Google Scholar] [CrossRef]
- Cairncross, G.; Wang, M.; Shaw, E.; Jenkins, R.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperriere, N.; Curran, W.; et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J. Clin. Oncol. 2013, 31, 337–343. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Brandes, A.A.; Taphoorn, M.J.; Kros, J.M.; Kouwenhoven, M.C.; Delattre, J.Y.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W.; et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 2013, 31, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Roth, P.; Hartmann, C.; Hau, P.; Nakamura, M.; Stockhammer, F.; Sabel, M.C.; Wick, A.; Koeppen, S.; Ketter, R.; et al. Long-term analysis of the NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with PCV or temozolomide. Neuro Oncol. 2016, 18, 1529–1537. [Google Scholar] [CrossRef] [Green Version]
- van den Bent, M.J.; Baumert, B.; Erridge, S.C.; Vogelbaum, M.A.; Nowak, A.K.; Sanson, M.; Brandes, A.A.; Clement, P.M.; Baurain, J.F.; Mason, W.P.; et al. Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: A phase 3, randomised, open-label intergroup study. Lancet 2017, 390, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Fisher, B.J.; Pugh, S.L.; Macdonald, D.R.; Chakravatri, A.; Lesser, G.J.; Fox, S.; Rogers, C.L.; Werner-Wasik, M.; Doyle, T.; Bahary, J.P.; et al. Phase 2 Study of a Temozolomide-Based Chemoradiation Therapy Regimen for High-Risk, Low-Grade Gliomas: Long-Term Results of Radiation Therapy Oncology Group 0424. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 720–725. [Google Scholar] [CrossRef]
- Strowd, R.E.; Abuali, I.; Ye, X.; Lu, Y.; Grossman, S.A. The role of temozolomide in the management of patients with newly diagnosed anaplastic astrocytoma: A comparison of survival in the era prior to and following the availability of temozolomide. J. Neurooncol. 2016, 127, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Jaeckle, K.A.; Ballman, K.V.; van den Bent, M.; Giannini, C.; Galanis, E.; Brown, P.D.; Jenkins, R.B.; Cairncross, J.G.; Wick, W.; Weller, M.; et al. CODEL: Phase III study of RT, RT + TMZ, or TMZ for newly diagnosed 1p/19q codeleted oligodendroglioma. Analysis from the initial study design. Neuro-Oncol. 2020. [Google Scholar] [CrossRef]
- Darlix, A.; Mandonnet, E.; Freyschlag, C.F.; Pinggera, D.; Forster, M.T.; Voss, M.; Steinbach, J.; Loughrey, C.; Goodden, J.; Banna, G.; et al. Chemotherapy and diffuse low-grade gliomas: A survey within the European Low-Grade Glioma Network. Neurooncol. Pract. 2019, 6, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutras, G.; Bélanger, K.; Letarte, N.; Adam, J.P.; Roberge, D.; Lemieux, B.; Lemieux-Blanchard, É.; Masucci, L.; Ménard, C.; Bahary, J.P.; et al. Procarbazine, lomustine and vincristine toxicity in low-grade gliomas. Curr. Oncol. 2018, 25, e33–e39. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, S.M.; Newlands, E.S.; Glaser, M.G.; Brampton, M.; Rice-Edwards, J.M.; Illingworth, R.D.; Richards, P.G.; Kennard, C.; Colquhoun, I.R.; Lewis, P.; et al. Temozolomide: A new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur. J. Cancer 1993, 29a, 940–942. [Google Scholar] [CrossRef]
- Newlands, E.S.; Stevens, M.F.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef]
- Loveless, A. Possible relevance of O-6 alkylation of deoxyguanosine to the mutagenicity and carcinogenicity of nitrosamines and nitrosamides. Nature 1969, 223, 206–207. [Google Scholar] [CrossRef]
- Mitra, G.; Pauly, G.T.; Kumar, R.; Pei, G.K.; Hughes, S.H.; Moschel, R.C.; Barbacid, M. Molecular analysis of O6-substituted guanine-induced mutagenesis of ras oncogenes. Proc. Natl. Acad. Sci. USA 1989, 86, 8650. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharm. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Catapano, C.V.; Broggini, M.; Erba, E.; Ponti, M.; Mariani, L.; Citti, L.; D’Incalci, M. In vitro and in vivo methazolastone-induced DNA damage and repair in L-1210 leukemia sensitive and resistant to chloroethylnitrosoureas. Cancer Res. 1987, 47, 4884–4889. [Google Scholar]
- Roos, W.P.; Batista, L.F.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Thuijl, H.F.; Mazor, T.; Johnson, B.E.; Fouse, S.D.; Aihara, K.; Hong, C.; Malmström, A.; Hallbeck, M.; Heimans, J.J.; Kloezeman, J.J.; et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 2015, 129, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823. [Google Scholar] [CrossRef] [Green Version]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef] [Green Version]
- Schold, S.C., Jr.; Kokkinakis, D.M.; Chang, S.M.; Berger, M.S.; Hess, K.R.; Schiff, D.; Robins, H.I.; Mehta, M.P.; Fink, K.L.; Davis, R.L.; et al. O6-benzylguanine suppression of O6-alkylguanine-DNA alkyltransferase in anaplastic gliomas. Neuro Oncol. 2004, 6, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Wedge, S.R.; Newlands, E.S. O6-benzylguanine enhances the sensitivity of a glioma xenograft with low O6-alkylguanine-DNA alkyltransferase activity to temozolomide and BCNU. Br. J. Cancer 1996, 73, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187. [Google Scholar] [CrossRef]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase I trial of temozolomide plus O6-benzylguanine 5-day regimen with recurrent malignant glioma. Neuro Oncol. 2009, 11, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Friedman, H.S.; Kokkinakis, D.M.; Pluda, J.; Friedman, A.H.; Cokgor, I.; Haglund, M.M.; Ashley, D.M.; Rich, J.; Dolan, M.E.; Pegg, A.E.; et al. Phase I trial of O6-benzylguanine for patients undergoing surgery for malignant glioma. J. Clin. Oncol. 1998, 16, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E.; Gururangan, S.; Geyer, J.R.; McLendon, R.E.; Poussaint, T.Y.; Wallace, D.; Balis, F.M.; Berg, S.L.; Packer, R.J.; Goldman, S.; et al. A phase II study of O6-benzylguanine and temozolomide in pediatric patients with recurrent or progressive high-grade gliomas and brainstem gliomas: A Pediatric Brain Tumor Consortium study. J. Neurooncol. 2012, 106, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Jascur, T.; Boland, C.R. Structure and function of the components of the human DNA mismatch repair system. Int. J. Cancer 2006, 119, 2030–2035. [Google Scholar] [CrossRef] [PubMed]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Müller-Goebel, J.; Weik, A.S.; Hoffer, K.; et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef] [Green Version]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A.; Piccart, M. An update on PARP inhibitors—moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.L.; Johnson, S.P.; Keir, S.T.; Quinn, J.A.; Ali-Osman, F.; Szabo, C.; Li, H.; Salzman, A.L.; Dolan, M.E.; Modrich, P.; et al. Poly(ADP-ribose) polymerase-1 inhibition reverses temozolomide resistance in a DNA mismatch repair–deficient malignant glioma xenograft. Mol. Cancer Ther. 2005, 4, 1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaldi, A.P.; Lima, S.C.G.; Godoy, P.; Xavier, D.J.; Sakamoto-Hojo, E.T. PARP-1 inhibition sensitizes temozolomide-treated glioblastoma cell lines and decreases drug resistance independent of MGMT activity and PTEN proficiency. Oncol. Rep. 2020, 44, 2275–2287. [Google Scholar] [CrossRef]
- Yuan, A.L.; Ricks, C.B.; Bohm, A.K.; Lun, X.; Maxwell, L.; Safdar, S.; Bukhari, S.; Gerber, A.; Sayeed, W.; Bering, E.A.; et al. ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PLoS ONE 2018, 13, e0202860. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Guo, Y.; Liu, Y.; Wang, H.; Gong, W.; Liu, Y.; Wang, X.; Gao, Y.; Yu, F.; Su, D.; et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.S.; Rau, R.E.; Berg, S.L.; Liu, X.; Minard, C.G.; Bishop, A.J.R.; Romero, J.C.; Hicks, M.J.; Nelson, M.D., Jr.; Voss, S.; et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatric Blood Cancer 2020, 67, e28073. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Snape, T.J. A therapeutic update on PARP inhibitors: Implications in the treatment of glioma. Drug Discov. Today 2020. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Mukherjee, J.; See, W.L.; Pieper, R.O. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014, 74, 4836–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.; Khanna, K.K.; Wiegmans, A.P. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat. Rev. 2015, 41, 35–45. [Google Scholar] [CrossRef]
- Chabalier-Taste, C.; Brichese, L.; Racca, C.; Canitrot, Y.; Calsou, P.; Larminat, F. Polo-like kinase 1 mediates BRCA1 phosphorylation and recruitment at DNA double-strand breaks. Oncotarget 2016, 7, 2269–2283. [Google Scholar] [CrossRef] [Green Version]
- Yata, K.; Lloyd, J.; Maslen, S.; Bleuyard, J.Y.; Skehel, M.; Smerdon, S.J.; Esashi, F. Plk1 and CK2 act in concert to regulate Rad51 during DNA double strand break repair. Mol. Cell 2012, 45, 371–383. [Google Scholar] [CrossRef]
- Koncar, R.F.; Chu, Z.; Romick-Rosendale, L.E.; Wells, S.I.; Chan, T.A.; Qi, X.; Bahassi, E.M. PLK1 inhibition enhances temozolomide efficacy in IDH1 mutant gliomas. Oncotarget 2017, 8, 15827–15837. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Hu, G.; Wang, H.; Li, Z.; Guo, Z. PLK1 inhibitor facilitates the suppressing effect of temozolomide on human brain glioma stem cells. J. Cell Mol. Med. 2018, 22, 5300–5310. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Sun, S.; Xu, H.; Zhao, Z.; Han, Z.; Jia, J.; Wu, D.; Lu, J.; Liu, H.; Yu, R. Combined Delivery of Temozolomide and siPLK1 Using Targeted Nanoparticles to Enhance Temozolomide Sensitivity in Glioma. Int. J. Nanomed. 2020, 15, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R.; Kastan, M.B. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Cai, J.; Tan, Z.; Meng, X.; Li, R.; Li, Y.; Jiang, C. Mutant IDH1 Enhances Temozolomide Sensitivity via Regulation of the ATM/CHK2 Pathway in Glioma. Cancer Res. Treat. 2021, 53, 367–377. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hänggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro-Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, A.-L.; Heiland, D.H.; Evert, B.O.; Almeida, F.R.; Behringer, S.P.; Dolf, A.; Güresir, Á.; Güresir, E.; Joseph, K.; Pietsch, T.; et al. Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide. Cancers 2019, 11, 858. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.; Vollmer, L.; Potthoff, A.L.; Ravi, V.M.; Evert, B.O.; Rahman, M.A.; Sarowar, S.; Kueckelhaus, J.; Will, P.; Zurhorst, D.; et al. Meclofenamate causes loss of cellular tethering and decoupling of functional networks in glioblastoma. Neuro Oncol. 2021. [Google Scholar] [CrossRef]
- Ricard, D.; Kaloshi, G.; Amiel-Benouaich, A.; Lejeune, J.; Marie, Y.; Mandonnet, E.; Kujas, M.; Mokhtari, K.; Taillibert, S.; Laigle-Donadey, F.; et al. Dynamic history of low-grade gliomas before and after temozolomide treatment. Ann. Neurol. 2007, 61, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, C.; Alentorn, A.; Idbaih, A.; Simó, M.; Kaloshi, G.; Ricard, D.; Barritault, M.; Meyronet, D.; Bruna, J.; Honnorat, J.; et al. Long-term impact of temozolomide on 1p/19q-codeleted low-grade glioma growth kinetics. J. Neurooncol. 2018, 136, 533–539. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [Green Version]
- McFaline-Figueroa, J.L.; Braun, C.J.; Stanciu, M.; Nagel, Z.D.; Mazzucato, P.; Sangaraju, D.; Cerniauskas, E.; Barford, K.; Vargas, A.; Chen, Y.; et al. Minor Changes in Expression of the Mismatch Repair Protein MSH2 Exert a Major Impact on Glioblastoma Response to Temozolomide. Cancer Res. 2015, 75, 3127–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Deng, L.; Bai, H.X.; Sun, J.; Neale, N.; Wu, J.; Wang, Y.; Chang, K.; Huang, R.Y.; Zhang, P.J.; et al. Reduced expression of DNA repair genes and chemosensitivity in 1p19q codeleted lower-grade gliomas. J. Neurooncol. 2018, 139, 563–571. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Jonsson, P.; Lin, A.L.; Young, R.J.; DiStefano, N.M.; Hyman, D.M.; Li, B.T.; Berger, M.F.; Zehir, A.; Ladanyi, M.; Solit, D.B.; et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin. Cancer Res. 2019, 25, 5537–5547. [Google Scholar] [CrossRef] [Green Version]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, J.M.; Travis, L.B. Mechanisms of therapy-related carcinogenesis. Nat. Rev. Cancer 2005, 5, 943–955. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrugala, M.M.; Chamberlain, M.C. Mechanisms of Disease: Temozolomide and glioblastoma—look to the future. Nat. Clin. Pract. Oncol. 2008, 5, 476–486. [Google Scholar] [CrossRef]
- Woo, P.; Li, Y.; Chan, A.; Ng, S.; Loong, H.; Chan, D.; Wong, G.; Poon, W.-S. A multifaceted review of temozolomide resistance mechanisms in glioblastoma beyond O-6-methylguanine-DNA methyltransferase. Glioma 2019, 2, 68–82. [Google Scholar] [CrossRef]
- Cohen, A.; Sato, M.; Aldape, K.; Mason, C.C.; Alfaro-Munoz, K.; Heathcock, L.; South, S.T.; Abegglen, L.M.; Schiffman, J.D.; Colman, H. DNA copy number analysis of Grade II-III and Grade IV gliomas reveals differences in molecular ontogeny including chromothripsis associated with IDH mutation status. Acta Neuropathol. Commun. 2015, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhou, C.; Yi, L.; Xu, L.; Xu, M. Effect and molecular mechanism of mTOR inhibitor rapamycin on temozolomide-induced autophagic death of U251 glioma cells. Oncol. Lett. 2018, 15, 2477–2484. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Guo, G.; Guo, L.; Wang, Z.; Chen, Z.; Nan, Y.; Cao, Y.; Li, R.; Yang, X.; Dong, J.; et al. Amlexanox Enhances Temozolomide-Induced Antitumor Effects in Human Glioblastoma Cells by Inhibiting IKBKE and the Akt-mTOR Signaling Pathway. ACS Omega 2021, 6, 4289–4299. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.; Sottero, T.; Yang, X.; Mueller, S.; James, C.D.; Weiss, W.A.; Polley, M.-Y.; Ozawa, T.; Berger, M.S.; Aftab, D.T.; et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-Oncol. 2011, 13, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C.; et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015, 17, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, B.G.; Peng, S.; Sereduk, C.P.; Sodoma, A.M.; Kitange, G.J.; Loftus, J.C.; Sarkaria, J.N.; Tran, N.L. Inhibition of phosphatidylinositol 3-kinase by PX-866 suppresses temozolomide-induced autophagy and promotes apoptosis in glioblastoma cells. Mol. Med. 2019, 25, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro Reily Rocha, C.; Reily Rocha, A.; Molina Silva, M.; Rodrigues Gomes, L.; Teatin Latancia, M.; Andrade Tomaz, M.; de Souza, I.; Karolynne Seregni Monteiro, L.; Frederico Martins Menck, C. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, W.; Yu, J.; Lu, Z.; Yu, J. Inhibition of Nrf2 might enhance the anti-tumor effect of temozolomide in glioma cells via inhibition of Ras/Raf/MEK signaling pathway. Int. J. Neurosci. 2020, 1–9. [Google Scholar] [CrossRef]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef]
- Li, K.; Ouyang, L.; He, M.; Luo, M.; Cai, W.; Tu, Y.; Pi, R.; Liu, A. IDH1 R132H mutation regulates glioma chemosensitivity through Nrf2 pathway. Oncotarget 2017, 8, 28865–28879. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.-Q.; Liu, A.-S.; Zhang, M.-J.; Liu, H.-J.; Meng, X.-L.; Qian, H.-P.; Wan, J.-H. Identifying Predictive Gene Expression and Signature Related to Temozolomide Sensitivity of Glioblastomas. Front. Oncol. 2020, 10, 669. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.S.; da Costa Rosa, M.; Borodovsky, A.; Festuccia, W.T.; Chan, T.; Riggins, G.J. Demethylation and epigenetic modification with 5-azacytidine reduces IDH1 mutant glioma growth in combination with temozolomide. Neuro Oncol. 2019, 21, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.E.; Taub, R.N.; Matushansky, I.; Uldrick, T.S.; Khandker, M.; Bressler, Y.; Wang, Y. A phase I/II study of azacitidine in combination with temozolomide in patients with unresectable or metastatic soft tissue sarcoma or malignant mesothelioma. J. Clin. Oncol. 2014, 32, 10560. [Google Scholar] [CrossRef]
- Cui, Y.; Naz, A.; Thompson, D.H.; Irudayaraj, J. Decitabine nanoconjugate sensitizes human glioblastoma cells to temozolomide. Mol. Pharm. 2015, 12, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Gallitto, M.; Cheng He, R.; Inocencio, J.F.; Wang, H.; Zhang, Y.; Deikus, G.; Wasserman, I.; Strahl, M.; Smith, M.; Sebra, R.; et al. Epigenetic preconditioning with decitabine sensitizes glioblastoma to temozolomide via induction of MLH1. J. Neuro-Oncol. 2020, 147, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawbi, H.; Beumer, J.; Tarhini, A.; Moschos, S.; Buch, S.; Egorin, M.; Lin, Y.; Christner, S.; Kirkwood, J.M. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: A phase I/II study and pharmacokinetic analysis. Ann. Oncol. 2013, 24, 1112–1119. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Marciniak, M.P.; Jackl, J.; Kaminska, B. Pre-treatment or Post-treatment of Human Glioma Cells With BIX01294, the Inhibitor of Histone Methyltransferase G9a, Sensitizes Cells to Temozolomide. Front. Pharm. 2018, 9, 1271. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef]
- Banelli, B.; Daga, A.; Forlani, A.; Allemanni, G.; Marubbi, D.; Pistillo, M.P.; Profumo, A.; Romani, M. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget 2017, 8, 34896–34910. [Google Scholar] [CrossRef] [Green Version]
- Romani, M.; Daga, A.; Forlani, A.; Pistillo, M.P.; Banelli, B. Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells. Cancers 2019, 11, 878. [Google Scholar] [CrossRef] [Green Version]
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Sampath, D.; Lang, F.F.; Prabhu, S.; Rao, G.; Fuller, G.N.; Liu, Y.; Puduvalli, V.K. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. J. Neuro-Oncol. 2011, 105, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Puduvalli, V.K.; Reid, J.M.; Kuhn, J.G.; Lamborn, K.R.; Cloughesy, T.F.; Chang, S.M.; Drappatz, J.; Yung, W.K.A.; Gilbert, M.R.; et al. Phase I study of vorinostat in combination with temozolomide in patients with high-grade gliomas: North American Brain Tumor Consortium Study 04-03. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6032–6039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nifterik, K.A.; Van den Berg, J.; Slotman, B.J.; Lafleur, M.V.M.; Sminia, P.; Stalpers, L.J.A. Valproic acid sensitizes human glioma cells for temozolomide and γ-radiation. J. Neuro-Oncol. 2012, 107, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother Pharm. 2014, 74, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoral-Theys, D.; Le Mercier, M.; Le Calvé, B.; Rynkowski, M.A.; Bruyère, C.; Decaestecker, C.; Haibe-Kains, B.; Bontempi, G.; Dubois, J.; Lefranc, F.; et al. Long-term temozolomide treatment induces marked amino metabolism modifications and an increase in TMZ sensitivity in Hs683 oligodendroglioma cells. Neoplasia 2010, 12, 69–79. [Google Scholar] [CrossRef]
- Subramani, E.; Radoul, M.; Najac, C.; Batsios, G.; Molloy, A.R.; Hong, D.; Gillespie, A.M.; Santos, R.D.; Viswanath, P.; Costello, J.F.; et al. Glutamate Is a Noninvasive Metabolic Biomarker of IDH1-Mutant Glioma Response to Temozolomide Treatment. Cancer Res. 2020, 80, 5098–5108. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Sehm, T.; Rauh, M.; Wiendieck, K.; Buchfelder, M.; Eyüpoglu, I.Y.; Savaskan, N.E. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget 2016, 7, 74630–74647. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizilbash, S.H.; Burgenske, D.M.; McBrayer, S.; Devarajan, S.; Gupta, S.K.; Hitosugi, T.; He, L.; Schroeder, M.A.; Carlson, B.L.; Gelman, M.; et al. Abstract 3870: The addition of CB-839 to temozolomide significantly reduces glioma aspartate and glutamate in an IDH mutated patient derived glioma xenograft model. Cancer Res. 2019, 79, 3870. [Google Scholar] [CrossRef]
- Kizilbash, S.H.; McBrayer, S.; Port, J.; Reid, J.M.; Lanza, I.; Allred, J.B.; Chakravarti, A.; Kunos, C.; Adjei, A.A. A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). J. Clin. Oncol. 2019, 37, TPS2075. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, K.; Higuchi, F.; Miller, J.J.; Koerner, M.V.A.; Lelic, N.; Shankar, G.M.; Tanaka, S.; Fisher, D.E.; Batchelor, T.T.; Iafrate, A.J.; et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1-Mutant Cancers and Potentiates NAD(+) Depletion-Mediated Cytotoxicity. Cancer Res. 2017, 77, 4102–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waitkus, M.S.; Yan, H. Targeting Isocitrate Dehydrogenase Mutations in Cancer: Emerging Evidence and Diverging Strategies. Clin. Cancer Res. 2021, 27, 383. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, H.; Lee, C.K.; Tateishi, K.; Higuchi, F.; Subramanian, M.; Rafferty, S.; Melamed, L.; Miller, J.J.; Wakimoto, H.; Cahill, D.P. Poly(ADP-ribose) Glycohydrolase Inhibition Sequesters NAD(+) to Potentiate the Metabolic Lethality of Alkylating Chemotherapy in IDH-Mutant Tumor Cells. Cancer Discov. 2020, 10, 1672–1689. [Google Scholar] [CrossRef]
- Le Calvé, B.; Rynkowski, M.; Le Mercier, M.; Bruyère, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, A.; Nato, G.; Parmigiani, E.; Garello, F.; Buffo, A.; Magrassi, L. Inhibitors of GLUT/SLC2A Enhance the Action of BCNU and Temozolomide against High-Grade Gliomas. Neoplasia 2017, 19, 364–373. [Google Scholar] [CrossRef]
- Guan, D.G.; Chen, H.M.; Liao, S.F.; Zhao, T.Z. Combination of temozolomide and Taxol exerts a synergistic inhibitory effect on Taxol-resistant glioma cells via inhibition of glucose metabolism. Mol. Med. Rep. 2015, 12, 7705–7711. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Mino, E.P.; Shaheen, M.F. Phase II trial of carboplatin, paclitaxel and temozolomide in metastatic melanoma. J. Clin. Oncol. 2016, 34, e21012. [Google Scholar] [CrossRef]
- Valtorta, S.; Lo Dico, A.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [Green Version]
- Allavena, G.; Del Bello, B.; Tini, P.; Volpi, N.; Valacchi, G.; Miracco, C.; Pirtoli, L.; Maellaro, E. Trehalose inhibits cell proliferation and amplifies long-term temozolomide-and radiation-induced cytotoxicity in melanoma cells: A role for autophagy and premature senescence. J. Cell. Physiol. 2019, 234, 11708–11721. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.P.C.; Kuo, J.S.; Chiang, H.-C.; Wang, H.-E.; Wang, Y.-S.; Huang, C.-C.; Huang, Y.-C.; Chi, M.-S.; Mehta, M.P.; Chi, K.-H. Temozolomide, sirolimus and chloroquine is a new therapeutic combination that synergizes to disrupt lysosomal function and cholesterol homeostasis in GBM cells. Oncotarget 2018, 9, 6883–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; et al. Considering the Experimental use of Temozolomide in Glioblastoma Research. Biomedicines 2020, 8, 151. [Google Scholar] [CrossRef]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [Green Version]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [Green Version]
- Stepanenko, A.A.; Chekhonin, V.P. On the Critical Issues in Temozolomide Research in Glioblastoma: Clinically Relevant Concentrations and MGMT-independent Resistance. Biomedicines 2019, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- 1Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. [Google Scholar] [CrossRef]
- Verheijen, M.; Lienhard, M.; Schrooders, Y.; Clayton, O.; Nudischer, R.; Boerno, S.; Timmermann, B.; Selevsek, N.; Schlapbach, R.; Gmuender, H.; et al. DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro. Sci. Rep. 2019, 9, 4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, C.S.; Newlands, E.S.; Wedge, S.R.; Bower, M.; Evans, H.; Colquhoun, I.; Roddie, M.; Glaser, M.; Brampton, M.H.; Rustin, G.J.S. Phase I Trial of Temozolomide Using an Extended Continuous Oral Schedule. Cancer Res. 1998, 58, 4363–4367. [Google Scholar]
- Korones, D.N.; Benita-Weiss, M.; Coyle, T.E.; Mechtler, L.; Bushunow, P.; Evans, B.; Reardon, D.A.; Quinn, J.A.; Friedman, H. Phase I study of temozolomide and escalating doses of oral etoposide for adults with recurrent malignant glioma. Cancer 2003, 97, 1963–1968. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Markossian, S., Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Stone, V.; Johnston, H.; Schins, R.P. Development of in vitro systems for nanotoxicology: Methodological considerations. Crit. Rev. Toxicol. 2009, 39, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Aslantürk, Ö.S. In Vitro Cytotoxicity and Cell Viability Assays: Principles, Advantages, and Disadvantages. In Genotoxicity—A Predictable Risk to Our Actual World; IntechOpen: London, UK, 2018. [Google Scholar]
- Halim, A.B. Do We have a Satisfactory Cell Viability Assay? Review of the Currently Commercially-Available Assays. Curr. Drug Discov. Technol. 2020, 17, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Ozawa, T.; Hirose, Y.; Aldape, K.D.; McMahon, M.; Berger, M.S.; Pieper, R.O. Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 2001, 61, 4956–4960. [Google Scholar] [PubMed]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015, 75, 2999–3009. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, R.; Sun, Y.; Xu, C.; Wang, Y.; Xiong, J.; Chen, Q.; Liu, Y. IDH1 mutation decreases the invasiveness of glioma by downregulating the expression and activity of TAZ. Glioma 2019, 2, 37. [Google Scholar] [CrossRef]
- Khurshed, M.; Aarnoudse, N.; Hulsbos, R.; Hira, V.V.V.; van Laarhoven, H.W.M.; Wilmink, J.W.; Molenaar, R.J.; van Noorden, C.J.F. IDH1-mutant cancer cells are sensitive to cisplatin and an IDH1-mutant inhibitor counteracts this sensitivity. FASEB J. 2018, 32, fj201800547R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Lu, Y.; Li, A.; Celiku, O.; Han, S.; Qian, M.; Yang, C. mTORC2/Rac1 Pathway Predisposes Cancer Aggressiveness in IDH1-Mutated Glioma. Cancers 2020, 12, 787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.E.; Hilz, S.; Grimmer, M.R.; Mazor, T.; Najac, C.; Mukherjee, J.; McKinney, A.; Chow, T.; Pieper, R.O.; Ronen, S.M.; et al. Patient-derived cells from recurrent tumors that model the evolution of IDH-mutant glioma. Neurooncol. Adv. 2020, 2, vdaa088. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Zhang, Y.; Shu, C.; Tsujiuchi, T.; Banu, M.A.; Garcia, F.; Roth, K.A.; Bruce, J.N.; et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat. Commun. 2017, 8, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, M.L.; Mathieu, V.; Haibe-Kains, B.; Bontempi, G.; Mijatovic, T.; Decaestecker, C.; Kiss, R.; Lefranc, F. Knocking Down Galectin 1 in Human Hs683 Glioblastoma Cells Impairs Both Angiogenesis and Endoplasmic Reticulum Stress Responses. J. Neuropathol. Exp. Neurol. 2008, 67, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blough, M.D.; Al-Najjar, M.; Chesnelong, C.; Binding, C.E.; Rogers, A.D.; Luchman, H.A.; Kelly, J.J.; Fliegel, L.; Morozova, O.; Yip, S. DNA hypermethylation and 1p Loss silence NHE-1 in oligodendroglioma. Ann. Neurol. 2012, 71, 845–849. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Kelly, J.J.P.; Blough, M.D.; Stechishin, O.D.M.; Chan, J.A.W.; Beauchamp, D.; Perizzolo, M.; Demetrick, D.J.; Steele, L.; Auer, R.N.; Hader, W.J.; et al. Oligodendroglioma cell lines containing t(1;19)(q10;p10). Neuro-Oncol. 2010, 12, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Luchman, H.A.; Stechishin, O.D.; Dang, N.H.; Blough, M.D.; Chesnelong, C.; Kelly, J.J.; Nguyen, S.A.; Chan, J.A.; Weljie, A.M.; Cairncross, J.G.; et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012, 14, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Borodovsky, A.; Meeker, A.K.; Kirkness, E.F.; Zhao, Q.; Eberhart, C.G.; Gallia, G.L.; Riggins, G.J. A model of a patient-derived IDH1 mutant anaplastic astrocytoma with alternative lengthening of telomeres. J. Neuro-Oncol. 2015, 121, 479–487. [Google Scholar] [CrossRef]
- Dao Trong, P.; Jungwirth, G.; Yu, T.; Pusch, S.; Unterberg, A.; Herold-Mende, C.; Warta, R. Large-Scale Drug Screening in Patient-Derived IDH(mut) Glioma Stem Cells Identifies Several Efficient Drugs among FDA-Approved Antineoplastic Agents. Cells 2020, 9, 1389. [Google Scholar] [CrossRef]
- Dexter, J.P.; Ward, P.S.; Dasgupta, T.; Hosios, A.M.; Gunawardena, J.; Vander Heiden, M.G. Lack of evidence for substrate channeling or flux between wildtype and mutant isocitrate dehydrogenase to produce the oncometabolite 2-hydroxyglutarate. J. Biol. Chem. 2018, 293, 20051–20061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piaskowski, S.; Bienkowski, M.; Stoczynska-Fidelus, E.; Stawski, R.; Sieruta, M.; Szybka, M.; Papierz, W.; Wolanczyk, M.; Jaskolski, D.J.; Liberski, P.P.; et al. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br. J. Cancer 2011, 104, 968–970. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Tang, Z.; Li, Y.; Yin, G.; Liu, Z.; Gao, J.; Chen, Y.; Chen, F. Patient-derived xenografts of different grade gliomas retain the heterogeneous histological and genetic features of human gliomas. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, K.G.; D’Arcangelo, E.; Tsao, M.S. Patient-derived cell line, xenograft and organoid models in lung cancer therapy. Transl. Lung Cancer Res. 2020, 9, 2214–2232. [Google Scholar] [CrossRef]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S. A patient-derived glioblastoma organoid model and biobank recapitulates inter-and intra-tumoral heterogeneity. Cell 2020, 180, 188–204.e122. [Google Scholar] [CrossRef]
- Zhang, C.; Jin, M.; Zhao, J.; Chen, J.; Jin, W. Organoid models of glioblastoma: Advances, applications and challenges. Am. J. Cancer Res. 2020, 10, 2242–2257. [Google Scholar] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Epigenetic alterations induced by IDH mutations and potential drug targets for TMZ combination therapy. (a) Cellular epigenetic regulation without IDH mutation; (b) cellular epigenetic alterations in IDH-mutant gliomas. AZA, 5-azacitidine; DAC, Decitabine; DNMT1, DNA methyltransferase 1; HDAC, Histone deacetylase; 2-HG, D-(R)-2-hydroxyglutarate; HMT, Histone methyltransferase; KDM, Histone demethylase; TET, Ten-eleven translocation methylcytosine dioxygenases; VPA, Valproic acid; 5mC, 5-Mehylcytosine. Me: Methyl group; Ac, Acetyl group.
Figure 1.
Epigenetic alterations induced by IDH mutations and potential drug targets for TMZ combination therapy. (a) Cellular epigenetic regulation without IDH mutation; (b) cellular epigenetic alterations in IDH-mutant gliomas. AZA, 5-azacitidine; DAC, Decitabine; DNMT1, DNA methyltransferase 1; HDAC, Histone deacetylase; 2-HG, D-(R)-2-hydroxyglutarate; HMT, Histone methyltransferase; KDM, Histone demethylase; TET, Ten-eleven translocation methylcytosine dioxygenases; VPA, Valproic acid; 5mC, 5-Mehylcytosine. Me: Methyl group; Ac, Acetyl group.

Figure 2.
Metabolic vulnerabilities in IDH-mutant gliomas and potential targets for single treatment or combination treatment with TMZ. Actionable metabolic dependencies in IDH-mutant gliomas include interrupted redox balance, low basal levels of glutamate, NAD+ and lipid synthesis. ASNS, Asparagine synthetase; BAF, Bafilomycin A1; CQ, Chloroquine; 2DG, 2-Deoxy-D-glucose; DNMS, N,N-dimethylsphingosine; ER, Endoplasmic reticulum; GLS, Glutaminase; IDH, Isocitrate dehydrogenase; NAD+, Nicotinamide adenine dinucleotide; NAMPT, Nicotinamide phosphoribosyl transferase; NAPRT, Nicotinate phosphoribosyltransferase; NRF2, nuclear factor erythroid 2-related factor; PARP, Poly (ADP-ribose) polymerse; PARG, Poly (ADP-ribose) glycohydrolase.
Figure 2.
Metabolic vulnerabilities in IDH-mutant gliomas and potential targets for single treatment or combination treatment with TMZ. Actionable metabolic dependencies in IDH-mutant gliomas include interrupted redox balance, low basal levels of glutamate, NAD+ and lipid synthesis. ASNS, Asparagine synthetase; BAF, Bafilomycin A1; CQ, Chloroquine; 2DG, 2-Deoxy-D-glucose; DNMS, N,N-dimethylsphingosine; ER, Endoplasmic reticulum; GLS, Glutaminase; IDH, Isocitrate dehydrogenase; NAD+, Nicotinamide adenine dinucleotide; NAMPT, Nicotinamide phosphoribosyl transferase; NAPRT, Nicotinate phosphoribosyltransferase; NRF2, nuclear factor erythroid 2-related factor; PARP, Poly (ADP-ribose) polymerse; PARG, Poly (ADP-ribose) glycohydrolase.

Figure 3.
TMZ molecular structure, metabolism, toxicity, and resistance. (a) TMZ structure and metabolism, (b) DNA methylation upon TMZ, (c) DNA base mispair upon DNA methylation, (d) mechanism of TMZ toxicity with intact MMR, BER, and HR, (e) mechanism of TMZ resistance with functional MGMT and non-functional MMR. AIC, 4-Amino-5-imidazolecarboxamide; BER, Base-excision repair; HR, Homologous repair; MGMT, O6-methylguanine-DNA-methyltransferase; MMR, Mismatch repair; MLH, MutL homologue; MSH, MutS homologue; PMS, Post-meiotic segregation; TMZ, Temozolomide.
Figure 3.
TMZ molecular structure, metabolism, toxicity, and resistance. (a) TMZ structure and metabolism, (b) DNA methylation upon TMZ, (c) DNA base mispair upon DNA methylation, (d) mechanism of TMZ toxicity with intact MMR, BER, and HR, (e) mechanism of TMZ resistance with functional MGMT and non-functional MMR. AIC, 4-Amino-5-imidazolecarboxamide; BER, Base-excision repair; HR, Homologous repair; MGMT, O6-methylguanine-DNA-methyltransferase; MMR, Mismatch repair; MLH, MutL homologue; MSH, MutS homologue; PMS, Post-meiotic segregation; TMZ, Temozolomide.


{kind=link}
{kind=link}
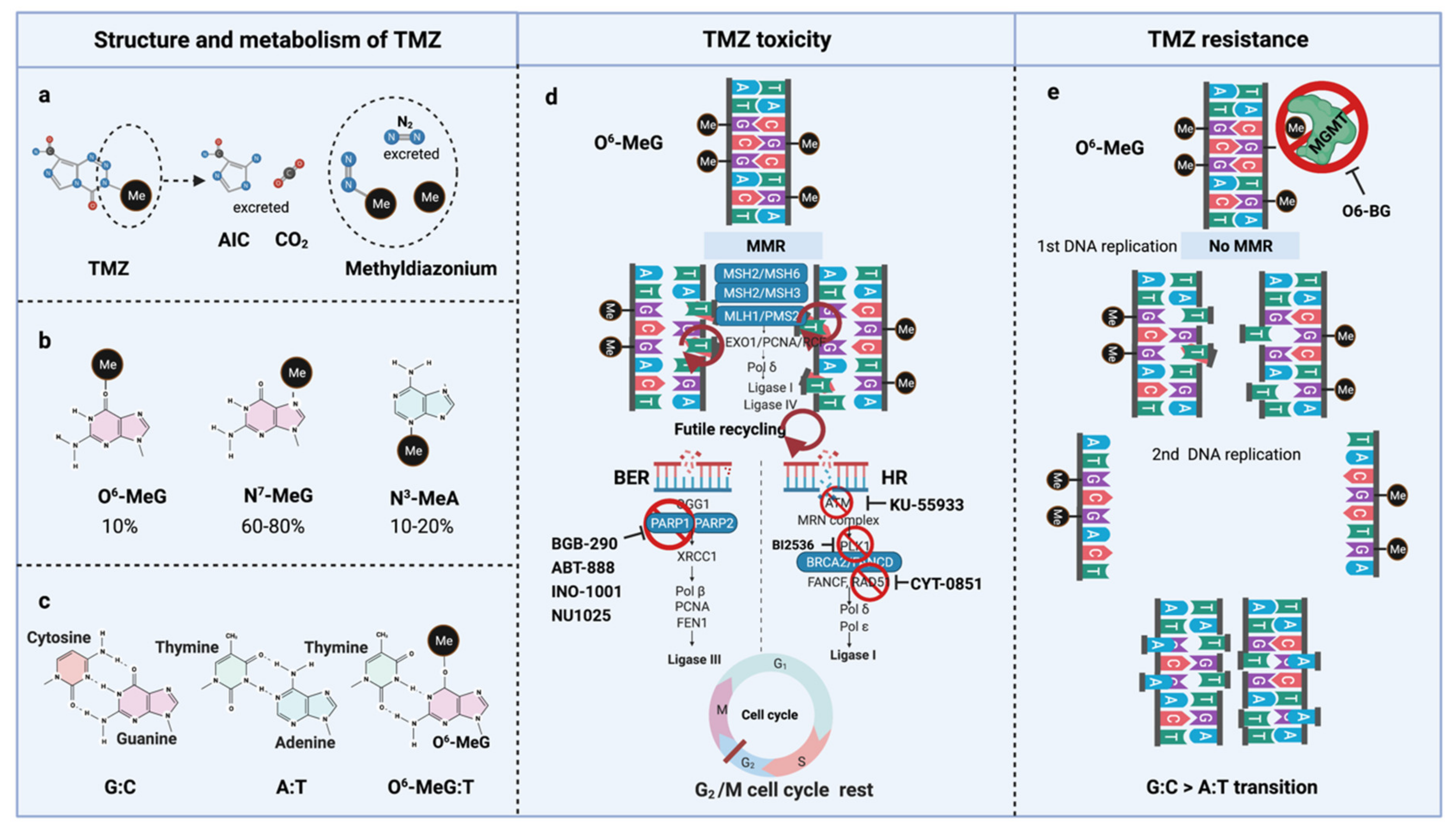{kind=link}
Table 1.
Molecular diagnostic markers and common genetic alterations in IDH-mutant glioma.
| Category | Alterations | Oligodendroglioma WHO Grade 2 | Oligodendroglioma WHO Grade 3 | Astrocytoma WHO Grade 2/3 | Astrocytoma WHO Grade 4 | |
|---|---|---|---|---|---|---|
| Diagnostic markers | IDH1 or IDH2 mutation | Present | Present | Present | Present | |
| G-CIMP | Present | Present | Present | Present | ||
| ATRX | Inactivated | Inactivated | ||||
| 1p (FUBP1) / 19q (CIC) codeletion | Present | Present | ||||
| TERT | Activated | Activated | ||||
| 9p21 (CDKN2A/B) | Inactivated | |||||
| Necrosis and/or microvascular proliferation | Present | |||||
| Other genomic alterations | TP53 | Inactivated | Inactivated | |||
| Myc | Activated | |||||
| TCF12 | Inactivated | |||||
| 10q (PTEN/MGMT) | Inactivated | |||||
| Signaling pathways | Activation of PI3K signaling through loss of PTEN and gain of mTOR | |||||
| Activation of cell cycle signaling through gain of CDK4, CDK6 and cyclin E2 | ||||||
ATRX, Alpha-thalassemia/mental retardation, X-linked; CDKN2A/B, Cyclin-dependent kinase inhibitor 2A/B; CDK, Cyclin-dependent kinases; CIC, Capicua transcriptional repressor; FUBP1, Far upstream element binding protein 1; G-CIMP, cytosine-phosphate-guanine (CpG) island methylator phenotype; IDH, Isocitrate dehydrogenase; MGMT, Methylguanine-DNA-Methyltransferase; PI3K, Phosphoinositide 3-kinase; PTEN, Phosphatase and tensin homolog; TCF12, Transcription factor 12; TERT, Telomerase reverse transcriptase.
Table 2.
Preclinical and clinical studies of combination therapy of TMZ with DNA damage repair pathway inhibitors.
Table 2.
Preclinical and clinical studies of combination therapy of TMZ with DNA damage repair pathway inhibitors.
| Targeting DNA Damage Repair | Synergistic with TMZ | Preclinical Model | Clinical Trial | Arms | Tumor Type | Phase | Year |
|---|---|---|---|---|---|---|---|
| MGMTi | O6BG + TMZ | GBM PDX [68]; astrocytoma or GBM patient [67] | NCT00006474 | O6-BG + TMZ | Astrocytoma | I | 2001–2004 |
| NCT00389090 | O6-BG + TMZ | Gliomas | II | (2006–2009)Terminated | |||
| NCT00613093 | O6-BG + TMZ | GBM | II | 2002–2008 | |||
| NCT00275002 | O6-BG + TMZ | Pediatrichigh-grade gliomas | II | 2006–2010 | |||
| PARPi | Olaparib + TMZ | U87-IDH mutant, U251-IDH mutant cell lines [81] | NCT03212742 | Olaparib + TMZ + IMRT | GBM | I/IIa | 2017–2022 |
| NCT04394858 | Olaparib + TMZ | Pheochromocytoma and paraganglioma | II | 2020–2023 | |||
| Veliparib (ABT-888)+TMZ | GBM BTICs and xenografts [80] | NCT01026493 | ABT-888 + TMZ | Recurrent GBM | I/II | 2010–2016 | |
| NCT01514201 | RT+ ABT-888 + TMZ | Children with newly diagnosed DIPG | I/II | 2012–2018 | |||
| NCT02152982 | veliparib + TMZ vs. placebo + TMZ | GBM | II/III | 2014–2020 | |||
| NCT03581292 | RT + TMZ + veliparib | GBM | II | 2018–2024 | |||
| Pamiparib (BGB-290) + TMZ | GBM, GL261 murine glioma cells xenografts [82] | NCT03150862 | BGB-290 + RT vs. BGB-290 + TMZ | GBM | 1b/2 | 2017–2021 | |
| NCT03914742 | BGB-290 + TMZ | Recurrent IDH mutant glioma | I/II | 2020–2023 | |||
| NCT03914742 | BGB-290 + TMZ | IDH mutant glioma | I | 2019–2027 |
BTICs, Brain tumor initiating cells; DIPG, Diffuse intrinsic pontine gliomas; GBM, Glioblastoma multiforme; MGMTi, Methylguanine-DNA-Methyltransferase inhibitor; PARPi, Poly (ADP-ribose) polymerase inhibitor; PDX, patient-derived xenograft; TMZ, Temozolomide.
Table 3.
Preclinical and clinical studies of combination therapy of epigenetic drug with TMZ.
| Epigenetic Target | Synergy with TMZ | Preclinical Model | Clinical Trial | Arms | Tumor Type | Phase | Year |
|---|---|---|---|---|---|---|---|
| DNMTi | AZA + TMZ | IDH-mutant glioma lines BT142, JHH273 [132] | NCT00629343 | AZA+TMZ | Soft tissue sarcoma or mesothelioma | I/II | 2007–2014 |
| DAC + TMZ | GBM PDXs [134] | NCT00715793 | DAC + TMZ | Metastatic melanoma | I/II | 2008–2015 | |
| HDACi | Vorinostat + TMZ | GBM cell lines, glioma slice culture [142] | NCT00731731 | RT + TMZ + Vorinostat | GBM | I | 2009–2014 |
| VPA + TMZ | astrocytoma grade III and GBM cell lines [144] | NCT00302159 | TMZ + VPA + RT | GBM | II | 2006–2014 | |
| DNMTi + HDACi | NCT00925132 | DAC + TMZ + Panobinostat | Melanoma | Ib/II | (2009–2016) Terminated | ||
AZA, 5-azacitidine; DAC, Decitabine; DNMTi, DNA methyltransferase inhibitor; GBM, Glioblastoma multiforme; HDACi, Histone deacetylase inhibitor; RT, Radiotherapy; TMZ, Temozolomide; VPA, Valproic acid.
Table 4.
Preclinical and clinical studies of TMZ combined with therapies targeting cancer metabolism
Table 4.
Preclinical and clinical studies of TMZ combined with therapies targeting cancer metabolism
| Metabolic Target | Combination Therapy | Preclinical Model | Clinical Trial | Arms | Tumor Type | Phase | Year |
|---|---|---|---|---|---|---|---|
| NAD+ | NAMPT inhibitor + TMZ | IDH1 mutant glioma lines [30] | NCT00724841 | GEM1777 + TMZ | Metastatic melanoma | I/II | 2008–2010 (Terminated) |
| Glutamine | CB-839 + TMZ | GBM164 (IDH mutant) and GBM6 (IDH wt) PDX [154] | NCT03528642 | CB-839 + RT + TMZ | Astrocytoma | 1b | 2018–2022 |
| Oxidative phosphorylation | MET + TMZ | GBM PDX [165] | NCT01430351 | MET + TMZ vs. mefloquine + TMZ vs. memantine + TMZ | GBM | I | 2011–2022 |
| Phospholipid | CQ + TMZ | GBM cell lines [156] | NCT02378532 | CQ + RT + TMZ | GBM | I | 2016–219 |
| Multiple metabolites | Paclitaxel + TMZ | GBM cell lines [163] | NCT01009515 | Carboplatin + Paclitaxel + TMZ | Metastatic melanoma | II | 2009–2015 (Terminated) |
| MET + CQ | NCT02496741 | MET + CQ | IDH mutant glioma | 1b | 2015–2019 |
CQ, Chloroquine; MET, Metformin; NAMPT, Nicotinamide phosphoribosyltransferase; PDX, Patient-derived xenografts; RT, Radiotherapy; TMZ, Temozolomide.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sun, X.; Turcan, S. From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas. Cells 2021, 10, 1225. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051225
AMA Style
Sun X, Turcan S. From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas. Cells. 2021; 10(5):1225. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051225
Chicago/Turabian StyleSun, Xueyuan, and Sevin Turcan. 2021. "From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas" Cells 10, no. 5: 1225. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051225
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.