
Loss of Motor Protein MYO1C Causes Rhodopsin Mislocalization and Results in Impaired Visual Function

 ,
,  , ,
, ,  , ,
, , 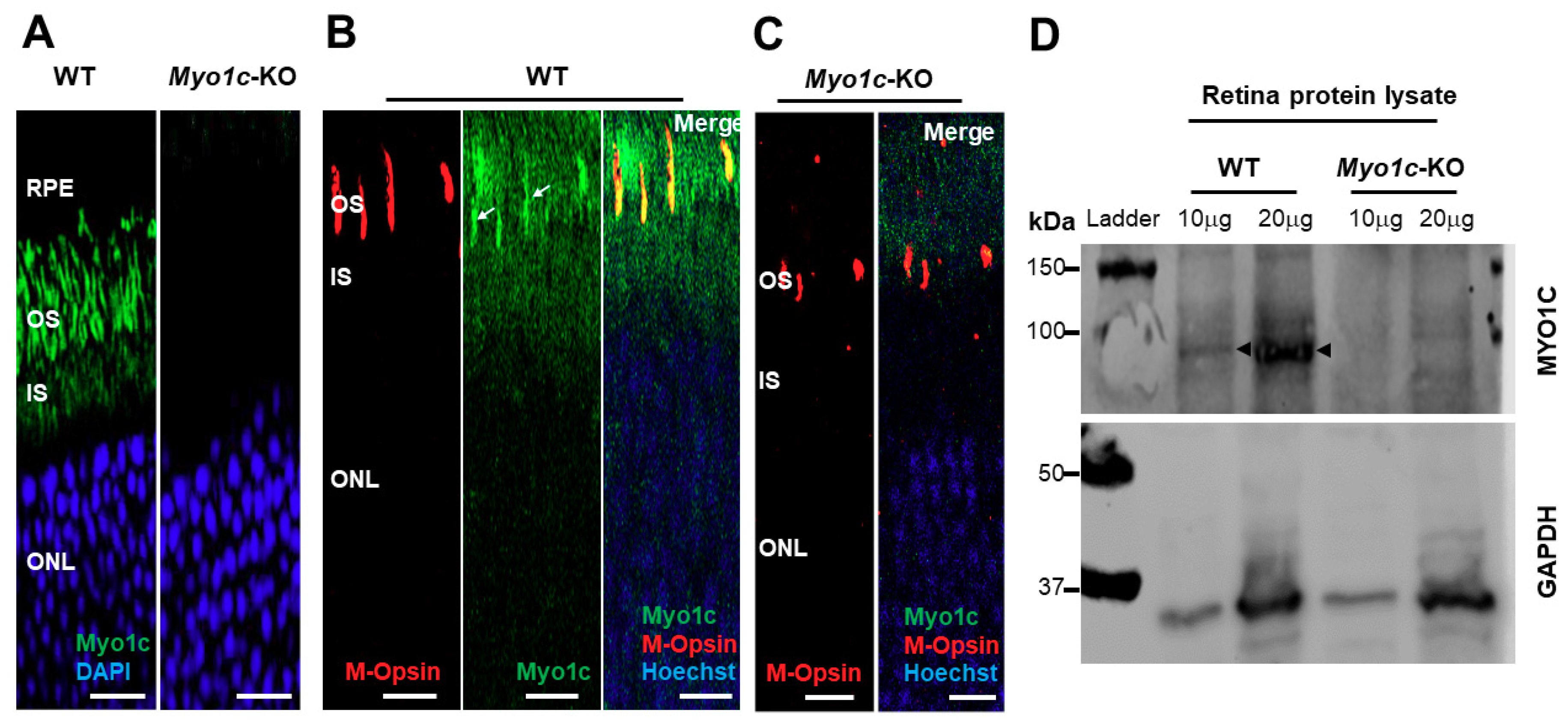{kind=link}
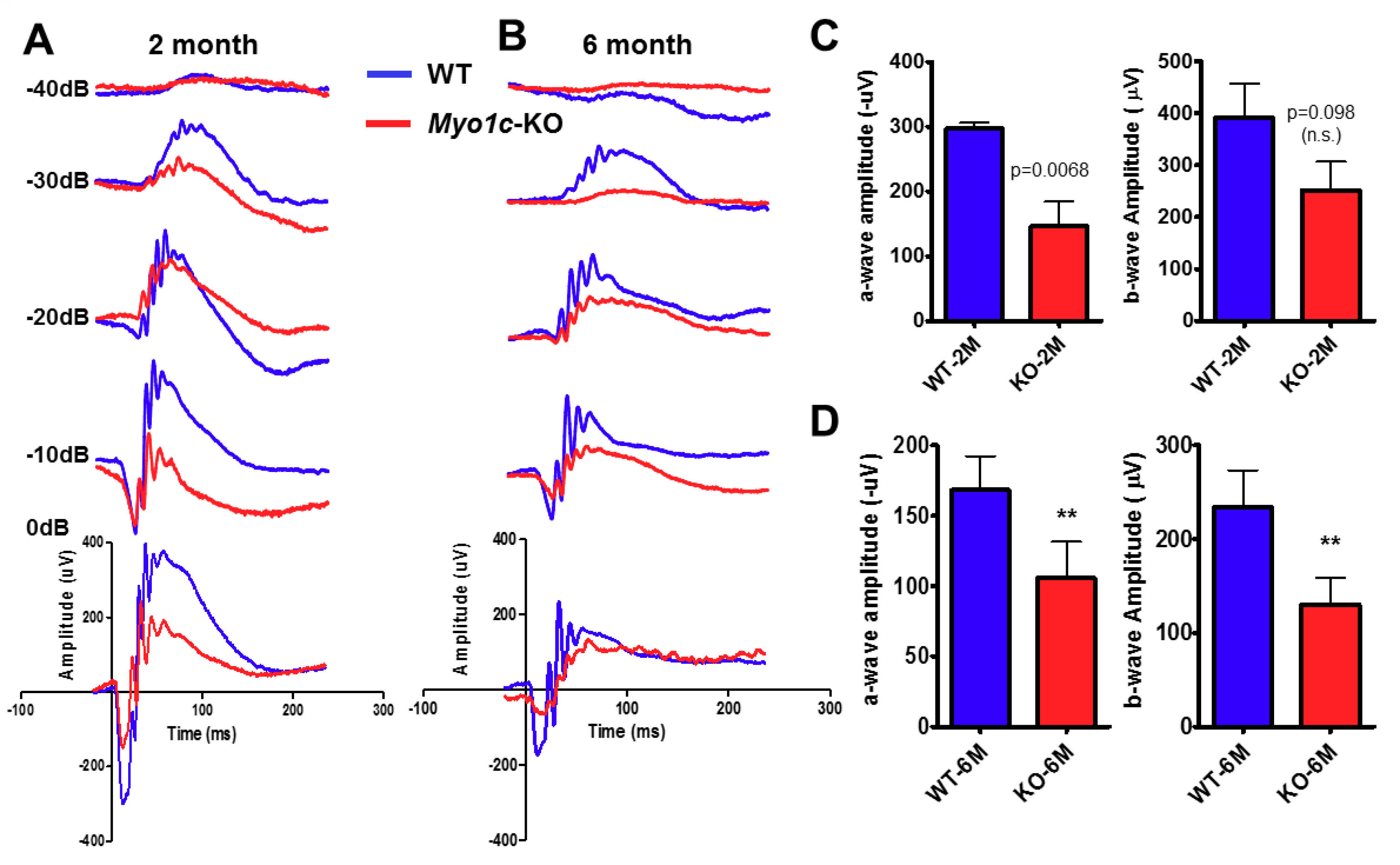{kind=link}
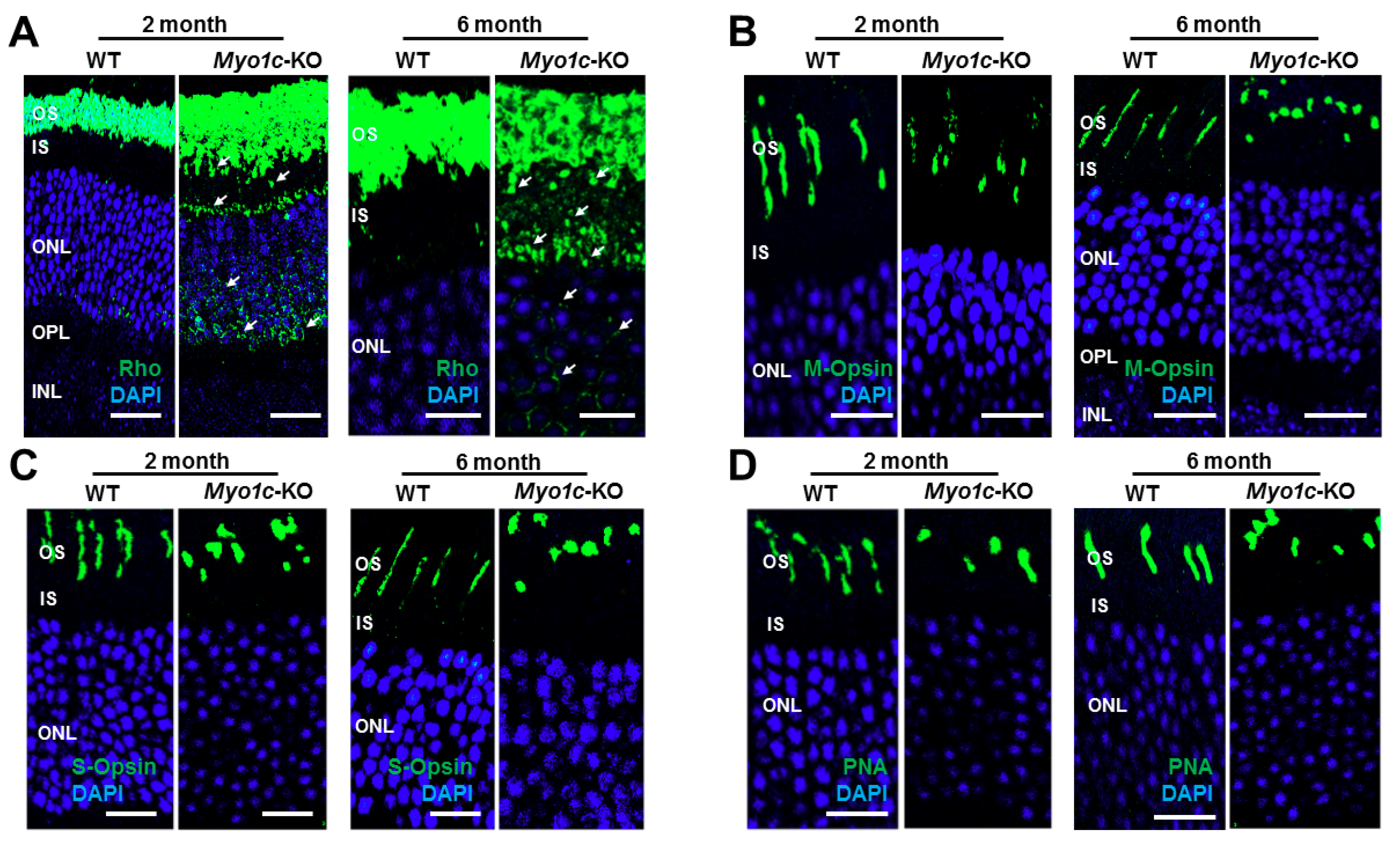{kind=link}
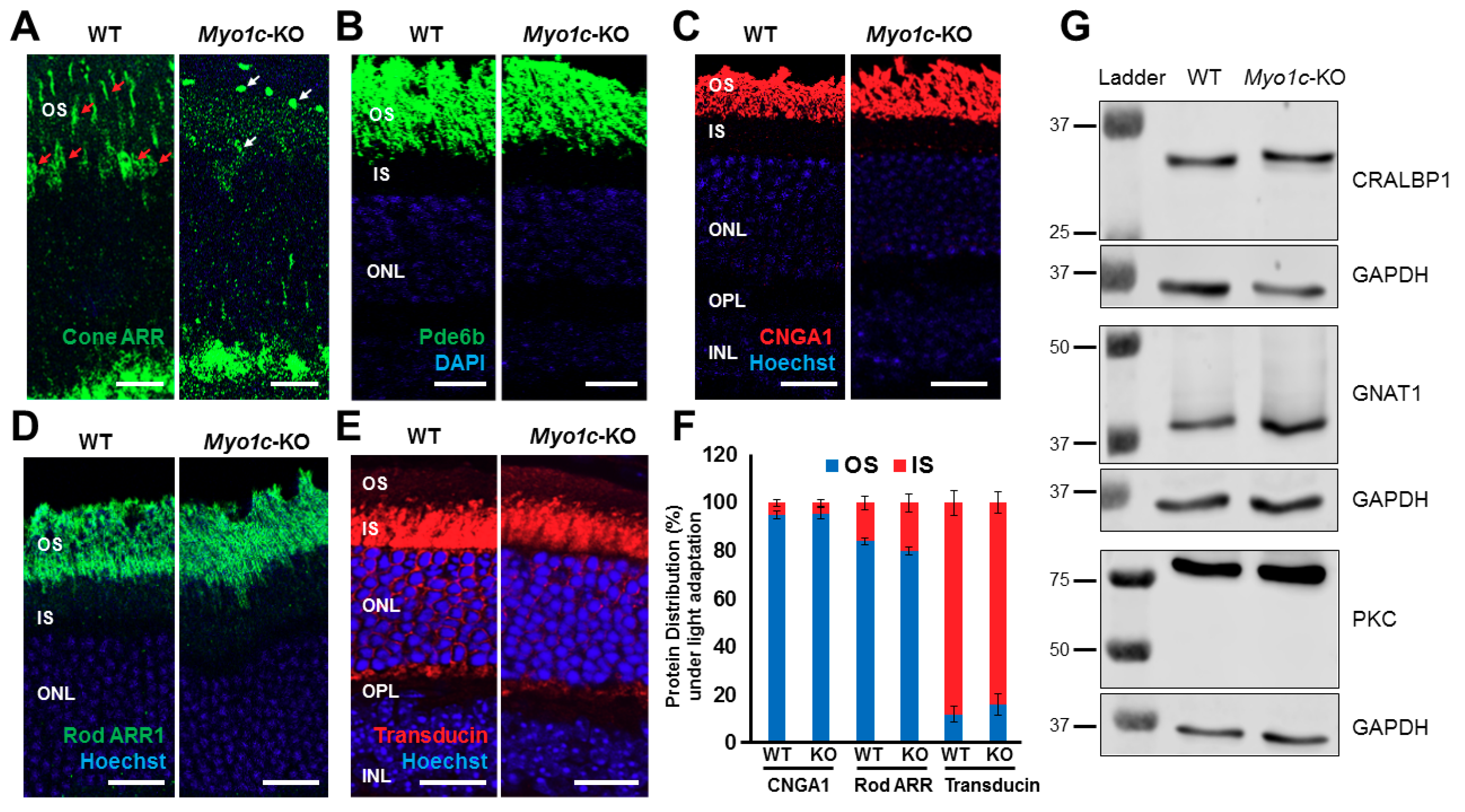{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedures
2.1. Materials
2.2. Myo1c-Knockout (Myo1c-KO) Mouse Model
2.3. Immunohistochemistry and Fluorescence Imaging
2.4. Measurement of Photoreceptor ONL Thickness and OS Lengths
2.5. ERG Analysis
2.6. TEM Analysis of Retinas
2.7. Western Blot Analysis and Densitometry
2.8. Co-Immunoprecipitation (Co-IP) Assays
2.9. Overlay Direct Binding Assay
2.10. ELISA
2.11. Quantitative Real-Time PCR
2.12. Liver Function Tests Using Alanine Aminotransferase (ALT) Assays
2.13. Heart Function Tests Using Echocardiographic (ECHO) Analyses
2.14. Statistical Analysis
3. Results
3.1. Construction and Validation of Myo1c Null Mice
3.2. Genetic Deletion of Myo1c Induced Visual Impairment in Mice
3.3. Localization of Rod and Cone Visual Pigments in Myo1c-KO Mice
3.4. Native Cre+ Mice Showed No Retinal Phenotypes
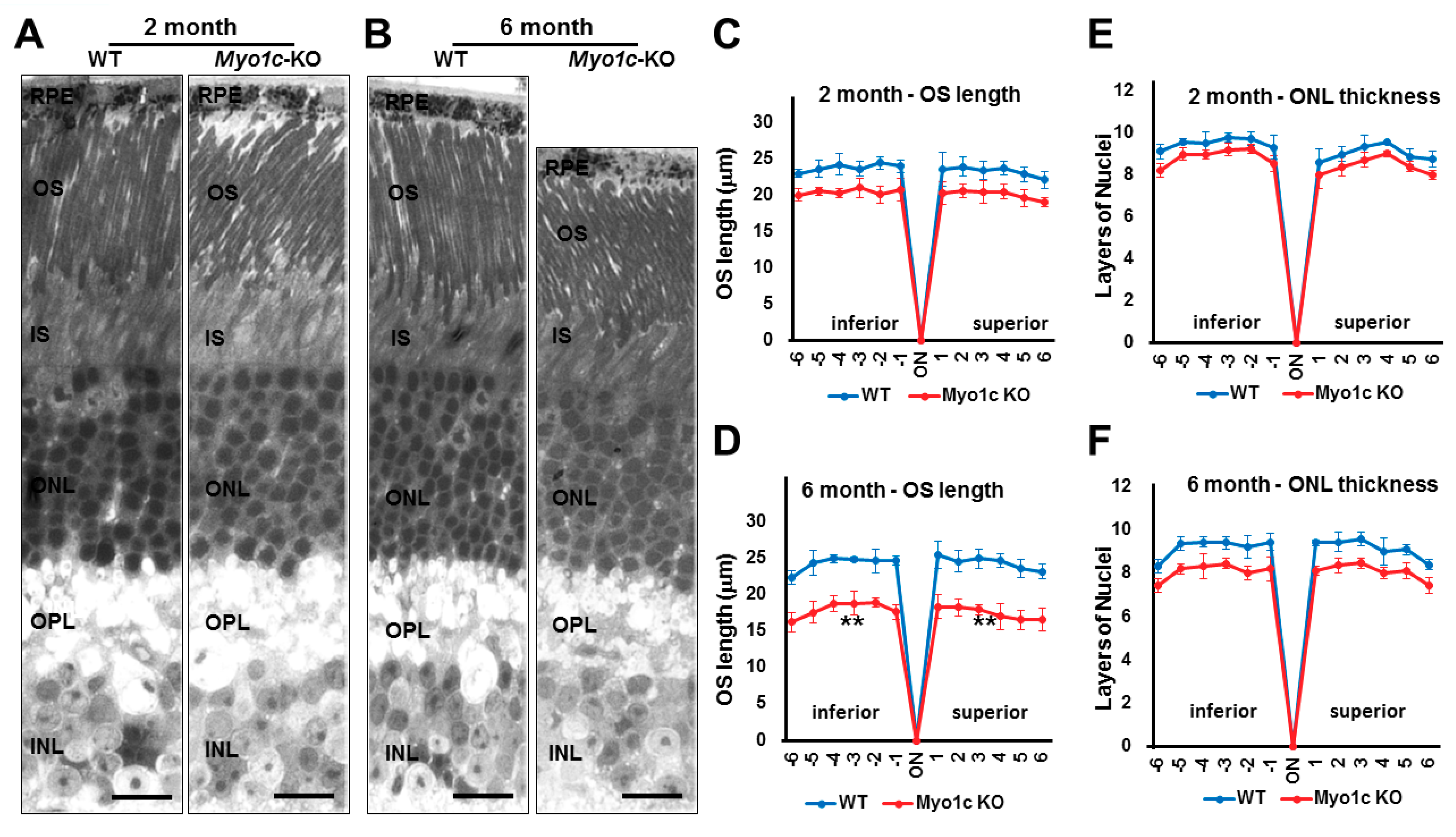
3.5. Myo1c-KO Mice Demonstrated Photoreceptor OS Loss
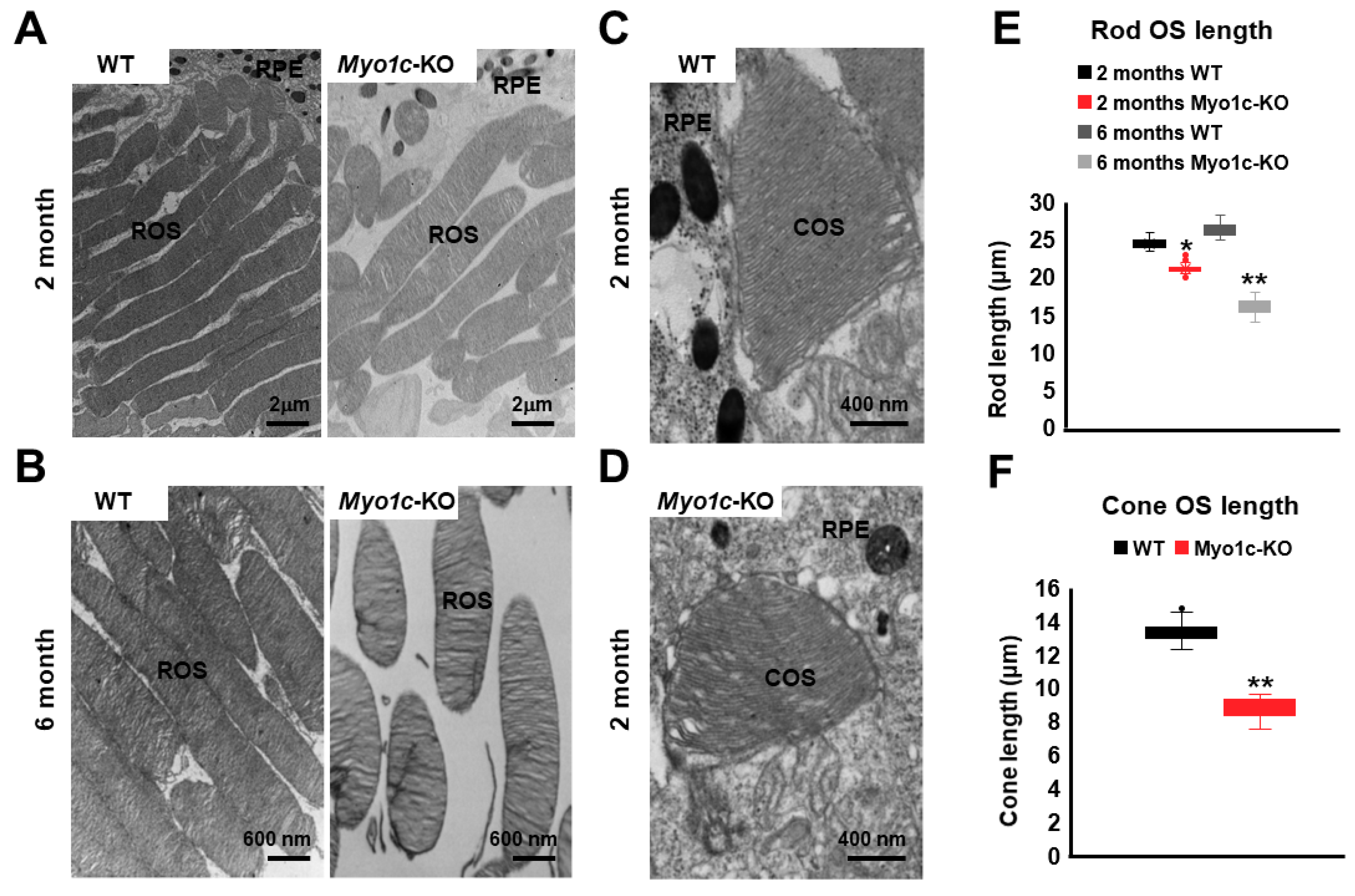
3.6. Ultrastructural TEM Analysis Showed Shorter Photoreceptor OS in Myo1c-KO Mice
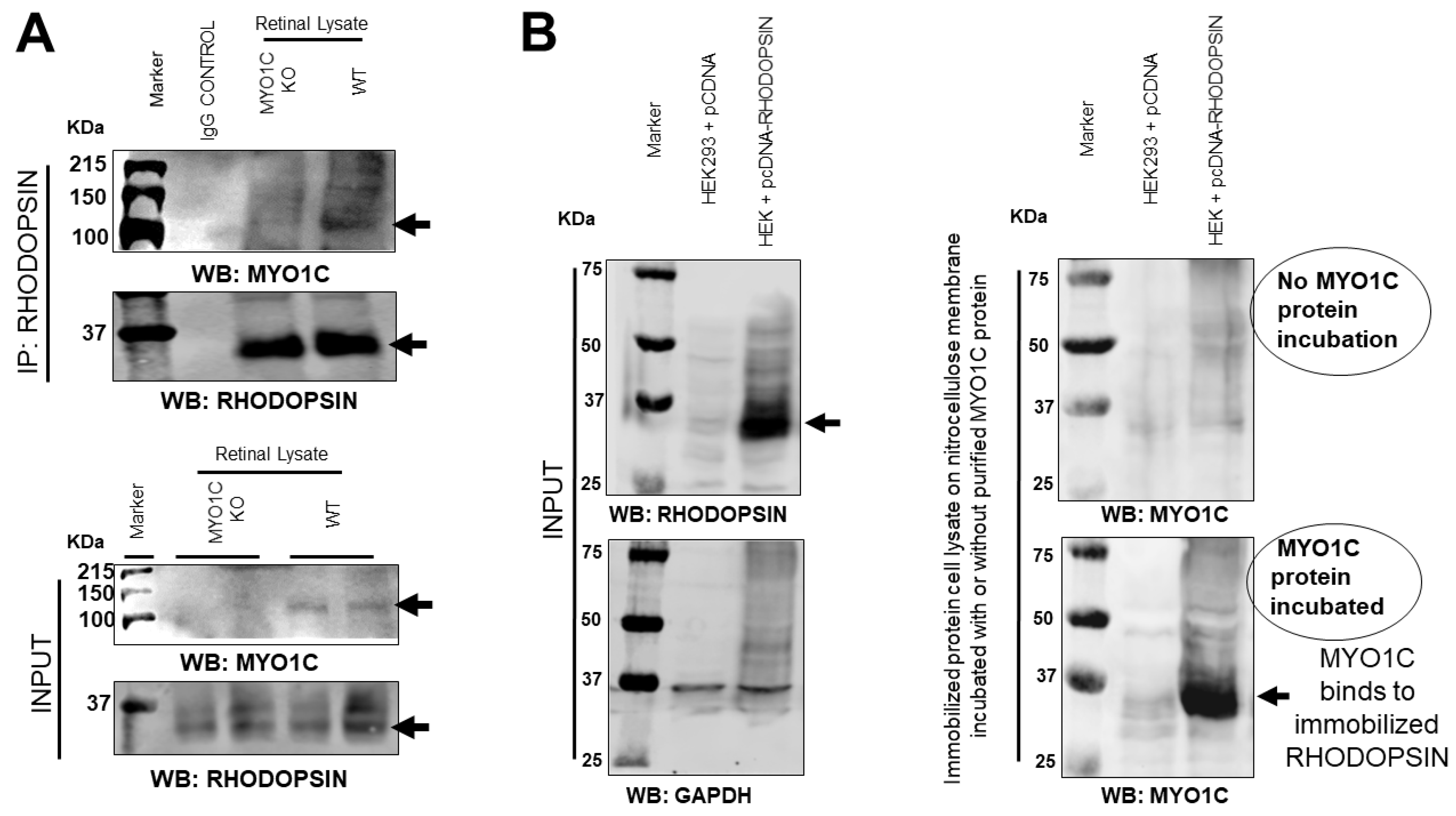
3.7. MYO1C Directly Interacted with Rhodopsin
3.8. Genetic Deletion of Myo1c Did Not Affect Systemic Organs in Mice
4. Discussion
4.1. MYO1C and Opsin Localization to Photoreceptor OS
4.2. MYO1C Contributed to Phototransduction and Retinal Homeostasis
4.3. Contributions from Other Motor Proteins in Proper Opsin Localization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baehr, W.; Karan, S.; Maeda, T.; Luo, D.G.; Li, S.; Bronson, J.D.; Watt, C.B.; Yau, K.W.; Frederick, J.M.; Palczewski, K. The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 2007, 282, 8837–8847. [Google Scholar] [CrossRef] [Green Version]
- Concepcion, F.; Chen, J. Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: A mouse model of Q344ter-induced retinal degeneration. PLoS ONE 2010, 5, e10904. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, T.J.; Gross, A.K. Defective trafficking of rhodopsin and its role in retinal degenerations. Int. Rev. Cell Mol. Biol. 2012, 293, 1–44. [Google Scholar]
- Grossman, G.H.; Watson, R.F.; Pauer, G.J.; Bollinger, K.; Hagstrom, S.A. Immunocytochemical evidence of Tulp1-dependent outer segment protein transport pathways in photoreceptor cells. Exp. Eye Res. 2011, 93, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Hüttl, S.; Michalakis, S.; Seeliger, M.; Luo, D.G.; Acar, N.; Geiger, H.; Hudl, K.; Mader, R.; Haverkamp, S.; Moser, M.; et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci. 2005, 25, 130–138. [Google Scholar] [CrossRef]
- Nishimura, D.Y.; Fath, M.; Mullins, R.F.; Searby, C.; Andrews, M.; Davis, R.; Andorf, J.L.; Mykytyn, K.; Swiderski, R.E.; Yang, B.; et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 16588–16593. [Google Scholar] [CrossRef] [Green Version]
- Pearring, J.N.; Salinas, R.Y.; Baker, S.A.; Arshavsky, V.Y. Protein sorting, targeting and trafficking in photoreceptor cells. Prog. Retin. Eye Res. 2013, 36, 24–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallory, D.P.; Gutierrez, E.; Pinkevitch, M.; Klinginsmith, C.; Comar, W.D.; Roushar, F.J.; Schlebach, J.P.; Smith, A.W.; Jastrzebska, B. The Retinitis Pigmentosa-Linked Mutations in Transmembrane Helix 5 of Rhodopsin Disrupt Cellular Trafficking Regardless of Oligomerization State. Biochemistry 2018, 57, 5188–5201. [Google Scholar] [CrossRef]
- Karan, S.; Zhang, H.; Li, S.; Frederick, J.M.; Baehr, W. A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vis. Res. 2008, 48, 442–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelkader, E.; Enani, L.; Schatz, P.; Safieh, L. Severe retinal degeneration at an early age in Usher syndrome type 1B associated with homozygous splice site mutations in MYO7A gene. Saudi J. Ophthalmol. 2018, 32, 119–125. [Google Scholar] [CrossRef]
- Cheng, L.; Yu, H.; Jiang, Y.; He, J.; Pu, S.; Li, X.; Zhang, L. Identification of a novel MYO7A mutation in Usher syndrome type 1. Oncotarget 2018, 9, 2295–2303. [Google Scholar] [CrossRef] [Green Version]
- Arif, E.; Wagner, M.C.; Johnstone, D.B.; Wong, H.N.; George, B.; Pruthi, P.A.; Lazzara, M.J.; Nihalani, D. Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol. Cell. Biol. 2011, 31, 2134–2150. [Google Scholar] [CrossRef] [Green Version]
- Woolner, S.; Bement, W.M. Unconventional myosins acting unconventionally. Trends Cell Biol. 2009, 19, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Bownds, D. Site of attachment of retinal in rhodopsin. Nature 1967, 216, 1178–1181. [Google Scholar] [CrossRef] [PubMed]
- Stenkamp, R.E.; Teller, D.C.; Palczewski, K. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef]
- Lee, E.S.; Flannery, J.G. Transport of truncated rhodopsin and its effects on rod function and degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- Wald, G.; Hubbard, R. The Synthesis of Rhodopsin from Vitamin A(1). Proc. Natl. Acad. Sci. USA 1950, 36, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.K.; McDowell, J.H.; Hargrave, P.A. Site of attachment of 11-cis-retinal in bovine rhodopsin. Biochemistry 1980, 19, 5111–5117. [Google Scholar] [CrossRef]
- McConnell, R.E.; Tyska, M.J. Leveraging the membrane—Cytoskeleton interface with myosin-1. Trends Cell Biol. 2010, 20, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Navinés-Ferrer, A.; Martín, M. Long-Tailed Unconventional Class I Myosins in Health and Disease. Int. J. Mol. Sci. 2020, 21, 2555. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Takemura, R. Biochemical and molecular characterization of diseases linked to motor proteins. Trends Biochem. Sci. 2003, 28, 558–565. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Retinitis Pigmentosa (Non-syndromic). Adv. Exp. Med. Biol. 2018, 1085, 125–130. [Google Scholar]
- Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants 2020, 9, 1158. [Google Scholar] [CrossRef]
- Jiang, M.; Volland, S.; Paniagua, A.E.; Wang, H.; Balaji, A.; Li, D.G.; Lopes, V.S.; Burgess, B.L.; Williams, D.S. Microtubule motor transport in the delivery of melanosomes to the actin-rich, apical domain of in the retinal pigment epithelium. J. Cell Sci. 2020, 133, 1233–1249. [Google Scholar] [CrossRef]
- Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Trovato Battagliola, E.; D’Angelo, R.; Sidoti, A.; Donato, L. Expression of Pro-Angiogenic Markers Is Enhanced by Blue Light in Human RPE Cells. Antioxidants 2020, 9, 1154. [Google Scholar] [CrossRef]
- Sripathi, S.R.; He, W.; Sylvester, O.; Neksumi, M.; Um, J.Y.; Dluya, T.; Bernstein, P.S.; Jahng, W.J. Altered Cytoskeleton as a Mitochondrial Decay Signature in the Retinal Pigment Epithelium. Protein J. 2016, 35, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Stürmer, K.; Baumann, O. Immunolocalization of a putative unconventional myosin on the surface of motile mitochondria in locust photoreceptors. Cell Tissue Res. 1998, 292, 219–227. [Google Scholar] [CrossRef]
- Gibbs, D.; Kitamoto, J.; Williams, D.S. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc. Natl. Acad. Sci. USA 2003, 100, 6481–6486. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.; Walsh, V.; Vreugde, S.; Hertzano, R.; Shahin, H.; Haika, S.; Lee, M.K.; Kanaan, M.; King, M.C.; Avraham, K.B. From flies’ eyes to our ears: Mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. USA 2002, 99, 7518–7523. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.U.; Bird, J.E.; Faridi, R.; Shahzad, M.; Shah, S.; Lee, K.; Khan, S.N.; Imtiaz, A.; Ahmed, Z.M.; Riazuddin, S.; et al. Mutational Spectrum of MYO15A and the Molecular Mechanisms of DFNB3 Human Deafness. Hum. Mutat. 2016, 37, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- Samuels, I.S.; Bell, B.A.; Sturgill-Short, G.; Ebke, L.A.; Rayborn, M.; Shi, L.; Nishina, P.M.; Peachey, N.S. Myosin 6 is required for iris development and normal function of the outer retina. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7223–7233. [Google Scholar] [CrossRef] [Green Version]
- Adamek, N.; Geeves, M.A.; Coluccio, L.M. Myo1c mutations associated with hearing loss cause defects in the interaction with nucleotide and actin. Cell. Mol. Life Sci. 2011, 68, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Greenberg, M.J.; Moore, J.R.; Ostap, E.M. A hearing loss-associated myo1c mutation (R156W) decreases the myosin duty ratio and force sensitivity. Biochemistry 2011, 50, 1831–1838. [Google Scholar] [CrossRef] [Green Version]
- Skeie, J.M.; Mahajan, V.B. Proteomic interactions in the mouse vitreous-retina complex. PLoS ONE 2013, 8, e82140. [Google Scholar] [CrossRef] [Green Version]
- Boguslavsky, S.; Chiu, T.; Foley, K.P.; Osorio-Fuentealba, C.; Antonescu, C.N.; Bayer, K.U.; Bilan, P.J.; Klip, A. Myo1c binding to submembrane actin mediates insulin-induced tethering of GLUT4 vesicles. Mol. Biol. Cell 2012, 23, 4065–4078. [Google Scholar] [CrossRef]
- Bose, A.; Robida, S.; Furcinitti, P.S.; Chawla, A.; Fogarty, K.; Corvera, S.; Czech, M.P. Unconventional myosin Myo1c promotes membrane fusion in a regulated exocytic pathway. Mol. Cell. Biol. 2004, 24, 5447–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Eswarappa, S.M.; Hitomi, M.; Fox, P.L. Myo1c facilitates G-actin transport to the leading edge of migrating endothelial cells. J. Cell Biol. 2012, 198, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Mattapallil, M.J.; Wawrousek, E.F.; Chan, C.C.; Zhao, H.; Roychoudhury, J.; Ferguson, T.A.; Caspi, R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2921–2927. [Google Scholar] [CrossRef] [Green Version]
- Lobo, G.P.; Fulmer, D.; Guo, L.; Zuo, X.; Dang, Y.; Kim, S.H.; Su, Y.; George, K.; Obert, E.; Fogelgren, B.; et al. The exocyst is required for photoreceptor ciliogenesis and retinal development. J. Biol. Chem. 2017, 292, 14814–14826. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.C.; Barylko, B.; Albanesi, J.P. Tissue distribution and subcellular localization of mammalian myosin I. J. Cell Biol. 1992, 119, 163–170. [Google Scholar] [CrossRef]
- Husain, S. Delta Opioids: Neuroprotective Roles in Preclinical Studies. J. Ocul. Pharmacol. Ther. 2018, 34, 119–128. [Google Scholar] [CrossRef]
- Husain, S.; Ahmad, A.; Singh, S.; Peterseim, C.; Abdul, Y.; Nutaitis, M.J. PI3K/Akt Pathway: A Role in delta-Opioid Receptor-Mediated RGC Neuroprotection. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6489–6499. [Google Scholar] [CrossRef] [Green Version]
- Brandstaetter, H.; Kishi-Itakura, C.; Tumbarello, D.A.; Manstein, D.J.; Buss, F. Loss of functional MYO1C/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome-lysosome fusion. Autophagy 2014, 10, 2310–2323. [Google Scholar] [CrossRef] [Green Version]
- Solanki, A.K.; Rathore, Y.S.; Badmalia, M.D.; Dhoke, R.R.; Nath, S.K.; Nihalani, D. Global shape and ligand binding efficiency of the HIV-1-neutralizing antibodies differ from those of antibodies that cannot neutralize HIV-1. J. Biol. Chem. 2014, 289, 34780–34800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, E.; Solanki, A.K.; Srivastava, P.; Rahman, B.; Tash, B.R.; Holzman, L.B.; Janech, M.G.; Martin, R.; Knölker, H.J.; Fitzgibbon, W.R.; et al. The motor protein Myo1c regulates transforming growth factor-beta-signaling and fibrosis in podocytes. Kidney Int. 2019, 96, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Crosson, C.E.; Mani, S.K.; Husain, S.; Alsarraf, O.; Menick, D.R. Inhibition of histone deacetylase protects the retina from ischemic injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3639–3645. [Google Scholar] [CrossRef]
- Husain, S.; Potter, D.E.; Crosson, C.E. Opioid receptor-activation: Retina protected from ischemic injury. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Krebs, W.; Kühn, H. Structure of isolated bovine rod outer segment membranes. Exp. Eye Res. 1977, 25, 511–526. [Google Scholar] [CrossRef]
- Bales, K.L.; Gross, A.K. Aberrant protein trafficking in retinal degenerations: The initial phase of retinal remodeling. Exp. Eye Res. 2016, 150, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Calvert, P.D.; Strissel, K.J.; Schiesser, W.E.; Pugh, E.N., Jr.; Arshavsky, V.Y. Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol. 2006, 16, 560–568. [Google Scholar] [CrossRef]
- Mustafi, D.; Kevany, B.M.; Genoud, C.; Bai, X.; Palczewski, K. Photoreceptor phagocytosis is mediated by phosphoinositide signaling. FASEB J. 2013, 27, 4585–4595. [Google Scholar] [CrossRef] [Green Version]
- Lopes, V.S.; Gibbs, D.; Libby, R.T.; Aleman, T.S.; Welch, D.L.; Lillo, C.; Jacobson, S.G.; Radu, R.A.; Steel, K.P.; Williams, D.S. The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum. Mol. Genet. 2011, 20, 2560–2570. [Google Scholar] [CrossRef] [Green Version]
- Young, R.W. The renewal of photoreceptor cell outer segments. J. Cell Biol. 1967, 33, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Young, R.W.; Droz, B. The renewal of protein in retinal rods and cones. J. Cell Biol. 1968, 39, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findlay, J.B.; Pappin, D.J. The opsin family of proteins. Biochem. J. 1986, 238, 625–642. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Au, A.; Kiser, P.D.; Hagstrom, S.A. Involvement of Endoplasmic Reticulum Stress in TULP1 Induced Retinal Degeneration. PLoS ONE 2016, 11, e0151806. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Rohrer, B.; Frederick, J.M.; Baehr, W.; Crouch, R.K. Rpe65−/− and Lrat−/− mice: Comparable models of leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Shi, Y.; Obert, E.; Rahman, B.; Rohrer, B.; Lobo, G.P. The Retinol Binding Protein Receptor 2 (Rbpr2) is required for Photoreceptor Outer Segment Morphogenesis and Visual Function in Zebrafish. Sci. Rep. 2017, 7, 16207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanki, A.; Kondkar, A.A.; Fogerty, J.; Su, Y.; Kim, S.H.; Lipschutz, J.H.; Nihalani, D.; Perkins, B.D.; Lobo, G.P. A Functional Binding Domain in the Rbpr2 Receptor Is Required for Vitamin A Transport, Ocular Retinoid Homeostasis, and Photoreceptor Cell Survival in Zebrafish. Cells 2020, 9, 1099. [Google Scholar] [CrossRef] [PubMed]
- Lobo, G.P.; Pauer, G.; Lipschutz, J.H.; Hagstrom, S.A. The Retinol-Binding Protein Receptor 2 (Rbpr2) Is Required for Photoreceptor Survival and Visual Function in the Zebrafish. Adv. Exp. Med. Biol. 2018, 1074, 569–576. [Google Scholar] [PubMed]
- Beech, P.L.; Pagh-Roehl, K.; Noda, Y.; Hirokawa, N.; Burnside, B.; Rosenbaum, J.L. Localization of kinesin superfamily proteins to the connecting cilium of fish photoreceptors. J. Cell Sci. 1996, 109, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Muresan, V.; Bendala-Tufanisco, E.; Hollander, B.A.; Besharse, J.C. Evidence for kinesin-related proteins associated with the axoneme of retinal photoreceptors. Exp. Eye Res. 1997, 64, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Chaitin, M.H.; Coelho, N. Immunogold localization of myosin in the photoreceptor cilium. Investig. Ophthalmol. Vis. Sci. 1992, 33, 3103–3108. [Google Scholar] [PubMed]
- Williams, D.S.; Hallett, M.A.; Arikawa, K. Association of myosin with the connecting cilium of rod photoreceptors. J. Cell Sci. 1992, 103, 183–190. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solanki, A.K.; Biswal, M.R.; Walterhouse, S.; Martin, R.; Kondkar, A.A.; Knölker, H.-J.; Rahman, B.; Arif, E.; Husain, S.; Montezuma, S.R.; et al. Loss of Motor Protein MYO1C Causes Rhodopsin Mislocalization and Results in Impaired Visual Function. Cells 2021, 10, 1322. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061322
Solanki AK, Biswal MR, Walterhouse S, Martin R, Kondkar AA, Knölker H-J, Rahman B, Arif E, Husain S, Montezuma SR, et al. Loss of Motor Protein MYO1C Causes Rhodopsin Mislocalization and Results in Impaired Visual Function. Cells. 2021; 10(6):1322. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061322
Chicago/Turabian StyleSolanki, Ashish K., Manas R. Biswal, Stephen Walterhouse, René Martin, Altaf A. Kondkar, Hans-Joachim Knölker, Bushra Rahman, Ehtesham Arif, Shahid Husain, Sandra R. Montezuma, and et al. 2021. "Loss of Motor Protein MYO1C Causes Rhodopsin Mislocalization and Results in Impaired Visual Function" Cells 10, no. 6: 1322. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061322