
Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy


1
Oscar-Langendorff-Institute of Physiology, Rostock University Medical Center, 18057 Rostock, Germany
2
Center for Transdisciplinary Neurosciences Rostock, University of Rostock, 18147 Rostock, Germany
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(5), 1226; https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051226
Submission received: 12 April 2021
/
Revised: 12 May 2021
/
Accepted: 13 May 2021
/
Published: 17 May 2021
(This article belongs to the Special Issue Molecular Biology in Glioblastoma Multiforme Treatment)
Abstract
:The progression of glioblastomas is associated with a variety of neurological impairments, such as tumor-related epileptic seizures. Seizures are not only a common comorbidity of glioblastoma but often an initial clinical symptom of this cancer entity. Both, glioblastoma and tumor-associated epilepsy are closely linked to one another through several pathophysiological mechanisms, with the neurotransmitter glutamate playing a key role. Glutamate interacts with its ionotropic and metabotropic receptors to promote both tumor progression and excitotoxicity. In this review, based on its physiological functions, our current understanding of glutamate receptors and glutamatergic signaling will be discussed in detail. Furthermore, preclinical models to study glutamatergic interactions between glioma cells and the tumor-surrounding microenvironment will be presented. Finally, current studies addressing glutamate receptors in glioma and tumor-related epilepsy will be highlighted and future approaches to interfere with the glutamatergic network are discussed.
1. Introduction
Glioblastomas (WHO grade Ⅳ gliomas) represent the most common tumors of the central nervous system, and with an overall 5-year survival of 6.8%, this tumor disease has one of the worst prognoses in the entire oncological spectrum [1]. The median age of onset is 64 years, with an increase at the age of 75–84 years [1]. Primary glioblastomas arise de novo, whereas secondary glioblastomas derive from less-malignant precursor lesions [2]. The majority of glioblastomas is defined as primary, mostly with wild-type isocitrate dehydrogenase 1 (IDH1), whereas most of the secondary cases harbor mutations in IDH1 [3]. Mutations in IDH1 are associated with an increased patient survival [4].
Depending on the genetic and epigenetic alterations [5], four subtypes can be defined [6]. Genes encoding members of the signaling pathways phosphatidylinositol-3-kinase (PI3K) or mitogen-activated protein kinase (MAPK) and the corresponding receptor tyrosine kinases-like epidermal growth factor receptor (EGFR) or platelet-derived growth factor receptor A (PDGFRA) are altered in up to 90% of cases [7,8]. Among the tumor suppressors, loss of function of phosphatase and tensin homolog (PTEN) is the most frequent mutation (30–40%) [6,9]. In approximately 10% of cases, neurofibromin 1 (NF1) is mutated or deregulated [8,9]. With respect to cell cycle control, loss-of-function mutations of cyclin dependent kinase inhibitor 2A/B (CDKN2A/B) can be found in half of all glioblastoma specimens, and cyclin-dependent kinase 4 (CDK4) is often highly amplified. Furthermore, like in other major cancer types [10], tumor protein P53 (TP53) is also subject to mutations.
Despite the knowledge gained on tumorigenesis and the progression of glioblastomas in the last decades, these have so far only insufficiently contributed to improving treatment options. Standard therapy includes resection of the tumor mass followed by adjuvant irradiation and chemotherapy with the alkylating drug temozolomide (TMZ) [11]. This regimen prolongs the median overall survival to 15–18 months (versus 3–4 months without treatment [12]) [11,13,14]. The status of O6-methylguanine-DNA methyltransferase (MGMT) promotor methylation is an established prognostic marker of the treatment with TMZ [13,15,16,17], albeit in a Spanish study including more than 300 patients, contrary to previous studies, no beneficial effect of MGMT promotor methylation status could be detected [18]. Combination of TMZ with lomustine, also an alkylating agent, may prolong the survival of patients with methylated MGMT promotors [19]. However, the findings of Herrlinger et al. in 2019 were based on a relatively small cohort (129 patients), and survival successes came at the price of additional side effects. Another add-on therapy is tumor-treating fields that mediate antimitotic effects by alternating electric fields, which can prolong the survival of patients with glioblastoma [20,21]. However, the low level of acceptance of this method has limited its widespread use [22,23].
Glioblastomas are associated with a variety of neurological symptoms and signs including headaches, changes in cognition and personality, gait imbalance, visual deficits and tumor-associated epilepsy [12]. The occurrence of seizures is not only a frequent comorbidity in the course of glioma progression [24] but is often the initial manifestation of the tumor disease [25]. In low-grade gliomas, 70–90% of the patients suffer from seizures at the time of diagnosis, whereas in glioblastoma, seizures appear to be less frequent (up to 60%) [25,26]. Epileptogenesis in the peritumoral tissue is a multifactorial process [27]. Based on numerous preclinical, but also some clinical, studies, there is increasing evidence that the neurotransmitter glutamate plays an important role in the development of seizures and also in the progression of the tumor disease.
Herein, we review our current understanding of the glutamatergic processes at the molecular level that drive glioma progression and presentation of an epileptic phenotype. Based on this, open research questions are presented that could serve to reach a better understanding of the disease in the future.
2. Glutamatergic Mechanisms of Glioma Progression and Tumor-Associated Epilepsy
Glioblastoma and tumor-associated epilepsy share pathophysiological mechanisms that drive both tumor progression and the generation of seizures [28]. One major pathological mechanism is aberrant glutamate signaling within the tumor tissue and its microenvironment. In the glioma-surrounding tissue, extracellular glutamate levels were found to be elevated up to 100-times higher than in unaffected brains [29,30]. On the one hand, high levels of glutamate stimulate proliferation and invasion of glioma cells [31,32,33] and, on the other hand, may lead to epileptic discharges, excitotoxicity and therefore allow for tumor bulk expansion (Figure 1) [34,35].
Primarily, glutamate is released by glioblastoma cells via cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11 or xCT) [36]. To counteract oxidative stress, cystine, an essential precursor for glutathione synthesis, is imported in exchange for glutamate. In glioblastoma cells, expression of xCT is often upregulated [37,38,39,40]. This may be in part due to hypoxic conditions in fast-growing glioblastomas [41]. It was reported that the expression levels of xCT were found to be associated with seizures [42,43] and identified as an independent biomarker for glioma-associated seizures [43], also in a cohort of IDH1-wildtype glioblastoma patients [44]. With respect to glioblastoma, no correlation with survival was found [42,43].
Furthermore, the re-uptake of glutamate from the extracellular space is also impaired in glioma tissue, as expression of glutamate transporter 1 (GLT-1), also known as excitatory amino acid transporter 2 (EAAT2), is downregulated, or carriers are mislocalized, and therefore sodium-dependent re-uptake of glutamate is reduced [37,45,46]. The situation may be exacerbated by an overexpression of branched-chain amino acid transaminase 1 (BCAT1) [47]. Again, upregulation may in part be driven by hypoxic conditions, in which HIF-1α was identified as an essential transcription factor [48]. BCAT1 transfers an α-amino group from branched-chain amino acids to α-ketoglutarate, thereby producing glutamate and the respective branched-chain α-ketoacid. This mechanism may contribute to an elevated glutamate level within the cytoplasm of the tumor cells and subsequently an increased glutamate release [47]. In glioblastomas, high expression of BCAT1 was associated with poor survival [49,50].
Isocitrate dehydrogenase 1 (IDH1) mutations are common in low-grade gliomas (>80%) and secondary glioblastomas (73%) but are rare in primary glioblastomas (3.7%) [3]. Instead of catalysis of isocitrate to α-ketoglutarate in the Krebs cycle, in IDH1-mutant glioma cells, D-2-hydroxyglutarate (D-2HG) is generated [51]. D-2HG shares a steric analogy with glutamate and therefore may act as an additional glutamate receptor agonist. Interestingly, D-2HG also affects BCAT1, possibly due to direct inhibition of enzyme activity [52] or DNA hypermethylation within the main promoter region of BCAT1 in IDH1-mutant gliomas [47]. Furthermore, McBrayer et al. showed that in IDH1-mutant gliomas, an additional inhibition of glutaminase (hydrolyzes glutamine to glutamate) by glutaminase inhibitor CB-839 led to an increased cell death under hypoxic conditions in vitro and increased susceptibility to irradiation in vivo [52]. The facilitatory role of IDH1 mutations in epileptogenesis has also been shown in patient-derived samples [53,54].
In terms of the surrounding neurons, excitotoxicity via glutamate-driven hyperexcitation contributes to tumor growth [55]. High levels of this neurotransmitter lead to receptor-mediated Na+/Ca2+-dependent depolarization that eventually end up in abnormally high levels of intracellular Ca2+, which in turn may trigger key players contributing to cell death [56]. At the same time, high levels of extracellular glutamate may attenuate cystine uptake via xCT, thereby cutting off the tumor cells from cysteine as a precursor for the synthesis of the antioxidant glutathione and hence contribute to cellular damage by reactive oxygen species [55]. In addition to glutamate, GABAergic mechanisms with altered intracellular chloride concentration [57] and microglia dysfunction [58] were also described as relevant contributors to epileptogenicity in the tumor-surrounding tissue. Hence, the epileptic phenotype has a multifactorial pathogenesis, and here we will focus on glutamatergic mechanisms.
Glutamate in the synaptic cleft may activate glutamate receptors on tumor cells themselves in an autocrine manner or on proximate neurons and astrocytes in a paracrine manner. The role of ionotropic and metabotropic glutamate receptors will be discussed separately in the following sections.
2.1. Ionotropic Glutamate Receptors
Glutamate receptors are subdivided into ionotropic and metabotropic receptors (Figure 2). Ionotropic receptors have been named after the prototypical glutamate agonists α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA receptors), kainic acid (kainate receptors) and N-methyl-D-aspartate (NMDA receptors).
2.1.1. AMPA Receptors
AMPA receptors (AMPARs) are tetrameric complexes composed of GluA1, GluA2, GluA3, or GluA4 subunits in various combinations and predominantly responsible for fast excitatory transmission in the central nervous system [59]. All subunits, albeit GluA1 to a lesser extent, have been detected in glioblastoma cell lines and patient-derived specimens [60,61]. Whereas immature neurons express low levels of GluA2-containing AMPARs, which confers Ca2+-permeability and an inwardly rectifying current–voltage relationship, GluA2 expression increases during postnatal development [62,63]. At adult synapses, however, GluA2 subunits typically show full adenosine-to-inosine mRNA editing by virtue of the enzyme ADAR (adenosine deaminase acting on RNA) at the Q/R-site leading to a translational modification from glutamine (Q) to arginine (R) [64]. In contrast, unedited GluA2 subunits also form Ca2+-permeable AMPARs and seem to be frequently expressed in glioma tissues [61,65,66,67], and importantly, these receptors appear to promote proliferation and migration [68]. Since the discovery of glutamatergic synapses between neurons and glioma cells (Section 2.3), Ca2+-permeable AMPARs have been regarded as attractive candidates for specific antagonists to interfere with the tumor-promoting effects of glutamate [67].
Since the non-competitive AMPAR antagonist, perampanel (PER), has been developed to treat patients with partial and generalized seizures [69,70], this compound has attracted much interest in treating glioma-associated seizures due to the hypothesis that glutamate released from glioma cells could not only activate surrounding neurons to cause seizures and lead to excitotoxicity but could also promote glioma progression [67]. Along these lines, PER could be effective in both seizure control and tumor repression (Section 2.4).
2.1.2. Kainate Receptors
Kainate receptors (KARs) are tetrameric transmembrane proteins composed of three low-affinity subunits, GluK1 to GluK3 (glutamate receptor, kainate-subtype, formerly named GluR5 to GluR7), and two high-affinity subunits, GluK4 (KA1) and GluK5 (KA2), in homo- and heteromeric combinations [71,72]. Our notion on the expression of KARs in gliomas is scarce, but at least in pediatric glioblastoma, GluK3 and GluK4 were found to be expressed at levels higher than in the human brain, whereas low-grade astrocytomas only showed increased expression of GluK4 [61]. This is consistent with the high expression of GluK4 in U373 glioma cells [73] and U87MG cells [60] as well as in HAP-1 glial precursor cells [74]. However, the role of KAR expression on glioma cells in tumor progression needs to be established, since glutamate released from glioma cells might activate KARs along with AMPARs in an autocrine fashion [32]. On the other hand, neuronal KAR activation by astrocyte-released glutamate can be highly specific, since glutamatergic effects on interneurons in the CA1 stratum radiatum were found to be confined to GluK1-containing KARs [75]. Using the highly selective GluK1-inhibitor topiramate (TPM) [76] or establishing novel subunit-specific KAR inhibitors such as piperazine-2,3-dicarboxylic acid derivatives [77], future studies are needed to focus on the role of KARs in glioma progression and glioma-associated epilepsy.
2.1.3. NMDA Receptors
NMDA receptors are tetrameric transmembrane proteins composed of two glycine/D-serine-binding GluN1 (glutamate receptor, NMDA-subtype) subunits and two glutamate-binding GluN2 or GluN3 subunits [59,78]. While there is only one GluN1 gene with diverse splice variants, there are four GluN2 subunits, named GluN2A-D, and two GluN3 subunits [78]. The vast majority of literature on physiologically expressed native NMDA receptors (NMDARs) has concentrated on the subunits GluN2A and GluN2B. During physiological ontogenesis, there is a shift from GluN2B-containing NMDARs in the immature brain towards GluN2A-containing isoforms in adult mammals [79]. This also has functional implications, since GluN2B-containing NMDARs show slower decay times compared with GluN2A-containing receptors, allowing for an increased Ca2+ influx in the open state [80]. On the network level, the NMDAR subunit composition governs synaptic plasticity [81,82], and GluN2B-containing receptors predominantly expressed at extrasynaptic sites can laterally diffuse to modulate synaptic plasticity [83]. Interestingly, the developmental shift from GluN2B to GluN2A seems to be reversed in chronically epileptic tissues [84,85]. Intriguingly, recent data showed a conversion of the upregulated GluN2B tyrosine(1472) phosphorylation in the pilocarpine model of chronic temporal lobe epilepsy by the AMPAR antagonists PER and GYKI 52,466 in responders, but not non-responders [86]. This effect has been attributed to increased Pten expression and decreased activity of the Src family-casein kinase 2 signaling pathway [86]. Human astrocytes express NMDARs, including GluN2B [87,88], and PTEN deficiency in the human glioblastoma U87MG cell line was associated with increased proliferation [89], but NMDARs seemed to be less abundant in glioma specimens [90,91]. In particular, GluN1 seemed to be underexpressed [60,61]. Nonetheless, since increased GluN2B serine(1303) phosphorylation was detected in human perigliomal tissue [92], and given that PER is quite effective in suppressing glioma-associated seizures (Table 1) [93,94,95,96,97,98], whether the efficacy of PER in brain tumors could partially rely on PTEN-mediated reversal of upregulated GluN2B tyrosine(1472) phosphorylation is an attractive hypothesis.
The atypical amino acid D-serine was identified as a substitute ligand at the till-then called strychnine-insensitive glycine-binding site of NMDARs [99] and subsequently detected in the cerebral cortex, representing 20–30% of total brain serine [100]. D-serine is produced from L-serine by the glial enzyme serine racemase [101], and the uneven distribution of D-serine in the brain [100] has been found to be reciprocal to the expression of its degrading enzyme D-amino acid oxidase (DAAO) described two decades earlier [102]. In turn, D-serine uptake into glial cells is due to the alanine-serine-cysteine amino acid transporter-2 (ASCT2) [103,104]. For more than 20 years, D-serine has been accepted as the endogenous coagonist of NMDARs [105,106]. With respect to gliomal D-serine metabolism, many groups have studied glioma cell lines, using them as surrogates for forebrain astrocytes. While DAAO appears to be underexpressed in C6 [107] and U87MG glioma cells [108], there is an important interaction between D-serine and nitric oxide (NO), which has been found in U87MG cells: NO suppressed D-serine abundance due to both serine racemase inhibition [109] and DAAO activation [110]. Together with the established upregulation of inducible NO synthase (iNOS) in human glioblastoma specimens [111], this may point to a global D-serine downregulation in glioma tissues. One plausible explanation for overall D-serine suppression in glioma tissues might be the potential cytotoxic effects of DAAO-mediated D-serine degradation leading to H2O2 production, which could be demonstrated to exert therapeutic relevance in C6 cells [112].
Taken together, GluN2B-mediated mechanisms might be related to epileptogenesis and, hence, specific antagonists against GluN2B-containing NMDARs could be effective in reducing glioma-associated epilepsy. In contrast, D-serine abundance appears to be downregulated in glioma tissues and therefore might be an anti-glioma strategy along with iNOS inhibition.
2.2. Metabotropic Glutamate Receptors
Metabotropic glutamate receptors (mGluR) are a family of G-protein-coupled receptors that, with respect to the central nervous system, are predominantly expressed in synapses [113]. A total of eight members (mGluR1-8) are known, which are divided into three groups based on sequence homology, pharmacological properties and intracellular signaling pathways (Figure 2, lower row). Upon the binding of glutamate, the receptors form dimers that are mostly homomers, but there is also evidence that heterodimers exist [114,115].
Group I mGluRs are coupled to Gq/G11. After receptor activation and G-protein dissociation, phospholipase C (PLC) hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3), leading to IP3-receptor-mediated Ca2+ release from the endoplasmatic reticulum. Finally, protein kinase C (PKC) is activated and may phosphorylate downstream targets. Both group Ⅱ and group Ⅲ receptors couple to Gi/o, and upon receptor activation, lyase activity of adenylate cyclase is attenuated. One major downstream pathway is the cyclic adenosine monophosphate (cAMP)-dependent activation/inactivation of protein kinase A (PKA). Both Gi/o- and Gq/G11-coupled receptors are linked to major signaling pathways like PI3K/AKT and MAPK, which are essential to the regulation of cell survival and proliferation [116].
Analyses of human tissue freshly taken from surgery and data of permanent lineages show that glioblastoma cells express all types of metabotropic glutamate receptors [60,61,117], with mGluR3 demonstrating the highest expression on average [117]. Hence, it is not surprising that most studies have focused on group 2 receptors.
2.2.1. Group I
Group I consists of mGluR1 and mGluR5. Little is known about the possible role of group I metabotropic receptors in tumorigeneses and glioblastoma progression. Studies employing permanent lineages could show that antagonists of mGluR1 mediated inhibitory effects with respect to cell proliferation and viability in vitro [118,119]. Accompanying immunoblot analysis displayed an overall lower phosphorylation of kinases of the PI3K/AKT pathway. In line with this finding, growth of U87MG xenografts in immunodeficient mice was also attenuated by mGluR1 antagonists [118]. Under hypoxic conditions, inhibition of mGluR5 contributed to an increase of cell death concomitant with a lower AKT phosphorylation [120].
2.2.2. Group II
The second group is formed by mGluR2 and mGluR3. A low expression of mGluR3 in surgery samples was correlated with an overall longer survival after diagnosis [117,121]. Cell culture studies employing primary glioblastoma cells and the U87MG cell line suggested that cell proliferation, survival and migration were dependent on functional mGluR2/3 proteins [122,123,124]. The antitumoral effects were confirmed in nude mice with subcutaneously localized glioma xenografts [122] or orthotopically-implanted tumors [125]. Inhibition of mGluR2/3 signaling may also increase susceptibility to an additional treatment with EGFR antagonist gefitinib in vitro [124].
Furthermore, low mGluR3 expression in patients suffering from glioblastoma is also associated with a higher response to treatment with TMZ [121]. This is consistent with the finding that patients with low mGluR3 and MGMT promotor methylation have the longest survival. Notably, no effect of GluR2/3 inhibition on sensitivity to TMZ was observed in permanent glioblastoma cells, whereas glioma stem-like cells were prone to combined treatment [117]. Unfortunately, experiments of xenografts with patient-derived glioma stem-like cells based on these findings showed only limited effects of GluR2/3 inhibitors on tumor size and no impact on survival at all [117].
2.2.3. Group III
Group Ⅲ consists of mGluR4, mGluR6, mGluR7, and mGluR8. Like group I, our knowledge of their possible function in the context of glioblastoma is scarce. Perturbation of mGluR4-mediated signaling may affect tumor cell growth [126]. The authors showed that activation of mGluR4 by selective agonist VU0155041 reduced proliferation and promoted apoptosis in the LN229 glioblastoma cell line in vitro. These data are in line with the effects of N-phenyl-7-(hydroxyimino) cyclopropa[b]chromen-1a-carboxamide (PHCCC) in medulloblastoma cell lines D283 Med, D341 Med, and DAOY [127]. Activation of mGluR4 by PHCCC reduced tumor cell growth in vitro and in immunodeficient mice [127]. A similar role could be played by mGluR8, as this receptor was identified to suppress glioblastoma cell growth and mediate susceptibility to chemotherapeutics in vitro [128].
To the best of our knowledge, no biological data has been published so far for mGluR6 and mGluR7. The expression of mGluR6, best known for its function in the ON-bipolar cells of the retina, was found to be associated with high-grade pediatric CNS tumors [61].
2.3. Neurogliomal Synapse
Since glutamate receptors are expressed on glioblastoma cells, the question of how glutamate may reach these cells was until recently believed to be purely spillover transmission from the same or neighboring glioma cells (autocrine and paracrine transmission). This scenario was dramatically extended by the ultrastructural discovery of synaptic contacts between neurons and glioma cells, referred to as neurogliomal synapses [66,67]. Whether all glutamate receptors expressed by glioma cells (i.e., postsynaptic in nature), as outlined in Section 2.1. and Section 2.2., mediate glutamate effects from these neurogliomal synapses (Figure 1) is an open question, but inhibitors of Ca2+-permeable AMPARs may yield intriguing candidates to interfere with glutamatergic transmission at these synapses [67].
2.4. Therapeutic Strategies
The current oncological treatment of glioblastoma patients has been excellently reviewed elsewhere [129]. Although temozolomide might exert some anticonvulsant effects [130], here we will focus on pharmacological means of addressing glioma-associated seizures since there is an increasing understanding of the shared mechanisms of tumor progression and epileptogenicity, suggesting that certain compounds could establish novel strategies for the treatment of glioblastoma patients presenting epileptic seizures.
2.4.1. Sulfasalazin
Sulfasalazin (SAS) is an inhibitor of the glutamate-cystine exchanger xCT and was therefore regarded as a potential anti-tumor drug, counteracting glutamate release by glioma cells [34,131]. Unfortunately, however, a human clinical trial adding SAS to radiochemotherapy revealed prolonged seizure-free survival in SAS-coadministered patients, but SAS was associated with hematologic side effects [132]. The multi-kinase inhibitor sorafenib was tested in glioblastoma therapy with limited success [133,134,135,136]. Nonetheless, the effects of sorafenib have to be reconciled since this compound was also identified as an xCT inhibitor [137,138].
2.4.2. Anticonvulsants
The initial enthusiastic observation of a survival benefit in glioblastoma patients treated with adjunctive valproic acid (VPA) to standard radiochemotherapy [139,140,141,142,143] was not corroborated in grade Ⅱ/Ⅱ glioma patients [144] and was then shown from a new perspective by a recent meta-analysis, raising concerns in comparing historical cohorts [145]. On the pathophysiological level, the VPA effect has been attributed to the inhibition of histone deacetylase [146], but again, this cell culture-based in vitro finding was not confirmed in patient-derived glioma tissue specimens [147] or patient-derived low-passage cell lines [148].
Despite there being no specific guidelines for the anticonvulsant choice regarding brain tumor-associated seizures, there is ongoing interest in identifying compounds with anti-tumoral in vitro effects such as levetiracetam (LEV) [149]; see [148]. Among more contemporary anticonvulsants, most patients receive LEV for glioma-associated epileptic seizures, and some recent studies have observed a benefit in overall survival [150,151,152]. Although the mechanism of this survival benefit has not been directly addressed, it may partly be due to a reduced glutamate-to-GABA ratio [153,154].
Although topiramate (TPM) was also considered as the anti-seizure medication of choice in the past [155], its use has been almost abandoned due to side effects and the introduction of LEV. However, since the KAR inhibitor TPM may have anti-proliferative effects in glioblastoma cells [156], it is probably worth reconsidering in patients with insufficient seizure control by LEV and VPA.
2.4.3. Talampanel
Talampanel is an orally bioavailable, noncompetitive antagonist of the AMPA subtype of ionotropic glutamate receptors. In a phase Ⅱ trial [157], it was added to radiotherapy (30-times 0.2 Gy) and concomitant TMZ (75 mg/m2 for 6 weeks), subsequently followed by adjuvant TMZ (150-200 mg/m2 for 5 days every 28 days for 6 months). In a sub-cohort of 60 patients age-matched to the European Organisation for Research and Treatment of Cancer (EORTC) population, the median survival was significantly prolonged, suggesting that pharmacological inhibition of AMPA receptors could be regarded as a promising therapeutic strategy [157]. However, this effect was only seen in combination with radiotherapy and TMZ, whereas talampanel alone had no significant effect on tumor progression or survival [158].
2.4.4. Perampanel
Perampanel is a structural analogue of talampanel and has been approved as an add-on-anticonvulsant for focal and generalized epilepsy. The encouraging data on talampanel have fostered studies with PER in glioma-associated epilepsy, and since the description of PER administered to a patient with IDH1-wildtype and MGMT-unmethylated glioblastoma who became seizure-free and survived for 18 months [159], a number of rather small studies have explored PER efficacy in glioma-associated seizures (Table 1). Taken together, the 81 patients enrolled, of whom 46 became seizure-free with PER (57%), had a responder rate (i.e., all patients with at least 50% seizure reduction) of 86%. Given the fact that PER was added to a so-far insufficient anti-seizure medication, PER appears to be substantially effective in these patients with glioma-associated epilepsy.
2.4.5. Memantine
3. Preclinical Models to Study Glutamate Interaction and Tumor-Associated Epilepsy
A variety of preclinical glioma models exist, ranging from cell cultures to in vivo models employing animal and human tumor cells. Experimental models that focus primary on tumors were recently critically discussed in a review by Lenting et al., 2017 [161]. In this section, we focus on models that are appropriated to investigate the role of glutamate.
3.1. Cell Culture Models
Cell culture studies have the decisive disadvantage that the interaction between the microenvironment and the immune system is neglected in principle (Table 3). Moreover, in cell lines like U87MG and U251, which were established several decades ago, genetic changes due to long-term cultivation in serum-containing conditions are likely [162,163]. Nevertheless, most basic mechanistic relationships are based on studies employing these permanent glioma cells (see previous section).
A superior alternative to these permanent cells is patient-derived cell lines in low passages. Published biobanks reflect all kinds of molecular subtypes of glioblastoma [174,175,176,177]. If the molecular status of the primary tumor is known and the corresponding clinical patient data are available, the correspondingly derived cell lines are of particular value for preclinical studies. Employing low-passage cell lines could be used to investigate drug effects on glutamatergic mechanisms in glioblastoma [117,148].
In vitro, glioblastoma cells can also be cultivated as 3D spheroids or organoids [165,166,167,178]. As in 2D cultures, the tumor-surrounding microenvironment is absent. Co-culture models and approaches with limited immune components exist, but these may only reflect selected components and are no full replacement for in vivo conditions [166]. With respect to glutamate-driven processes in glioblastoma, the main focus with these models is on drug screening and as a source for injection in in vivo models or in organotypic brain slices.
3.2. Organotypic Brain Slice Cultures
With respect to the 3R principle (replacement, reduction, and refinement), organotypic brain slices can be a valuable link between cell culture studies (2D/3D) and animal experiments. Organotypic brain slice cultures are primarily used to study glioblastoma invasion and angiogenesis [179,180,181,182]. Interestingly, Savaskan et al. (2011) found no direct effect on glioma cells by AMPA receptor inhibition in vitro [168]. However, in the same study on organotypic slices harboring glioma cells, tumor growth was attenuated, and neuronal cell death was alleviated, which highlights the close interaction of glioma cells and peritumoral tissue [168]. Organotypic brain slice cultures may also be used to functionally characterize synaptic interactions, which will gain additional importance with the recent discovery of neurogliomal synapses (see Section 2.3.) [66,67].
Here, we show that C6-bearing slices based on brain tissue from 6–8 days-old Fischer rats with C6 cells also represent a feasible tool to investigate the network activity (Figure 3A). A high synaptic synchrony of cortical neurons that declines as the incubation progresses was estimated, which was expected for slice cultures without glioma [183].
3.3. In Vivo Models
Only models with orthotopic glioma are useful models to investigate glioma-associated epilepsy. In order to reliably verify tumor-associated seizures, EEG recordings are required, supported by video recordings. To the best of our knowledge, only a small number of in vivo rodent models with glioma-associated epilepsy are published so far [184].
A well-established in vivo model is the implantation of orthotopically-growing F98 rat glioma cells in Fischer rats [185]. Depending on the number of cells injected, the animals survive for about 2–5 weeks after glioma implantation [169,185,186,187]. Recently, the model was reported to develop a tumor-associated seizure during glioma progression [169,170]. In line with own data from video-EEG analyses (Figure 3B), Bouchaert et al. (2020) observed seizures in all tumor-bearing animals in their study. However, the model shows a heterogeneous distribution of the seizures, with some animals suffering from more seizures per day and others having only a few seizures over the entire period of the experiment [169,170]. Between the seizures, interictal events can be found in the EEG (black traces in Figure 3B). These spikes become more frequent as tumor growth is proceeding. Interestingly, we found a decrease in spike rates during the pre-moribund state prior sacrificing the animals, presumably due to neuronal death, increasing intracranial pressure and the loss of network integrity (data not shown).
C6 glioma cells have been used in a variety of experimental studies in the past [188]. Like the F98 model, the glioma-based epileptiform activity of C6 glioma is verified [171,172]. Following external auditory stimuli, the authors recorded seizures in EEG analyses. Additionally, spontaneous seizures could be documented in our video EEG recordings, although these are less frequent compared with the F98 glioma model (data not shown). In brain slices with C6 tumors, field potential records revealed that epileptiform discharges are glutamatergic [164].
Recently, Hatcher et al. (2020) published a CRISPR-based mice model [58]. Fetal deletion of Pten, Nf1, and Trp53 leads to an onset of glioblastoma with an epileptiform phenotype. Like the rat model of F98 glioma, abnormalities become visible in the EEG in the mice model, and from P60, seizures are present. Another mouse model with a glioma-associated epileptiform phenotype is also based on Pten and TrP53 deletions [173]. In the late stages of tumor progression, the animals exhibit interictal events and seizures. Both models combine the great advantage that the tumors are based on alterations of human glioblastomas.
In addition to immunocompetent models of rodent glioma, primary human glioblastoma cells could be used in nude mice. By implanting primary human glioblastoma cells (GBM12 or GBM22), Buckingham and colleagues recorded epileptiform events in tumor-bearing C.B-17 SCID mice [34]. The advantage of using human glioblastoma cells in immunodeficient animals compared with the immunocompetent rodent models of glioma must be weighed up according to the scientific question.
4. Conclusions and Future Perspectives
A number of glutamatergic mechanisms have been revealed to be involved in both glioma progression and glioma-associated epilepsy, ultimately culminating in the discovery of the neurogliomal synapse. Hence, pharmacological interventions in these mechanisms are promising strategies in addressing both tumor progression and epilepsy. Findings on AMPAR antagonists and glutamate transporter inhibitors have revealed potentially beneficial effects in a variety of glioma models, but human studies are rare, and being easily underpowered, which is an inherent challenge. However, a number of players in this glutamatergic interplay, such as metabotropic glutamate receptors and D-serine, have only been marginally addressed so far and certainly merit more attention in future studies. Thus, it is conceivable that combined interventional strategies rather than individual clinical proving may be helpful in promoting new therapies [189].
In the absence of glioma-specific guidelines for treating symptomatic seizures, glioma-associated epilepsy was controlled with LEV and VPA [25,190], but further compounds were also effective [191]. The AMPAR antagonist PER may offer the opportunity to address the two faces of glutamatergic effects in glioma, i.e., progression and epileptogenicity. Currently, there are further ongoing clinical trials evaluating PER efficacy and, in addition, brivaracetam and lacosamide are increasingly applied due to their parenteral formulations. Since the latter two are assumed to reduce glutamate release not only from neurons, but also from astroglia [192,193], the glutamatergic interplay in the glioma-surrounding network will reasonably attract more interest in future studies.
Author Contributions
Conceptualization, F.L. and T.K.; writing—original draft preparation, F.L. and T.K.; writing—review and editing, F.L. and T.K.; data curation, J.H., F.L. and T.K.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| aCSF | artificial cerebrospinal fluid |
| AKT | protein kinase B |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPAR | AMPA receptor |
| ASCT2 | alanine-serine-cysteine amino acid transporter-2 |
| BCAT1 | branched-chain amino acid transaminase 1 |
| cAMP | cyclic adenosine monophosphate |
| CDKN2A/B | cyclin dependent kinase inhibitor 2A/B |
| CDK4 | cyclin-dependent kinase 4 |
| DAAO | D-amino acid oxidase |
| DAG | diacylglycerol |
| D-2HG | D-2-hydroxyglutarate |
| EAAT2 | excitatory amino acid transporter 2 |
| GLT-1 | glutamate transporter 1 |
| IDH1 | isocitrate dehydrogenase 1 |
| iNOS | inducible NO synthase |
| IP3 | inositol-1,4,5-trisphosphate |
| KAR | kainate receptor |
| LEV | levetiracetam |
| MAPK | mitogen-activated protein kinase |
| MGMT | O6-methylguanine-DNA methyltransferase |
| mGluR | metabotropic glutamate receptor |
| Nf1 | neurofibromin 1 |
| NMDA | N-methyl-D-aspartate |
| NMDAR | NMDA receptor |
| PDGFRA | platelet-derived growth factor receptor A |
| PER | perampanel |
| PI3K | phosphatidylinositol-3-kinase |
| PKA | protein kinase A |
| PLC | phospholipase C |
| PTEN | phosphatase and tensin homolog |
| SAS | sulfasalazine |
| SV2A | synaptic vesicle glycoprotein 2A |
| TMZ | temozolomide |
| TPM | topiramate |
| TP53 | tumor protein P53 |
| VPA | valproic acid |
| xCT | solute carrier family 7 member 11 |
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino, M.dC.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Egaña, L.; Auzmendi-Iriarte, J.; Andermatten, J.; Villanua, J.; Ruiz, I.; Elua-Pinin, A.; Aldaz, P.; Querejeta, A.; Sarasqueta, C.; Zubia, F.; et al. Methylation of MGMT promoter does not predict response to temozolomide in patients with glioblastoma in Donostia Hospital. Sci. Rep. 2020, 10, 18445. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs. Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, D.; Guillermo Prieto Eibl, M.D.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassman, A.B.; Joanta-Gomez, A.E.; Pan, P.C.; Wick, W. Current usage of tumor treating fields for glioblastoma. Neurooncol. Adv. 2020, 2, vdaa069. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.L.; Palmisano, S.; Schwartzbaum, J.A.; Svensson, T.; Lönn, S. Comorbid conditions associated with glioblastoma. J. Neurooncol. 2014, 116, 585–591. [Google Scholar] [CrossRef] [PubMed]
- van Breemen, M.S.M.; Wilms, E.B.; Vecht, C.J. Epilepsy in patients with brain tumours: Epidemiology, mechanisms, and management. Lancet Neurol. 2007, 6, 421–430. [Google Scholar] [CrossRef]
- Kerkhof, M.; Vecht, C.J. Seizure characteristics and prognostic factors of gliomas. Epilepsia 2013, 54, 12–17. [Google Scholar] [CrossRef]
- Armstrong, T.S.; Grant, R.; Gilbert, M.R.; Lee, J.W.; Norden, A.D. Epilepsy in glioma patients: Mechanisms, management, and impact of anticonvulsant therapy. Neuro-Oncology 2016, 18, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huberfeld, G.; Vecht, C.J. Seizures and gliomas—Towards a single therapeutic approach. Nat. Rev. Neurol. 2016, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Roslin, M.; Henriksson, R.; Bergström, P.; Ungerstedt, U.; Bergenheim, A.T. Baseline levels of glucose metabolites, glutamate and glycerol in malignant glioma assessed by stereotactic microdialysis. J. Neurooncol. 2003, 61, 151–160. [Google Scholar] [CrossRef]
- Marcus, H.J.; Carpenter, K.L.H.; Price, S.J.; Hutchinson, P.J. In vivo assessment of high-grade glioma biochemistry using microdialysis: A study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 2010, 97, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Lin, J.H.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiuchi, S.; Yoshida, Y.; Sugawara, K.; Aihara, M.; Ohtani, T.; Watanabe, T.; Saito, N.; Tsuzuki, K.; Okado, H.; Miwa, A.; et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 2007, 27, 7987–8001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.I.; Morokoff, A.P.; Bjorksten, A.; D’Abaco, G.; Paradiso, L.; Finch, S.; Wong, D.; Reid, C.A.; Powell, K.L.; Drummond, K.J.; et al. Glutamate is associated with a higher risk of seizures in patients with gliomas. Neurology 2012, 79, 883–889. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.-Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.J.; Lyons, S.A.; Nelson, G.M.; Hamza, H.; Gladson, C.L.; Gillespie, G.Y.; Sontheimer, H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005, 25, 7101–7110. [Google Scholar] [CrossRef] [PubMed]
- Savaskan, N.E.; Heckel, A.; Hahnen, E.; Engelhorn, T.; Doerfler, A.; Ganslandt, O.; Nimsky, C.; Buchfelder, M.; Eyüpoglu, I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008, 14, 629–632. [Google Scholar] [CrossRef]
- Takeuchi, S.; Wada, K.; Toyooka, T.; Shinomiya, N.; Shimazaki, H.; Nakanishi, K.; Nagatani, K.; Otani, N.; Osada, H.; Uozumi, Y.; et al. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 2013, 72, 33–41. [Google Scholar] [CrossRef]
- Long, Y.; Tao, H.; Karachi, A.; Grippin, A.J.; Jin, L.; Chang, Y.E.; Zhang, W.; Dyson, K.A.; Hou, A.Y.; Na, M.; et al. Dysregulation of Glutamate Transport Enhances Treg Function That Promotes VEGF Blockade Resistance in Glioblastoma. Cancer Res. 2020, 80, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 289ra86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, M.F.; Heimisdóttir, S.B.; Sørensen, M.D.; Mellegaard, C.S.; Wohlleben, H.; Kristensen, B.W.; Beier, C.P. High expression of cystine-glutamate antiporter xCT (SLC7A11) is an independent biomarker for epileptic seizures at diagnosis in glioma. J. Neurooncol. 2018, 138, 49–53. [Google Scholar] [CrossRef]
- Dührsen, L.; Sauvigny, T.; Ricklefs, F.L.; Mende, K.-C.; Schaper, M.; Matschke, J.; Goebell, E.; Westphal, M.; Martens, T. Seizures as presenting symptom in patients with glioblastoma. Epilepsia 2019, 60, 149–154. [Google Scholar] [CrossRef] [Green Version]
- de Groot, J.F.; Liu, T.J.; Fuller, G.; Yung, W.K.A. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 2005, 65, 1934–1940. [Google Scholar] [CrossRef] [Green Version]
- Buccoliero, A.M.; Caporalini, C.; Scagnet, M.; Mussa, F.; Giordano, F.; Sardi, I.; Migliastro, I.; Moscardi, S.; Conti, V.; Barba, C.; et al. Angiocentric glioma-associated seizures: The possible role of EATT2, pyruvate carboxylase and glutamine synthetase. Seizure 2021, 86, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. 2021, 78, 195–206. [Google Scholar] [CrossRef]
- Cho, H.R.; Jeon, H.; Park, C.K.; Park, S.H.; Kang, K.M.; Choi, S.H. BCAT1 is a New MR Imaging-related Biomarker for Prognosis Prediction in IDH1-wildtype Glioblastoma Patients. Sci. Rep. 2017, 7, 17740. [Google Scholar] [CrossRef]
- Yi, L.; Fan, X.; Li, J.; Yuan, F.; Zhao, J.; Nistér, M.; Yang, X. Enrichment of branched chain amino acid transaminase 1 correlates with multiple biological processes and contributes to poor survival of IDH1 wild-type gliomas. Aging 2021, 13, 3645–3660. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mao, Q.; Wang, X.; Liu, Y.; Mao, Y.; Zhou, Q.; Luo, J. An analysis of 170 glioma patients and systematic review to investigate the association between IDH-1 mutations and preoperative glioma-related epilepsy. J. Clin. Neurosci. 2016, 31, 56–62. [Google Scholar] [CrossRef]
- Feyissa, A.M.; Worrell, G.A.; Tatum, W.O.; Chaichana, K.L.; Jentoft, M.E.; Cazares, H.G.; Ertekin-Taner, N.; Rosenfeld, S.S.; ReFaey, K.; Quinones-Hinojosa, A. Potential influence of IDH1 mutation and MGMT gene promoter methylation on glioma-related preoperative seizures and postoperative seizure control. Seizure 2019, 69, 283–289. [Google Scholar] [CrossRef]
- Noch, E.; Khalili, K. Molecular mechanisms of necrosis in glioblastoma: The role of glutamate excitotoxicity. Cancer Biol. Ther. 2009, 8, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Cresto, N.; Baulac, M.; Duyckaerts, C.; Kourdougli, N.; Chazal, G.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatcher, A.; Yu, K.; Meyer, J.; Aiba, I.; Deneen, B.; Noebels, J.L. Pathogenesis of peritumoral hyperexcitability in an immunocompetent CRISPR-based glioblastoma model. J. Clin. Investig. 2020, 130, 2286–2300. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Stepulak, A.; Luksch, H.; Gebhardt, C.; Uckermann, O.; Marzahn, J.; Sifringer, M.; Rzeski, W.; Staufner, C.; Brocke, K.S.; Turski, L.; et al. Expression of glutamate receptor subunits in human cancers. Histochem. Cell Biol. 2009, 132, 435–445. [Google Scholar] [CrossRef]
- Brocke, K.S.; Staufner, C.; Luksch, H.; Geiger, K.D.; Stepulak, A.; Marzahn, J.; Schackert, G.; Temme, A.; Ikonomidou, C. Glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol. Ther. 2010, 9, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Pickard, L.; Noël, J.; Henley, J.M.; Collingridge, G.L.; Molnar, E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J. Neurosci. 2000, 20, 7922–7931. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.S.; Bacci, A.; Kharazia, V.; Huguenard, J.R. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 2002, 22, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.; Vissel, B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar] [CrossRef] [Green Version]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef] [PubMed]
- French, J.A.; Krauss, G.L.; Wechsler, R.T.; Wang, X.F.; DiVentura, B.; Brandt, C.; Trinka, E.; O’Brien, T.J.; Laurenza, A.; Patten, A.; et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology 2015, 85, 950–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piña-Garza, J.E.; Rosenfeld, W.; Saeki, K.; Villanueva, V.; Yoshinaga, H.; Patten, A.; Williams, B.; Malhotra, M. Efficacy and safety of adjunctive perampanel in adolescent patients with epilepsy: Post hoc analysis of six randomized studies. Epilepsy Behav. 2020, 104, 106876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Ann. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, D.J.; Pérez-Cerdá, F.; Matute, C. KA1-like kainate receptor subunit immunoreactivity in neurons and glia using a novel anti-peptide antibody. Brain Res. Mol. Brain Res. 2000, 81, 164–176. [Google Scholar] [CrossRef]
- Cauley, K.; Kukekov, V.; Young, D. Kainate/AMPA receptors expressed on human fetal astrocytes in long-term culture. J. Neurosci. Res. 1997, 47, 311–321. [Google Scholar] [CrossRef]
- Liu, Q.S.; Xu, Q.; Arcuino, G.; Kang, J.; Nedergaard, M. Astrocyte-mediated activation of neuronal kainate receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 3172–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryder, D.S.; Rogawski, M.A. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J. Neurosci. 2003, 23, 7069–7074. [Google Scholar] [CrossRef] [PubMed]
- Irvine, M.W.; Costa, B.M.; Dlaboga, D.; Culley, G.R.; Hulse, R.; Scholefield, C.L.; Atlason, P.; Fang, G.; Eaves, R.; Morley, R.; et al. Piperazine-2,3-dicarboxylic acid derivatives as dual antagonists of NMDA and GluK1-containing kainate receptors. J. Med. Chem. 2012, 55, 327–341. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T.R. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 2011, 70, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.C.; Morera-Herreras, T.; Cui, Y.; Du, K.; Sheehan, T.; Kotaleski, J.H.; Venance, L.; Blackwell, K.T. The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLoS Comput. Biol. 2012, 8, e1002493. [Google Scholar] [CrossRef]
- Philpot, B.D.; Sekhar, A.K.; Shouval, H.Z.; Bear, M.F. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 2001, 29, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuate, A.; Barria, A. Rapid exchange of synaptic and extrasynaptic NMDA receptors in hippocampal CA1 neurons. J. Neurophysiol. 2020, 123, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Hosseinmardi, N.; Mirnajafi-Zadeh, J.; Javan, M.; Semnanian, S.; Naghdi, N.; Fathollahi, Y. Long-term potentiation enhancing effect of epileptic insult in the CA1 area is dependent on prior-application of primed-burst stimulation. Exp. Brain Res. 2020, 238, 897–903. [Google Scholar] [CrossRef]
- Müller, L.; Tokay, T.; Porath, K.; Köhling, R.; Kirschstein, T. Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 2013, 54, 183–193. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, D.S.; Park, H.; Kang, T.C. Src/CK2/PTEN-Mediated GluN2B and CREB Dephosphorylations Regulate the Responsiveness to AMPA Receptor Antagonists in Chronic Epilepsy Rats. Int. J. Mol. Sci. 2020, 21, 9633. [Google Scholar] [CrossRef]
- Conti, F.; Barbaresi, P.; Melone, M.; Ducati, A. Neuronal and glial localization of NR1 and NR2A/B subunits of the NMDA receptor in the human cerebral cortex. Cereb. Cortex 1999, 9, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef]
- Xing, W.J.; Zou, Y.; Han, Q.L.; Dong, Y.C.; Deng, Z.L.; Lv, X.H.; Jiang, T.; Ren, H. Effects of epidermal growth factor receptor and phosphatase and tensin homologue gene expression on the inhibition of U87MG glioblastoma cell proliferation induced by protein kinase inhibitors. Clin. Exp. Pharmacol. Physiol. 2013, 40, 13–21. [Google Scholar] [CrossRef]
- Markert, J.M.; Fuller, C.M.; Gillespie, G.Y.; Bubien, J.K.; McLean, L.A.; Hong, R.L.; Lee, K.; Gullans, S.R.; Mapstone, T.B.; Benos, D.J. Differential gene expression profiling in human brain tumors. Physiol. Genom. 2001, 5, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Wei, B.; Wang, L.; Wang, L.; Kong, D.; Jin, Y.; Sun, Z. Analysis of gene expression profiles associated with glioma progression. Mol. Med. Rep. 2015, 12, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wang, H.; Cai, S.; Saadatzadeh, M.R.; Hanenberg, H.; Pollok, K.E.; Cohen-Gadol, A.A.; Chen, J. Phosphorylation of NMDA 2B at S1303 in human glioma peritumoral tissue: Implications for glioma epileptogenesis. Neurosurg. Focus 2014, 37, E17. [Google Scholar] [CrossRef] [Green Version]
- Vecht, C.; Duran-Peña, A.; Houillier, C.; Durand, T.; Capelle, L.; Huberfeld, G. Seizure response to perampanel in drug-resistant epilepsy with gliomas: Early observations. J. Neurooncol. 2017, 133, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Dunn-Pirio, A.M.; Woodring, S.; Lipp, E.; Herndon, J.E.; Healy, P.; Weant, M.; Randazzo, D.; Desjardins, A.; Friedman, H.S.; Peters, K.B. Adjunctive perampanel for glioma-associated epilepsy. Epilepsy Behav. Case Rep. 2018, 10, 114–117. [Google Scholar] [CrossRef]
- Izumoto, S.; Miyauchi, M.; Tasaki, T.; Okuda, T.; Nakagawa, N.; Nakano, N.; Kato, A.; Fujita, M. Seizures and Tumor Progression in Glioma Patients with Uncontrollable Epilepsy Treated with Perampanel. Anticancer Res. 2018, 38, 4361–4366. [Google Scholar] [CrossRef]
- Maschio, M.; Pauletto, G.; Zarabla, A.; Maialetti, A.; Ius, T.; Villani, V.; Fabi, A.; Koudriavtseva, T.; Giannarelli, D. Perampanel in patients with brain tumor-related epilepsy in real-life clinical practice: A retrospective analysis. Int. J. Neurosci. 2019, 129, 593–597. [Google Scholar] [CrossRef]
- Chonan, M.; Saito, R.; Kanamori, M.; Osawa, S.I.; Watanabe, M.; Suzuki, H.; Nakasato, N.; Tominaga, T. Experience of Low Dose Perampanel to Add-on in Glioma Patients with Levetiracetam-uncontrollable Epilepsy. Neurol. Med. Chir. 2020, 60, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, A.; Zarabla, A.; Maialetti, A.; Villani, V.; Koudriavtseva, T.; Russo, E.; Nozzolillo, A.; Sueri, C.; Belcastro, V.; Balestrini, S.; et al. Perampanel Confirms to Be Effective and Well-Tolerated as an Add-On Treatment in Patients with Brain Tumor-Related Epilepsy (PERADET Study). Front. Neurol. 2020, 11, 592. [Google Scholar] [CrossRef] [PubMed]
- Kleckner, N.W.; Dingledine, R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 1988, 241, 835–837. [Google Scholar] [CrossRef]
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Wolosker, H.; Blackshaw, S.; Snyder, S.H. Serine racemase: A glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA 1999, 96, 13409–13414. [Google Scholar] [CrossRef] [Green Version]
- Neims, A.H.; Zieverink, W.D.; Smilack, J.D. Distribution of D-amino acid oxidase in bovine and human nervous tissues. J. Neurochem. 1966, 13, 163–168. [Google Scholar] [CrossRef]
- Hayashi, F.; Takahashi, K.; Nishikawa, T. Uptake of D- and L-serine in C6 glioma cells. Neurosci. Lett. 1997, 239, 85–88. [Google Scholar] [CrossRef]
- Shao, Z.; Kamboj, A.; Anderson, C.M. Functional and immunocytochemical characterization of D-serine transporters in cortical neuron and astrocyte cultures. J. Neurosci. Res. 2009, 87, 2520–2530. [Google Scholar] [CrossRef]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O.J.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyle, J.T.; Balu, D.; Wolosker, H. D-Serine, the Shape-Shifting NMDA Receptor Co-agonist. Neurochem. Res. 2020, 45, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Sikka, P.; Walker, R.; Cockayne, R.; Wood, M.J.A.; Harrison, P.J.; Burnet, P.W.J. D-Serine metabolism in C6 glioma cells: Involvement of alanine-serine-cysteine transporter (ASCT2) and serine racemase (SRR) but not D-amino acid oxidase (DAO). J. Neurosci. Res. 2010, 88, 1829–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelletti, P.; Campomenosi, P.; Pollegioni, L.; Sacchi, S. The degradation (by distinct pathways) of human D-amino acid oxidase and its interacting partner pLG72--two key proteins in D-serine catabolism in the brain. FEBS J. 2014, 281, 708–723. [Google Scholar] [CrossRef] [Green Version]
- Shoji, K.; Mariotto, S.; Ciampa, A.R.; Suzuki, H. Regulation of serine racemase activity by D-serine and nitric oxide in human glioblastoma cells. Neurosci. Lett. 2006, 392, 75–78. [Google Scholar] [CrossRef]
- Shoji, K.; Mariotto, S.; Ciampa, A.R.; Suzuki, H. Mutual regulation between serine and nitric oxide metabolism in human glioblastoma cells. Neurosci. Lett. 2006, 394, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Cuny, E.; Loiseau, H.; Penchet, G.; Ellie, E.; Arsaut, J.; Vital, A.; Vincendeau, P.; Demotes-Mainard, J. Association of elevated glial expression of interleukin-1beta with improved survival in patients with glioblastomas multiforme. J. Neurosurg. 2002, 96, 294–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Shishido, Y.; Ichise-Shishido, S.; Kawazoe, T.; Ono, K.; Iwana, S.; Tomita, Y.; Yorita, K.; Sakai, T.; Fukui, K. Potential role for astroglial D-amino acid oxidase in extracellular D-serine metabolism and cytotoxicity. J. Biochem. 2006, 139, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Noetzel, M.J.; Johnson, K.A.; Zamorano, R.; Jalan-Sakrikar, N.; Gregory, K.J.; Conn, P.J.; Niswender, C.M. Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci. 2014, 34, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Munguba, H.; Gutzeit, V.A.; Singh, D.R.; Kristt, M.; Dittman, J.S.; Levitz, J. Defining the Homo- and Heterodimerization Propensities of Metabotropic Glutamate Receptors. Cell Rep. 2020, 31, 107605. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.-I. Aberrant signaling pathways in glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [Green Version]
- Wirsching, H.G.; Silginer, M.; Ventura, E.; Macnair, W.; Burghardt, I.; Claassen, M.; Gatti, S.; Wichmann, J.; Riemer, C.; Schneider, H.; et al. Negative allosteric modulators of metabotropic glutamate receptor 3 target the stem-like phenotype of glioblastoma. Mol. Ther. Oncolytics 2020, 20, 166–174. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, X.; Li, H.; Zhao, Z.; Liao, Y.; Wang, X.; Su, J.; Sang, S.; Liu, Q. Anti-cancer effect of metabotropic glutamate receptor 1 inhibition in human glioma U87 cells: Involvement of PI3K/Akt/mTOR pathway. Cell. Physiol. Biochem. 2015, 35, 419–432. [Google Scholar] [CrossRef]
- Dalley, C.B.; Wroblewska, B.; Wolfe, B.B.; Wroblewski, J.T. The Role of Metabotropic Glutamate Receptor 1 Dependent Signaling in Glioma Viability. J. Pharmacol. Exp. Ther. 2018, 367, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhao, S.; Qi, C.; Zhao, X.; Liu, B.; Hao, F.; Zhao, Z. Inhibition of metabotropic glutamate receptor 5 facilitates hypoxia-induced glioma cell death. Brain Res. 2019, 1704, 241–248. [Google Scholar] [CrossRef]
- Ciceroni, C.; Bonelli, M.; Mastrantoni, E.; Niccolini, C.; Laurenza, M.; Larocca, L.M.; Pallini, R.; Traficante, A.; Spinsanti, P.; Ricci-Vitiani, L.; et al. Type-3 metabotropic glutamate receptors regulate chemoresistance in glioma stem cells, and their levels are inversely related to survival in patients with malignant gliomas. Cell Death Differ. 2013, 20, 396–407. [Google Scholar] [CrossRef]
- Arcella, A.; Carpinelli, G.; Battaglia, G.; D’Onofrio, M.; Santoro, F.; Ngomba, R.T.; Bruno, V.; Casolini, P.; Giangaspero, F.; Nicoletti, F. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro-Oncology 2005, 7, 236–245. [Google Scholar] [CrossRef]
- D’Onofrio, M.; Arcella, A.; Bruno, V.; Ngomba, R.T.; Battaglia, G.; Lombari, V.; Ragona, G.; Calogero, A.; Nicoletti, F. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J. Neurochem. 2003, 84, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yelskaya, Z.; Carrillo, V.; Dubisz, E.; Gulzar, H.; Morgan, D.; Mahajan, S.S. Synergistic inhibition of survival, proliferation, and migration of U87 cells with a combination of LY341495 and Iressa. PLoS ONE 2013, 8, e64588. [Google Scholar] [CrossRef] [Green Version]
- Ciceroni, C.; Arcella, A.; Mosillo, P.; Battaglia, G.; Mastrantoni, E.; Oliva, M.A.; Carpinelli, G.; Santoro, F.; Sale, P.; Ricci-Vitiani, L.; et al. Type-3 metabotropic glutamate receptors negatively modulate bone morphogenetic protein receptor signaling and support the tumourigenic potential of glioma-initiating cells. Neuropharmacology 2008, 55, 568–576. [Google Scholar] [CrossRef]
- Zhang, Z.; Zheng, X.; Luan, Y.; Liu, Y.; Li, X.; Liu, C.; Lu, H.; Chen, X.; Liu, Y. Activity of Metabotropic Glutamate Receptor 4 Suppresses Proliferation and Promotes Apoptosis with Inhibition of Gli-1 in Human Glioblastoma Cells. Front. Neurosci. 2018, 12, 320. [Google Scholar] [CrossRef] [Green Version]
- Iacovelli, L.; Arcella, A.; Battaglia, G.; Pazzaglia, S.; Aronica, E.; Spinsanti, P.; Caruso, A.; De Smaele, E.; Saran, A.; Gulino, A.; et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J. Neurosci. 2006, 26, 8388–8397. [Google Scholar] [CrossRef]
- Jantas, D.; Grygier, B.; Gołda, S.; Chwastek, J.; Zatorska, J.; Tertil, M. An endogenous and ectopic expression of metabotropic glutamate receptor 8 (mGluR8) inhibits proliferation and increases chemosensitivity of human neuroblastoma and glioma cells. Cancer Lett. 2018, 432, 1–16. [Google Scholar] [CrossRef]
- Nguyen, H.-M.; Guz-Montgomery, K.; Lowe, D.B.; Saha, D. Pathogenetic Features and Current Management of Glioblastoma. Cancers 2021, 13, 856. [Google Scholar] [CrossRef]
- Climans, S.A.; Brandes, A.A.; Cairncross, J.G.; Ding, K.; Fay, M.; Laperriere, N.; Menten, J.; Nishikawa, R.; O’Callaghan, C.J.; Perry, J.R.; et al. Temozolomide and seizure outcomes in a randomized clinical trial of elderly glioblastoma patients. J. Neurooncol. 2020, 149, 65–71. [Google Scholar] [CrossRef]
- Garcia, C.G.; Kahn, S.A.; Geraldo, L.H.M.; Romano, I.; Domith, I.; E Silva, D.C.L.; Assunção, F.D.S.; Ferreira, M.J.; Portugal, C.C.; De Souza, J.M.; et al. Combination Therapy with Sulfasalazine and Valproic Acid Promotes Human Glioblastoma Cell Death Through Imbalance of the Intracellular Oxidative Response. Mol. Neurobiol. 2018, 55, 6816–6833. [Google Scholar] [CrossRef]
- Takeuchi, S.; Wada, K.; Nagatani, K.; Otani, N.; Osada, H.; Nawashiro, H. Sulfasalazine and temozolomide with radiation therapy for newly diagnosed glioblastoma. Neurol. India 2014, 62, 42–47. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef] [Green Version]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A. New Approaches to Brain Tumor Therapy Consortium. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro-Oncology 2013, 15, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Zustovich, F.; Landi, L.; Lombardi, G.; Porta, C.; Galli, L.; Fontana, A.; Amoroso, D.; Galli, C.; Andreuccetti, M.; Falcone, A.; et al. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: A phase II study. Anticancer Res. 2013, 33, 3487–3494. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Aissa, A.B.; Espeli, V.; Squiban, D.; Dunkel, N.; Vargas, M.I.; Hundsberger, T.; Mach, N.; Schaller, K.; Weber, D.C.; et al. Phase I study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Sehm, T.; Rauh, M.; Wiendieck, K.; Buchfelder, M.; Eyüpoglu, I.Y.; Savaskan, N.E. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget 2016, 7, 74630–74647. [Google Scholar] [CrossRef]
- Dahlmanns, M.; Yakubov, E.; Chen, D.; Sehm, T.; Rauh, M.; Savaskan, N.; Wrosch, J.K. Chemotherapeutic xCT inhibitors sorafenib and erastin unraveled with the synaptic optogenetic function analysis tool. Cell Death Discov. 2017, 3, 17030. [Google Scholar] [CrossRef]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; van den Bent, M.J.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthrie, G.D.; Eljamel, S. Impact of particular antiepileptic drugs on the survival of patients with glioblastoma multiforme. J. Neurosurg. 2013, 118, 859–865. [Google Scholar] [CrossRef]
- Barker, C.A.; Bishop, A.J.; Chang, M.; Beal, K.; Chan, T.A. Valproic acid use during radiation therapy for glioblastoma associated with improved survival. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Kerkhof, M.; Dielemans, J.C.; van Breemen, M.S.; Zwinkels, H.; Walchenbach, R.; Taphoorn, M.J.; Vecht, C.J. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro-Oncology 2013, 15, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redjal, N.; Reinshagen, C.; Le, A.; Walcott, B.P.; McDonnell, E.; Dietrich, J.; Nahed, B.V. Valproic acid, compared to other antiepileptic drugs, is associated with improved overall and progression-free survival in glioblastoma but worse outcome in grade II/III gliomas treated with temozolomide. J. Neurooncol. 2016, 127, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.M.; Texakalidis, P.; McDonald, K.L.; Mekary, R.A.; Smith, T.R. The survival effect of valproic acid in glioblastoma and its current trend: A systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2018, 174, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, S.; Frijlink, E.; Kroonen, J.; Spliet, W.G.M.; van Hecke, W.; Seute, T.; Snijders, T.J.; Robe, P.A. Effects of valproic acid on histone deacetylase inhibition in vitro and in glioblastoma patient samples. Neurooncol. Adv. 2019, 1, vdz025. [Google Scholar] [CrossRef]
- Lange, F.; Weßlau, K.; Porath, K.; Hörnschemeyer, J.; Bergner, C.; Krause, B.J.; Mullins, C.S.; Linnebacher, M.; Köhling, R.; Kirschstein, T. AMPA receptor antagonist perampanel affects glioblastoma cell growth and glutamate release in vitro. PLoS ONE 2019, 14, e0211644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engh, J.A. Anti-convulsants and gene expression in malignant gliomas. Neurosurgery 2010, 67, N24–N26. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.F.; Rojas, L.; Wills, B.; Bernal, L.; Ruiz-Patiño, A.; Arrieta, O.; Hakim, E.J.; Hakim, F.; Mejía, J.A.; Useche, N.; et al. Efficacy and safety of Levetiracetam vs. other antiepileptic drugs in Hispanic patients with glioblastoma. J. Neurooncol. 2018, 136, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Min, K.L.; Chang, M.J. Effect of anti-epileptic drugs on the survival of patients with glioblastoma multiforme: A retrospective, single-center study. PLoS ONE 2019, 14, e0225599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, T.H.; Moon, J.H.; Park, H.H.; Kim, E.H.; Hong, C.K.; Kim, S.H.; Kang, S.G.; Chang, J.H. Association between survival and levetiracetam use in glioblastoma patients treated with temozolomide chemoradiotherapy. Sci. Rep. 2020, 10, 10783. [Google Scholar] [CrossRef]
- Ueda, Y.; Doi, T.; Nagatomo, K.; Tokumaru, J.; Takaki, M.; Willmore, L.J. Effect of levetiracetam on molecular regulation of hippocampal glutamate and GABA transporters in rats with chronic seizures induced by amygdalar FeCl3 injection. Brain Res. 2007, 1151, 55–61. [Google Scholar] [CrossRef]
- Pichardo-Macías, L.A.; Ramírez-Mendiola, B.A.; Contreras García, I.J.; Zamudio Hernández, S.R.; Chávez Pacheco, J.L.; Sánchez Huerta, K.B.; Mendoza Torreblanca, J.G. Effect of levetiracetam on extracellular amino acid levels in the dorsal hippocampus of rats with temporal lobe epilepsy. Epilepsy Res. 2018, 140, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Vecht, C.J.; Wilms, E.B. Seizures in low- and high-grade gliomas: Current management and future outlook. Expert Rev. Anticancer Ther. 2010, 10, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Deneka-Hannemann, S.; Jarosz, B.; Zgrajka, W.; Stoma, F.; Trojanowski, T.; Turski, W.A.; Rzeski, W. Kynurenic acid inhibits proliferation and migration of human glioblastoma T98G cells. Pharmacol. Rep. 2014, 66, 130–136. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: A multicenter phase II trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, F.M.; Kreisl, T.N.; Kim, L.; Duic, J.P.; Butman, J.A.; Albert, P.S.; Fine, H.A. Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer 2010, 116, 1776–1782. [Google Scholar] [CrossRef]
- Rösche, J.; Piek, J.; Hildebrandt, G.; Grossmann, A.; Kirschstein, T.; Benecke, R. Perampanel in the treatment of a patient with glioblastoma multiforme without IDH1 mutation and without MGMT promotor methylation. Fortschr. Neurol. Psychiatr. 2015, 83, 286–289. [Google Scholar] [CrossRef]
- Müller-Längle, A.; Lutz, H.; Hehlgans, S.; Rödel, F.; Rau, K.; Laube, B. NMDA Receptor-Mediated Signaling Pathways Enhance Radiation Resistance, Survival and Migration in Glioblastoma Cells-A Potential Target for Adjuvant Radiotherapy. Cancers 2019, 11, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenting, K.; Verhaak, R.; Laan, M.T.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [Green Version]
- Torsvik, A.; Stieber, D.; Enger, P.Ø.; Golebiewska, A.; Molven, A.; Svendsen, A.; Westermark, B.; Niclou, S.P.; Olsen, T.K.; Enger, M.C.; et al. U-251 revisited: Genetic drift and phenotypic consequences of long-term cultures of glioblastoma cells. Cancer Med. 2014, 3, 812–824. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Mayer, J.; Kirschstein, T.; Resch, T.; Porath, K.; Krause, B.J.; Köhling, R.; Lange, F. Perampanel attenuates epileptiform phenotype in C6 glioma. Neurosci. Lett. 2020, 715, 134629. [Google Scholar] [CrossRef]
- Caragher, S.; Chalmers, A.J.; Gomez-Roman, N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers 2019, 11, 44. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.; Hau, A.C.; Oudin, A.; Golebiewska, A.; Niclou, S.P. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front. Oncol. 2020, 10, 604121. [Google Scholar] [CrossRef]
- Soubéran, A.; Tchoghandjian, A. Practical Review on Preclinical Human 3D Glioblastoma Models: Advances and Challenges for Clinical Translation. Cancers 2020, 12, 2347. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Seufert, S.; Hauke, J.; Tränkle, C.; Eyüpoglu, I.Y.; Hahnen, E. Dissection of mitogenic and neurodegenerative actions of cystine and glutamate in malignant gliomas. Oncogene 2011, 30, 43–53. [Google Scholar] [CrossRef]
- Lange, F.; Hartung, J.; Liebelt, C.; Boisserée, J.; Resch, T.; Porath, K.; Hörnschemeyer, J.; Reichart, G.; Sellmann, T.; Neubert, V.; et al. Perampanel Add-on to Standard Radiochemotherapy in vivo Promotes Neuroprotection in a Rodent F98 Glioma Model. Front. Neurosci. 2020, 14, 598266. [Google Scholar] [CrossRef]
- Bouckaert, C.; Germonpré, C.; Verhoeven, J.; Chong, S.A.; Jacquin, L.; Mairet-Coello, G.; André, V.M.; Leclercq, K.; Vanhove, C.; De Vos, F.; et al. Development of a Rat Model for Glioma-Related Epilepsy. Int. J. Mol. Sci. 2020, 21, 6999. [Google Scholar] [CrossRef] [PubMed]
- Köhling, R.; Senner, V.; Paulus, W.; Speckmann, E.J. Epileptiform activity preferentially arises outside tumor invasion zone in glioma xenotransplants. Neurobiol. Dis. 2006, 22, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Senner, V.; Köhling, R.; Püttmann-Cyrus, S.; Straub, H.; Paulus, W.; Speckmann, E.J. A new neurophysiological/neuropathological ex vivo model localizes the origin of glioma-associated epileptogenesis in the invasion area. Acta Neuropathol. 2004, 107, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Kim, S.H.; Dovas, A.; Zhao, H.T.; Goldberg, A.R.; Xu, W.; Yagielski, A.J.; Cambareri, M.K.; Patel, K.B.; Mela, A.; et al. Glioma-Induced Alterations in Neuronal Activity and Neurovascular Coupling during Disease Progression. Cell Rep. 2020, 31, 107500. [Google Scholar] [CrossRef]
- Mullins, C.S.; Schneider, B.; Lehmann, A.; Stockhammer, F.; Mann, S.; Classen, C.F.; Linnebacher, M. A comprehensive approach to patient-individual glioblastoma multiforme model establishment. J. Cancer Sci. Ther. 2014, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Stringer, B.W.; Day, B.W.; D’Souza, R.C.J.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgül, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 4902. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef]
- Ye, L.F.; Reznik, E.; Korn, J.M.; Lin, F.; Yang, G.; Malesky, K.; Gao, H.; Loo, A.; Pagliarini, R.; Mikkelsen, T.; et al. Patient-derived glioblastoma cultures as a tool for small-molecule drug discovery. Oncotarget 2020, 11, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Roman, N.; Stevenson, K.; Gilmour, L.; Hamilton, G.; Chalmers, A.J. A novel 3D human glioblastoma cell culture system for modeling drug and radiation responses. Neuro-Oncology 2017, 19, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Ghoochani, A.; Yakubov, E.; Sehm, T.; Fan, Z.; Hock, S.; Buchfelder, M.; Eyüpoglu, I.Y.; Savaskan, N.E. A versatile ex vivo technique for assaying tumor angiogenesis and microglia in the brain. Oncotarget 2016, 7, 1838–1853. [Google Scholar] [CrossRef]
- Chadwick, E.J.; Yang, D.P.; Filbin, M.G.; Mazzola, E.; Sun, Y.; Behar, O.; Pazyra-Murphy, M.F.; Goumnerova, L.; Ligon, K.L.; Stiles, C.D.; et al. A Brain Tumor/Organotypic Slice Co-culture System for Studying Tumor Microenvironment and Targeted Drug Therapies. J. Vis. Exp. 2015, 7, e53304. [Google Scholar] [CrossRef] [Green Version]
- Eisemann, T.; Costa, B.; Strelau, J.; Mittelbronn, M.; Angel, P.; Peterziel, H. An advanced glioma cell invasion assay based on organotypic brain slice cultures. BMC Cancer 2018, 18, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemann, A.; Hess, S.; Bhuwania, R.; Linder, S.; Kloppenburg, P.; Noegel, A.A.; Clemen, C.S. CRN2 enhances the invasiveness of glioblastoma cells. Neuro-Oncology 2013, 15, 548–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Leguizamón, C.R.; Elnagar, M.R.; Kristiansen, U.; Kohlmeier, K.A. Increasing cellular lifespan with a flow system in organotypic culture of the Laterodorsal Tegmentum (LDT). Sci. Rep. 2019, 9, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschstein, T.; Köhling, R. Animal models of tumour-associated epilepsy. J. Neurosci. Methods 2016, 260, 109–117. [Google Scholar] [CrossRef]
- Mathieu, D.; Lecomte, R.; Tsanaclis, A.M.; Larouche, A.; Fortin, D. Standardization and detailed characterization of the syngeneic Fischer/F98 glioma model. Can. J. Neurol. Sci. 2007, 34, 296–306. [Google Scholar] [CrossRef] [Green Version]
- von Eckardstein, K.L.; Patt, S.; Kratzel, C.; Kiwit, J.C.W.; Reszka, R. Local chemotherapy of F98 rat glioblastoma with paclitaxel and carboplatin embedded in liquid crystalline cubic phases. J. Neurooncol. 2005, 72, 209–215. [Google Scholar] [CrossRef]
- Schültke, E.; Bräuer-Krisch, E.; Blattmann, H.; Requardt, H.; Laissue, J.A.; Hildebrandt, G. Survival of rats bearing advanced intracerebral F 98 tumors after glutathione depletion and microbeam radiation therapy: Conclusions from a pilot project. Radiat. Oncol. 2018, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Giakoumettis, D.; Kritis, A.; Foroglou, N. C6 cell line: The gold standard in glioma research. Hippokratia 2018, 22, 105–112. [Google Scholar]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; O’Brien, B.J.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2019, 125, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Rosati, A.; Buttolo, L.; Stefini, R.; Todeschini, A.; Cenzato, M.; Padovani, A. Efficacy and safety of levetiracetam in patients with glioma: A clinical prospective study. Arch. Neurol. 2010, 67, 343–346. [Google Scholar] [CrossRef] [Green Version]
- Laghari, A.A.; Ahmed, S.I.; Qadeer, N.; Shamim, M.S. Choice of therapeutic anti-seizure medication in patients with brain tumour. J. Pak. Med. Assoc. 2019, 69, 442–444. [Google Scholar] [PubMed]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Brivaracetam prevents astroglial l-glutamate release associated with hemichannel through modulation of synaptic vesicle protein. Biomed. Pharmacother. 2021, 138, 111462. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Glutamatergic interaction in health and glioma. Details of the molecular processes are described in Section 2. AMPA receptor (AMPAR), alanine-serine-cysteine amino acid transporter-2 (ASCT2), branched-chain amino acid transaminase 1 (BCAT1), D-amino acid oxidase (DAAO), glutamate transporter 1 (GLT-1), isocitrate dehydrogenase 1 (IDH1), kainate receptor (KAR), levetiracetam (LEV), mGluR (metabotropic glutamate receptor), NMDA receptor (NMDAR), perampanel (PER), sulfasalazine (SAS), serine racemase (SR), synaptic vesicle glycoprotein 2A (SV2A), topiramate (TPM), cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11 or xCT).
Figure 1.
Glutamatergic interaction in health and glioma. Details of the molecular processes are described in Section 2. AMPA receptor (AMPAR), alanine-serine-cysteine amino acid transporter-2 (ASCT2), branched-chain amino acid transaminase 1 (BCAT1), D-amino acid oxidase (DAAO), glutamate transporter 1 (GLT-1), isocitrate dehydrogenase 1 (IDH1), kainate receptor (KAR), levetiracetam (LEV), mGluR (metabotropic glutamate receptor), NMDA receptor (NMDAR), perampanel (PER), sulfasalazine (SAS), serine racemase (SR), synaptic vesicle glycoprotein 2A (SV2A), topiramate (TPM), cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11 or xCT).

Figure 2.
Synopsis of ionotropic (upper row) and metabotropic (lower row) glutamate receptors. Subunits detected in glioblastomas are highlighted in red.
Figure 2.
Synopsis of ionotropic (upper row) and metabotropic (lower row) glutamate receptors. Subunits detected in glioblastomas are highlighted in red.

Figure 3.
Experimental models to study glutamatergic mechanisms in glioblastoma and epilepsy. (A) Coronal slices (350 µm) of 6–8 days-old Fischer rats were prepared to establish organotypic brain slices. The slices were cultured in 6-wells onto Millicell, cell culture inserts (0.4 µm pore size) in slice culture medium (composed of 49% MEM with GlutaMAX, 1% penicillin/streptomycin, 12.5% BME, 12% FCS, and 0.5% glucose). C6 glioma cells (5 × 103 cells) with stable expression of GFP (humanized Renilla reniformis green fluorescence protein) were placed onto the slices in cortical areas. For electrophysiological recordings, slices were exposed to artificial cerebrospinal fluid (aCSF) solution [164] without Mg2+, 8 mM KCl and 5 µM gabazine and network deflections were quantified in the last 20 min after two hours of perfusion. Data represent mean of 3–4 separate experiments ± SEM. At the same day, C6 glioma cell growth was estimated by laserscanning microscopy. (B) Orthotopically-growing F98 glioma in male Fischer rats exhibits an epileptiform phenotype. Therefore, 1 × 105 F98 glioma cells were injected in the frontal cortex of Fischer rats (for experimental details see [169]). Additionally, two electrodes were placed epidurally above the cortex, and continuous 24/7 video-EEG data (sample rate 500 Hz, low-pass filter 30 Hz) were recorded. The F98 model exhibit seizures (green traces) and interictal events (black traces). Representative parts of the EEG trace are enlarged on the right-hand side (boxes with 10-fold time resolution but identical amplitude scaling). Consistent with the orthotopic inoculation of F98 cells, both focal (upper row) and secondarily generalized seizures (lower row) can be detected.
Figure 3.
Experimental models to study glutamatergic mechanisms in glioblastoma and epilepsy. (A) Coronal slices (350 µm) of 6–8 days-old Fischer rats were prepared to establish organotypic brain slices. The slices were cultured in 6-wells onto Millicell, cell culture inserts (0.4 µm pore size) in slice culture medium (composed of 49% MEM with GlutaMAX, 1% penicillin/streptomycin, 12.5% BME, 12% FCS, and 0.5% glucose). C6 glioma cells (5 × 103 cells) with stable expression of GFP (humanized Renilla reniformis green fluorescence protein) were placed onto the slices in cortical areas. For electrophysiological recordings, slices were exposed to artificial cerebrospinal fluid (aCSF) solution [164] without Mg2+, 8 mM KCl and 5 µM gabazine and network deflections were quantified in the last 20 min after two hours of perfusion. Data represent mean of 3–4 separate experiments ± SEM. At the same day, C6 glioma cell growth was estimated by laserscanning microscopy. (B) Orthotopically-growing F98 glioma in male Fischer rats exhibits an epileptiform phenotype. Therefore, 1 × 105 F98 glioma cells were injected in the frontal cortex of Fischer rats (for experimental details see [169]). Additionally, two electrodes were placed epidurally above the cortex, and continuous 24/7 video-EEG data (sample rate 500 Hz, low-pass filter 30 Hz) were recorded. The F98 model exhibit seizures (green traces) and interictal events (black traces). Representative parts of the EEG trace are enlarged on the right-hand side (boxes with 10-fold time resolution but identical amplitude scaling). Consistent with the orthotopic inoculation of F98 cells, both focal (upper row) and secondarily generalized seizures (lower row) can be detected.


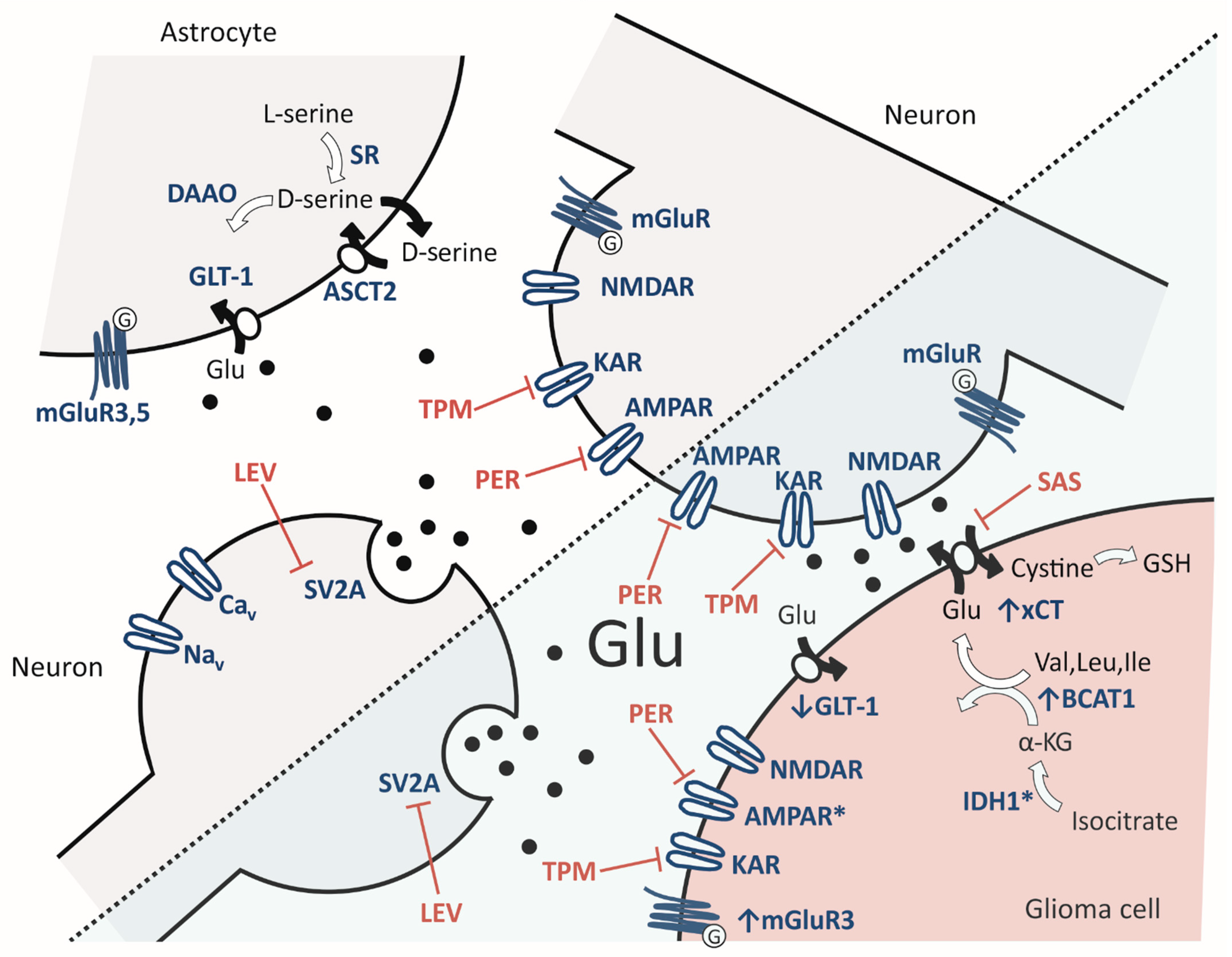{kind=link}
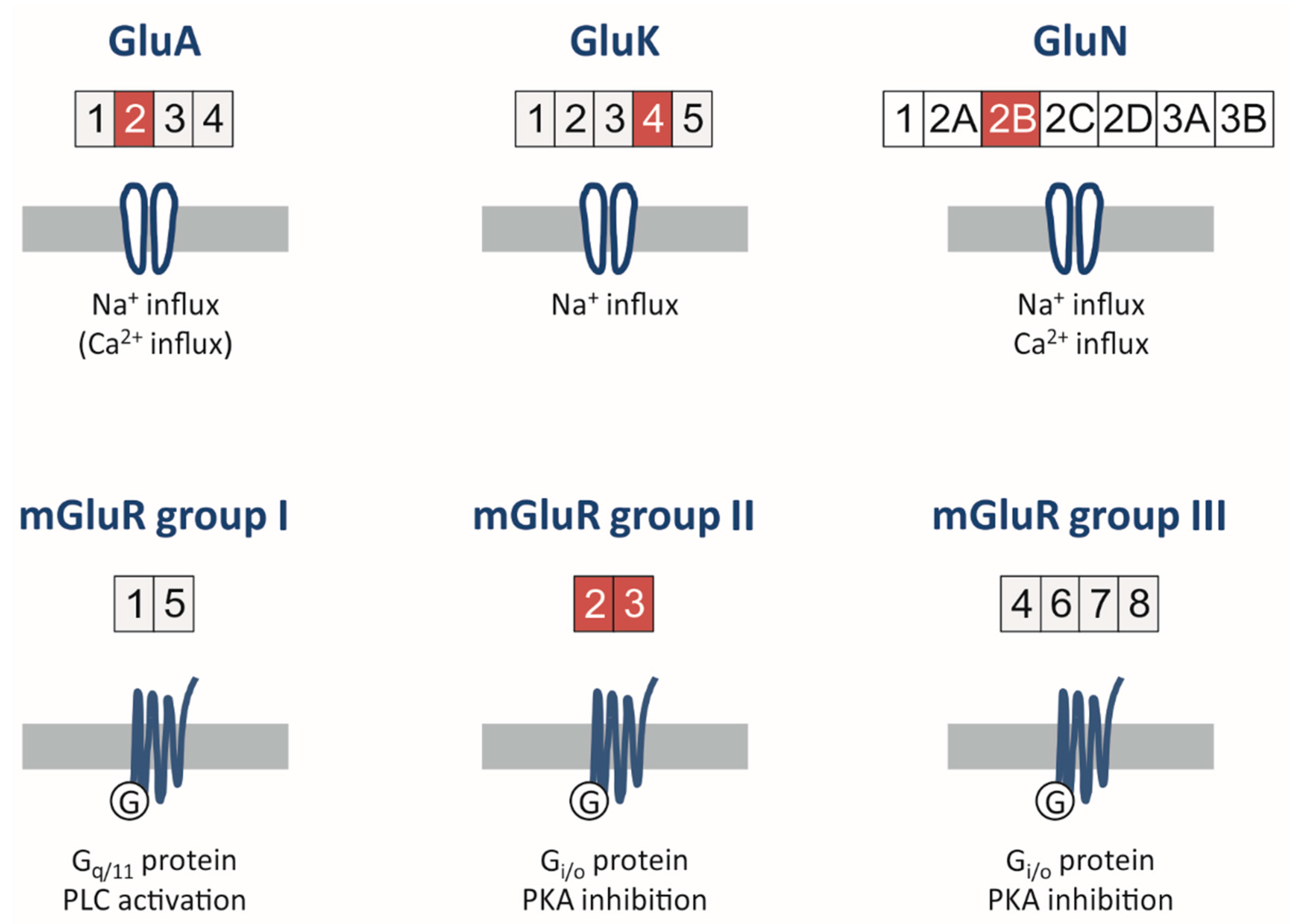{kind=link}
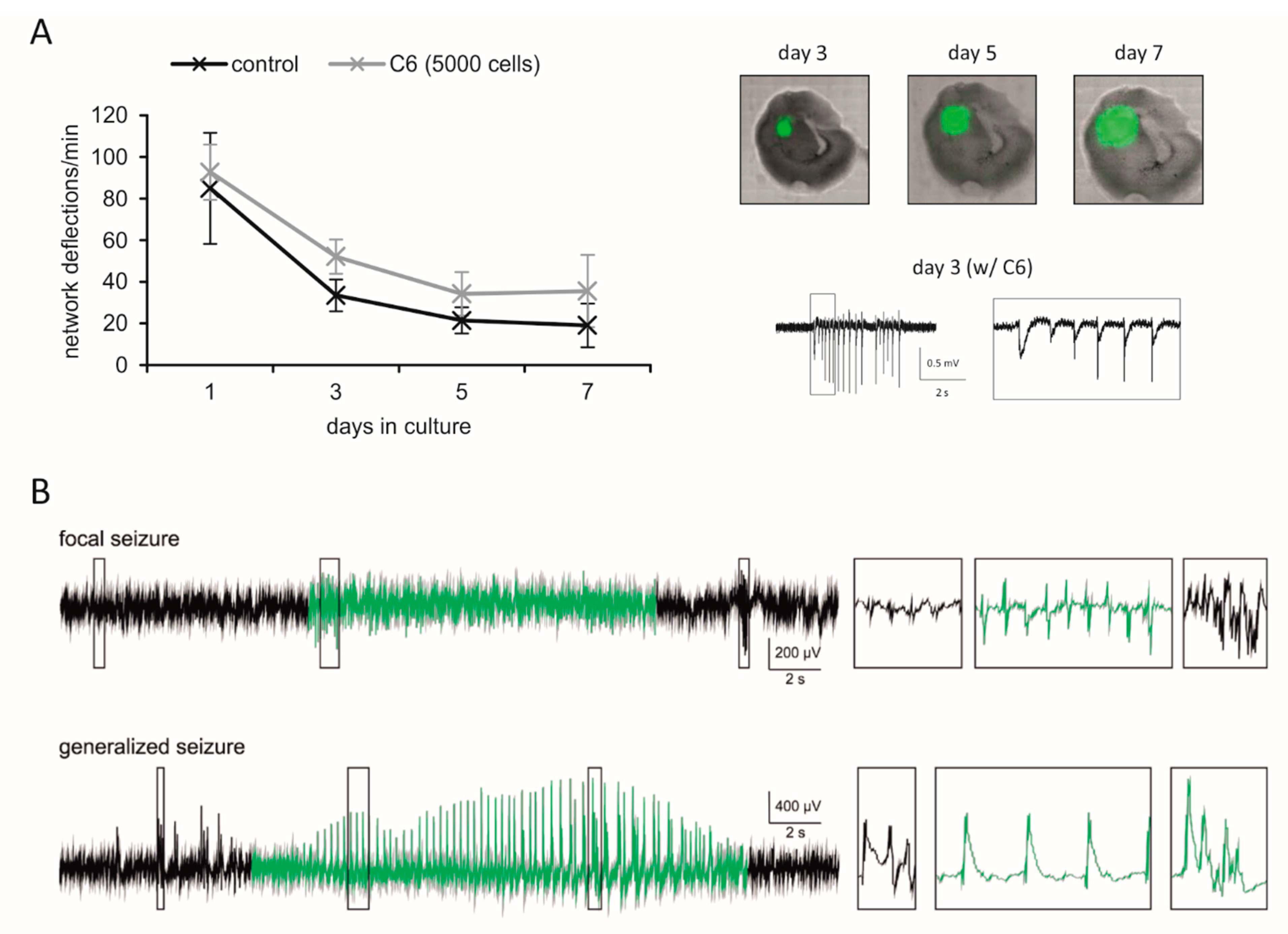{kind=link}
Table 1.
Studies with perampanel add-on therapy for glioma-associated seizures.
| Reference | Patients Enrolled | Perampanel Therapy | Seizure Reponse |
|---|---|---|---|
| Vecht et al., 2017 [93] | 12 patients | 2–12 mg/d | seizure-free = 6/12 |
| 9 male, 3 female | follow-up = 6 months | ≥50% reduction = 3/12 | |
| median = 41 years | responder rate = 75% | ||
| Dunn-Pirio et al., 2018 [94] | 8 patients | 2–8 mg/d | seizure-free = 5/8 |
| 6 male, 2 female | follow-up = 16 weeks | ≥50% reduction = 1/8 | |
| median = 45 years | responder rate = 75% | ||
| Izumoto et al., 2018 [95] | 10 patients | 4–8 mg/d | seizure-free = 6/10 |
| 6 male, 4 female | follow-up = 6 months | ≥50% reduction = 4/10 | |
| median = 59 years | responder rate = 100% | ||
| Maschio et al., 2019 [96] | 11 patients | 7.3 mg/d | seizure-free = 5/12 |
| 9 male, 2 female | follow-up = 12 months | ≥50% reduction = 4/12 | |
| median = 54 years | responder rate = 82% | ||
| Chonan et al., 2020 [97] | 18 patients | 2–4 mg/d | seizure-free = 17/18 |
| 9 male, 9 female | follow-up = 10.6 months | ≥50% reduction = 0/18 | |
| median = 49 years | responder rate = 94% | ||
| Coppola et al., 2020 1 [98] | 36 patients | 2–12 mg/d | seizure-free = 7/21 |
| 23 male, 13 female | follow-up = 12 months | ≥50% reduction = 12/21 | |
| median = 46 years | responder rate = 90% |
1 Patient data from intention-to-treat analysis, seizure response data from per-protocol analysis.
Table 2.
Studies with add-on drugs for glioma-associated seizures.
| Title (Trial) | Status | Interventions | Location |
|---|---|---|---|
| Perampanel for the reduction of seizure frequency in patients with high-grade glioma and focal epilepsy (NCT04650204) | Not yet recruiting | Perampanel | Jacksonville, FL, USA |
| Effect of perampanel on peritumoral hyperexcitability in HGG (NCT04497142) | Recruiting | Perampanel | Boston, MA, USA |
| Sulfasalazine and stereotactic radiosurgery for recurrent glioblastoma (NCT04205357) | Recruiting | Sulfasalazine | Bergen, Norway |
| Efficacy and Safety of perampanel in combination in glioma-refractory epilepsy (NCT03636958) | Recruiting | Perampanel | Marseille, France |
| Memantine for prevention of cognitive late effects in pediatric patients receiving cranial radiation therapy for localized brain tumors (NCT03194906) | Recruiting | Memantine | Memphis, TN, USA |
| Temozolomide, memantine hydrochloride, mefloquine, and metformin hydrochloride in treating patients with glioblastoma multiforme after radiation therapy (NCT01430351) | Active, not recruiting | Memantine, mefloquine, metformin | Houston, TX, USA |
Table 3.
Relevant Preclinical Models to Study Glutamatergic Mechanisms in Glioma and Tumor-Associated Epilepsy.
Table 3.
Relevant Preclinical Models to Study Glutamatergic Mechanisms in Glioma and Tumor-Associated Epilepsy.
| Level | Model | Glioma | Advantages/Disadvantages |
|---|---|---|---|
| in vitro | permanent cell lines | rodent and human cells [45,128,164] | (+) high throughput |
| (−) genetic drift | |||
| (−) no microenvironment | |||
| patient-derived cell lines | human (primary) glioblastoma cells [117,148] | (+) high throughput (+) genetic status of primary tumor and clinical data accessible (−) no microenvironment | |
| spheroids/organoids | human and rodent glioblastoma [165,166,167] | (+) median throughput (−) no microenvironment | |
| ex vivo | organotypic slice cultures with glio- blastoma cells | human or rodent glioblastoma [168] | (+) median throughput (+) genetic manipulation feasible (+) interaction with healthy brain tissue |
| (−) only short-time monitoring (1-3 weeks) | |||
| (−) microenvironment lacking immune system | |||
| (−) animal consuming research | |||
| in vivo 1 | orthotopic rat glioma | F98 and C6 rat [169,170,171,172] | (+) glioma-associated seizures (+) immunocompetent (−) ethical issues related to animal studies (−) low throughput (−) no genetic variances |
| orthotopic mice glioma | murine glioma [58,173] | (+) glioma-associated seizures (+) immunocompetent (+) genetic alterations based on human glioma (−) ethical issues related to animal studies (−) low throughput (−) low genetic variances | |
| orthotopic human glioblastoma | GBM12/GBM22 [34] | (+) glioma-associated seizures (−) ethical issues related to animal studies (−)low throughput (−) immunodeficient host (−) no genetic variances |
1 only models with documented epileptic seizures are included.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lange, F.; Hörnschemeyer, J.; Kirschstein, T. Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy. Cells 2021, 10, 1226. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051226
AMA Style
Lange F, Hörnschemeyer J, Kirschstein T. Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy. Cells. 2021; 10(5):1226. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051226
Chicago/Turabian StyleLange, Falko, Julia Hörnschemeyer, and Timo Kirschstein. 2021. "Glutamatergic Mechanisms in Glioblastoma and Tumor-Associated Epilepsy" Cells 10, no. 5: 1226. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051226
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.