
Extracellularly Released Calreticulin Induced by Endoplasmic Reticulum Stress Impairs Syncytialization of Cytotrophoblast Model BeWo Cells

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Human Tissue Collection and Sample Preparation
2.3. Immunohistochemistry
2.4. Cell Culture
2.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.6. Immunodepletion
2.7. Immunoblot Analysis
2.8. Cytotoxicity of Thapsigargin Treatment
2.9. Immunocytochemistry and Determination of Fusion Index
2.10. Statistical Analysis
3. Results
3.1. Upregulation of Serum CRT Levels in Patients with Preeclampsia
3.2. Thapsigargin-Induced ER Stress in BeWo Cells
3.3. Extracellular Release of CRT after Thapsigargin-Induced ER Stress in BeWo Cells
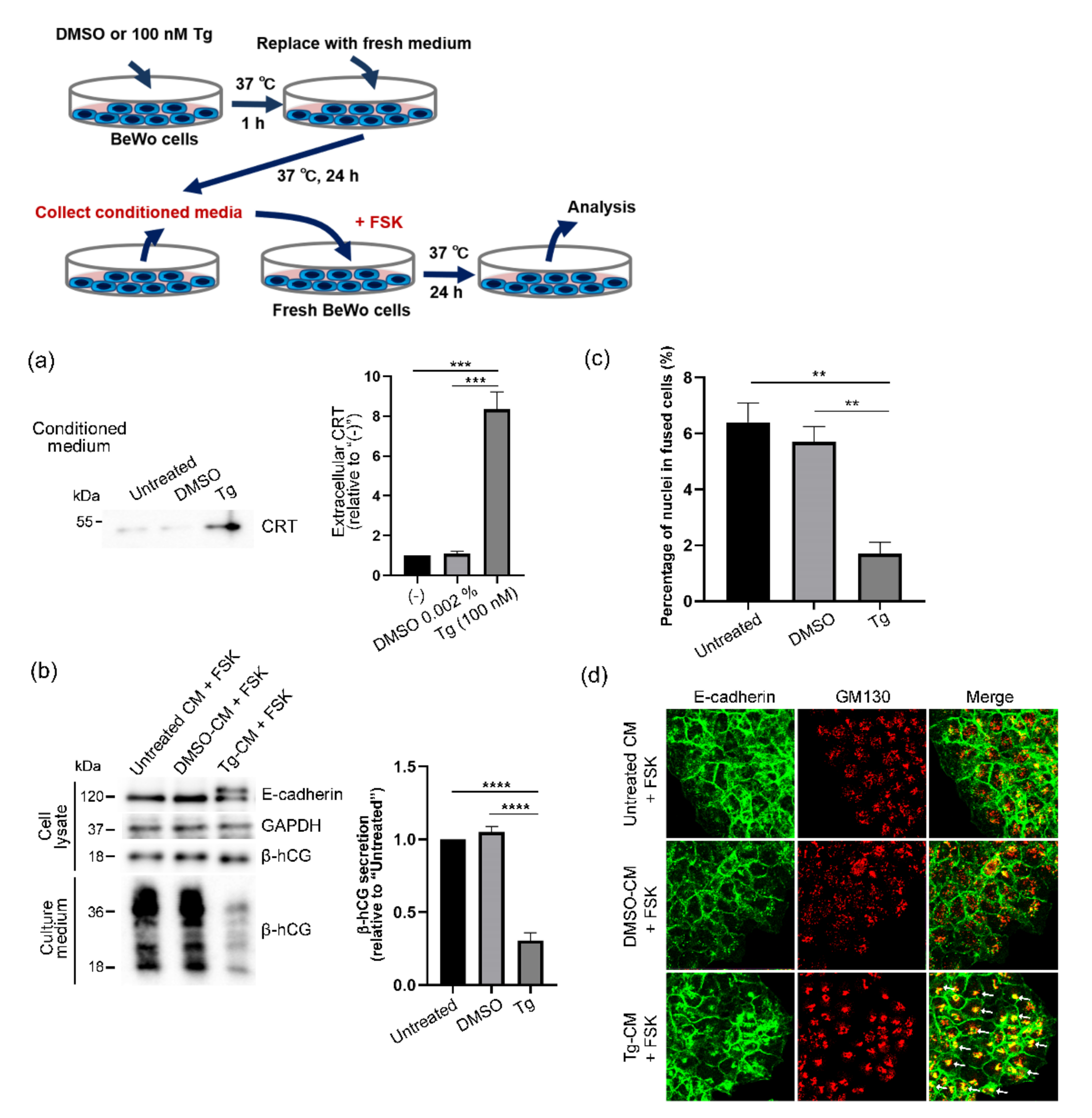
3.4. Forskolin-Induced Syncytialization of BeWo Cells Inhibited by Extracellular CRT
3.5. Elimination of Harmful Effects of Tg-CM on BeWo Cells by Means of CRT Immunodepletion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bischof, P.; Irminger-Finger, I. The human cytotrophoblastic cell, a mononuclear chameleon. Int. J. Biochem. Cell Biol. 2005, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Moser, G.; Orendi, K.; Gauster, M.; Siwetz, M.; Helige, C.; Huppertz, B. The art of identification of extravillous trophoblast. Placenta 2011, 32, 197–199. [Google Scholar] [CrossRef]
- Eaton, B.; Contractor, S. In Vitro Assessment of Trophoblast Receptors and Placental Transport Mechanisms; Blackwell Scientific Publication: London, UK, 1993; pp. 471–503. [Google Scholar]
- Ogren, L.; Talamentes, F. The Placenta as an Endocrine Organ: Polypeptides; Raven Press: New York, NY, USA, 1994; pp. 875–945. [Google Scholar]
- Gauster, M.; Moser, G.; Orendi, K.; Huppertz, B. Factors involved in regulating trophoblast fusion: Potential role in the development of preeclampsia. Placenta 2009, 30, S49–S54. [Google Scholar] [CrossRef]
- Acien, P.; Lloret, G.; Lloret, M. Perinatal morbidity and mortality in pregnancy hypertensive disorders: Prognostic value of the clinical and laboratory findings. Int. J. Gynaecol. Obs. 1990, 32, 229–235. [Google Scholar] [CrossRef]
- Brown, M.A.; Hague, W.M.; Higgins, J.; Lowe, S.; McCowan, L.; Oats, J.; Rowan, J.A.; Walters, B.N.J. The detection, investigation and management of hypertension in pregnancy: Executive summary. Aust. N. Z. J. Obs. Gynaecol. 2000, 40, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.A.; Løset, M.; Mundal, S.B.; Fenstad, M.H.; Johnson, M.P.; Eide, I.P.; Austgulen, R.; Bjørge, L.; Freed, K.A.; Moses, E.K. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta 2011, 32, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuuchi, M.; Cindrova-Davies, T.; Olovsson, M.; Charnock-Jones, D.S.; Burton, G.J.; Yung, H.W. Placental endoplasmic reticulum stress negatively regulates transcription of placental growth factor via ATF4 and ATF6beta: Implications for the pathophysiology of human pregnancy complications. J. Pathol. 2016, 238, 550–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.Y.; Wang, H.; Chen, Y.H.; Xia, M.Z.; Zhang, C.; Xu, D.X. N-acetylcysteine alleviates cadmium-induced placental endoplasmic reticulum stress and fetal growth restriction in mice. PLoS ONE 2018, 13, e0191667. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Zhao, L.; Wang, L.; Zhu, X. Expression of markers of endoplasmic reticulum stress-induced apoptosis in the placenta of women with early and late onset severe pre-eclampsia. Taiwan J. Obs. Gynecol. 2015, 54, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Hou, W.; Hua, X.; Zhang, X.; Liu, X.; Wang, X.; Wang, X. Overexpression of calreticulin in pre-eclamptic placentas: Effect on apoptosis, cell invasion and severity of pre-eclampsia. Cell Biochem. Biophys. 2012, 63, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Gu, V.Y.; Wong, M.H.; Stevenson, J.L.; Crawford, K.E.; Brennecke, S.P.; Gude, N.M. Calreticulin in human pregnancy and pre-eclampsia. Mol. Hum. Reprod. 2008, 14, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Yamamoto, M.; Ikezaki, M.; Toujima, S.; Iwahashi, N.; Mizoguchi, M.; Nanjo, S.; Minami, S.; Ihara, Y.; Ino, K. Calreticulin is involved in invasion of human extravillous trophoblasts through functional regulation of integrin beta1. Endocrinology 2017, 158, 3874–3889. [Google Scholar] [CrossRef]
- Iwahashi, N.; Ikezaki, M.; Matsuzaki, I.; Yamamoto, M.; Toujima, S.; Murata, S.I.; Ihara, Y.; Ino, K. Calreticulin regulates syncytialization through control of the synthesis and transportation of e-cadherin in bewo cells. Endocrinology 2019, 160, 359–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelfi, F. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Nanney, L.B.; Woodrell, C.D.; Greives, M.R.; Cardwell, N.L.; Pollins, A.C.; Bancroft, T.A.; Chesser, A.; Michalak, M.; Rahman, M.; Gold, L.I.; et al. Calreticulin enhances porcine wound repair by diverse biological effects. Am. J. Pathol. 2008, 173, 610–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppolino, M.G.; Dedhar, S. Ligand-specific, transient interaction between integrins and calreticulin during cell adhesion to extracellular matrix proteins is dependent upon phosphorylation/dephosphorylation events. Biochem. J. 1999, 340, 41–50. [Google Scholar] [CrossRef]
- Coppolino, M.G.; Woodside, M.J.; Demaurex, N.; Grinstein, S.; St-Arnaud, R.; Dedhar, S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 1997, 386, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Pedraza, C.E.; Pallero, M.A.; Elzie, C.A.; Goicoechea, S.; Strickland, D.K.; Murphy-Ullrich, J.E. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J. Cell Biol. 2003, 161, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.W.; Pallero, M.A.; Murphy-Ullrich, J.E. Thrombospondin stimulates focal adhesion disassembly through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J. Biol. Chem. 2002, 277, 20453–20460. [Google Scholar] [CrossRef] [Green Version]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, K.E.; Kalionis, B.; Stevenson, J.L.; Brennecke, S.P.; Gude, N.M. Calreticulin has opposing effects on the migration of human trophoblast and myometrial endothelial cells. Placenta 2012, 33, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Coutifaris, C.; Kao, L.C.; Sehdev, H.M.; Chin, U.; Babalola, G.O.; Blaschuk, O.W.; Strauss, J.F. E-cadherin expression during the differentiation of human trophoblasts. Development 1991, 113, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Kohlberger, P.D.; Breitenecker, F.; Kaider, A.; Losch, A.; Gitsch, G.; Breitenecker, G.; Kieback, D.G. Modified true-color computer-assisted image analysis versus subjective scoring of estrogen receptor expression in breast cancer: A comparison. Anticancer Res. 1999, 19, 2189–2193. [Google Scholar] [PubMed]
- Drewlo, S.; Baczyk, D.; Dunk, C.; Kingdom, J. Fusion assays and models for the trophoblast. Methods Mol. Biol. 2008, 475, 363–382. [Google Scholar] [CrossRef]
- Zamudio, S.; Plamer, S.K.; Regensteiner, J.G.; Moore, L.G. High altitude and hypertension during pregnancy. Am. J. Hum. Biol. 1995, 7, 183–193. [Google Scholar] [CrossRef]
- Soothill, P.W.; Nicolaides, K.H.; Rodeck, C.H. Effect of anaemia on fetal acid-base status. Br. J. Obs. Gynaecol. 1987, 94, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Yao, J.; Kitamura, M. Real-time detection and continuous monitoring of ER stress in vitro and in vivo by ES-TRAP: Evidence for systemic, transient ER stress during endotoxemia. Nucleic Acids Res. 2006, 34, e93. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef]
- Geng, F.; Zhu, W.; Anderson, R.A.; Leber, B.; Andrews, D.W. Multiple post-translational modifications regulate E-cadherin transport during apoptosis. J. Cell Sci. 2012, 125, 2615–2625. [Google Scholar] [CrossRef] [Green Version]
- Pidoux, G.; Gerbaud, P.; Tsatsaris, V.; Marpeau, O.; Ferreira, F.; Meduri, G.; Guibourdenche, J.; Badet, J.; Evain-Brion, D.; Frendo, J.L. Biochemical characterization and modulation of LH/CG-receptor during human trophoblast differentiation. J. Cell. Physiol. 2007, 212, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.J.; Lei, Z.M.; Rao, C.V.; Lin, J. Novel role of human chorionic gonadotropin in differentiation of human cytotrophoblasts. Endocrinology 1993, 132, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Calreticulin signaling in health and disease. Int. J. Biochem. Cell Biol. 2012, 44, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2021, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Gehring, K. Calnexin cycle—Structural features of the ER chaperone system. FEBS J. 2020, 287, 4322–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojrup, P.; Roepstorff, P.; Houen, G. Human placental calreticulin characterization of domain structure and post-translational modifications. Eur. J. Biochem. 2001, 268, 2558–2565. [Google Scholar] [CrossRef]
- Crawford, K.E.; Stevenson, J.L.; Wlodek, M.E.; Gude, N.M. No change in calreticulin with fetal growth restriction in human and rat pregnancies. Placenta 2013, 34, 1066–1071. [Google Scholar] [CrossRef]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.M.; Srivastava, P.K. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 2000, 12, 1539–1546. [Google Scholar] [CrossRef]
- Tong, M.; Kleffmann, T.; Pradhan, S.; Johansson, C.L.; DeSousa, J.; Stone, P.R.; James, J.L.; Chen, Q.; Chamley, L.W. Proteomic characterization of macro-, micro- and nano-extracellular vesicles derived from the same first trimester placenta: Relevance for feto-maternal communication. Hum. Reprod. 2016, 31, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Thery, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, G.J.; Yung, H.W.; Cindrova-Davies, T.; Charnock-Jones, D.S. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009, 30 (Suppl. SA), S43–S48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, H.W.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008, 173, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Pregnancy | Preeclampsia Alone | p Value | |
|---|---|---|---|
| Characteristic | (n = 22) | (n = 18) | |
| Maternal age (years) | 30.4 ± 5.5 | 32.8 ± 4.6 | ns |
| Prepregnancy body mass index (kg/m2) | 20.7 ± 2.6 | 25.5 ± 5.9 | <0.001 |
| Gestational age at delivery (weeks) | 34.7 ± 4.2 | 34.2 ± 2.6 | ns |
| Neonatal weight (g) | 2447 ± 712 | 2232 ± 614 | ns |
| Placental weight (g) | 528 ± 133 | 512 ± 129 | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwahashi, N.; Ikezaki, M.; Nishitsuji, K.; Yamamoto, M.; Matsuzaki, I.; Kato, N.; Takaoka, N.; Taniguchi, M.; Murata, S.-i.; Ino, K.; et al. Extracellularly Released Calreticulin Induced by Endoplasmic Reticulum Stress Impairs Syncytialization of Cytotrophoblast Model BeWo Cells. Cells 2021, 10, 1305. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061305
Iwahashi N, Ikezaki M, Nishitsuji K, Yamamoto M, Matsuzaki I, Kato N, Takaoka N, Taniguchi M, Murata S-i, Ino K, et al. Extracellularly Released Calreticulin Induced by Endoplasmic Reticulum Stress Impairs Syncytialization of Cytotrophoblast Model BeWo Cells. Cells. 2021; 10(6):1305. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061305
Chicago/Turabian StyleIwahashi, Naoyuki, Midori Ikezaki, Kazuchika Nishitsuji, Madoka Yamamoto, Ibu Matsuzaki, Naoki Kato, Naoyuki Takaoka, Mana Taniguchi, Shin-ichi Murata, Kazuhiko Ino, and et al. 2021. "Extracellularly Released Calreticulin Induced by Endoplasmic Reticulum Stress Impairs Syncytialization of Cytotrophoblast Model BeWo Cells" Cells 10, no. 6: 1305. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061305