
Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease


Abstract
:1. Introduction
2. Methods
2.1. Animals and Diets
2.2. Biochemical Analysis
2.3. Liver Histology
2.4. Cell Culture and Treatments
2.5. Fatty Acid and Very Low-Density Lipoprotein Uptake in Isolated Hepatocytes
2.6. Cell Viability Assay
2.7. Global DNA CpG Methylation Analysis
2.8. DNA Methyltransferase Activity
2.9. Analysis of PparγPromoter-Specific DNA Methylation
2.10. Immunoblotting and Protein Level Determination
2.11. Gene Expression and QPCR
2.12. Statistical Analysis
3. Results
3.1. Blood Parameter Analysis
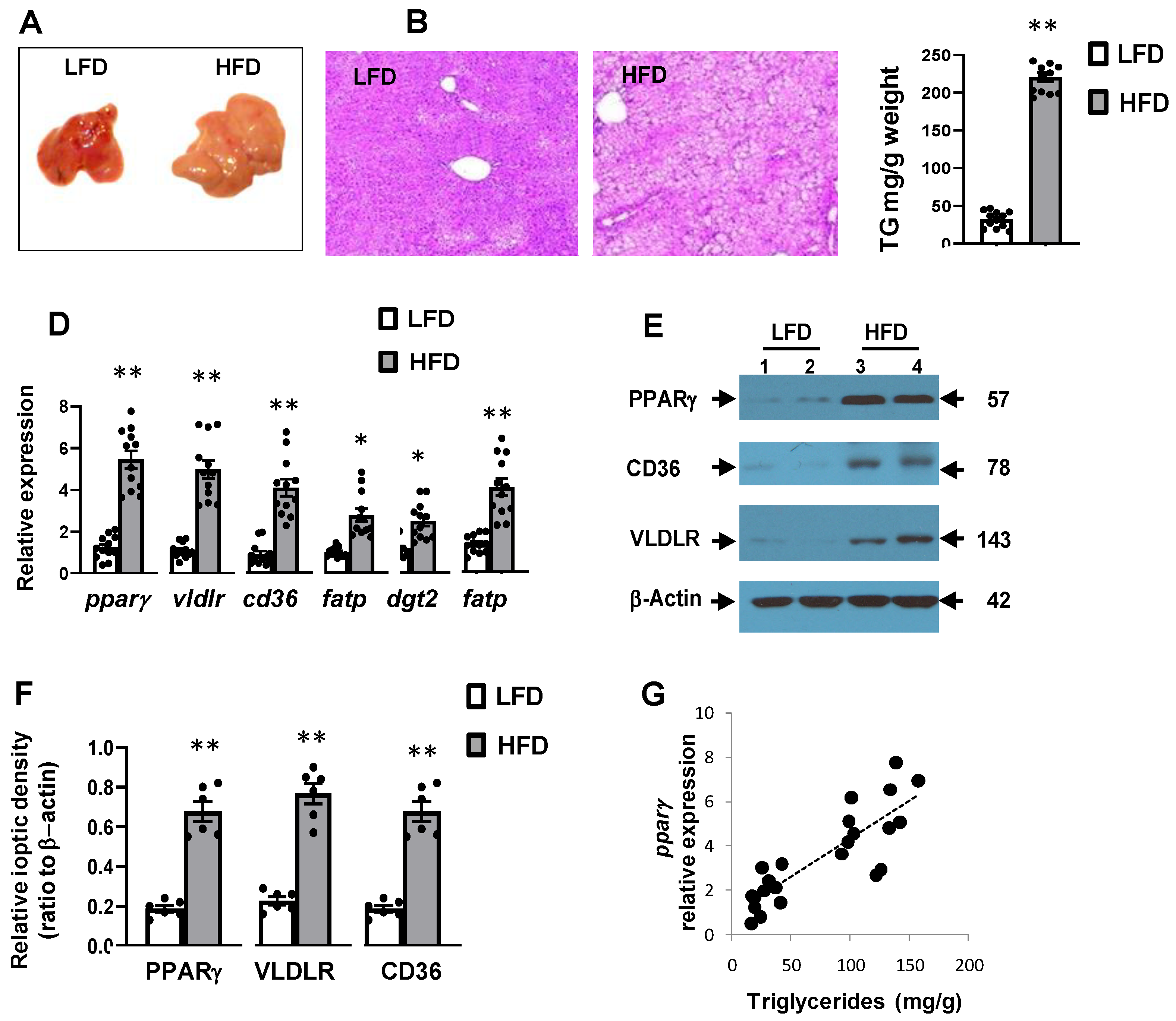
3.2. High-Fat Diet Was Associated with Hepatic Steatosis and Inflammation
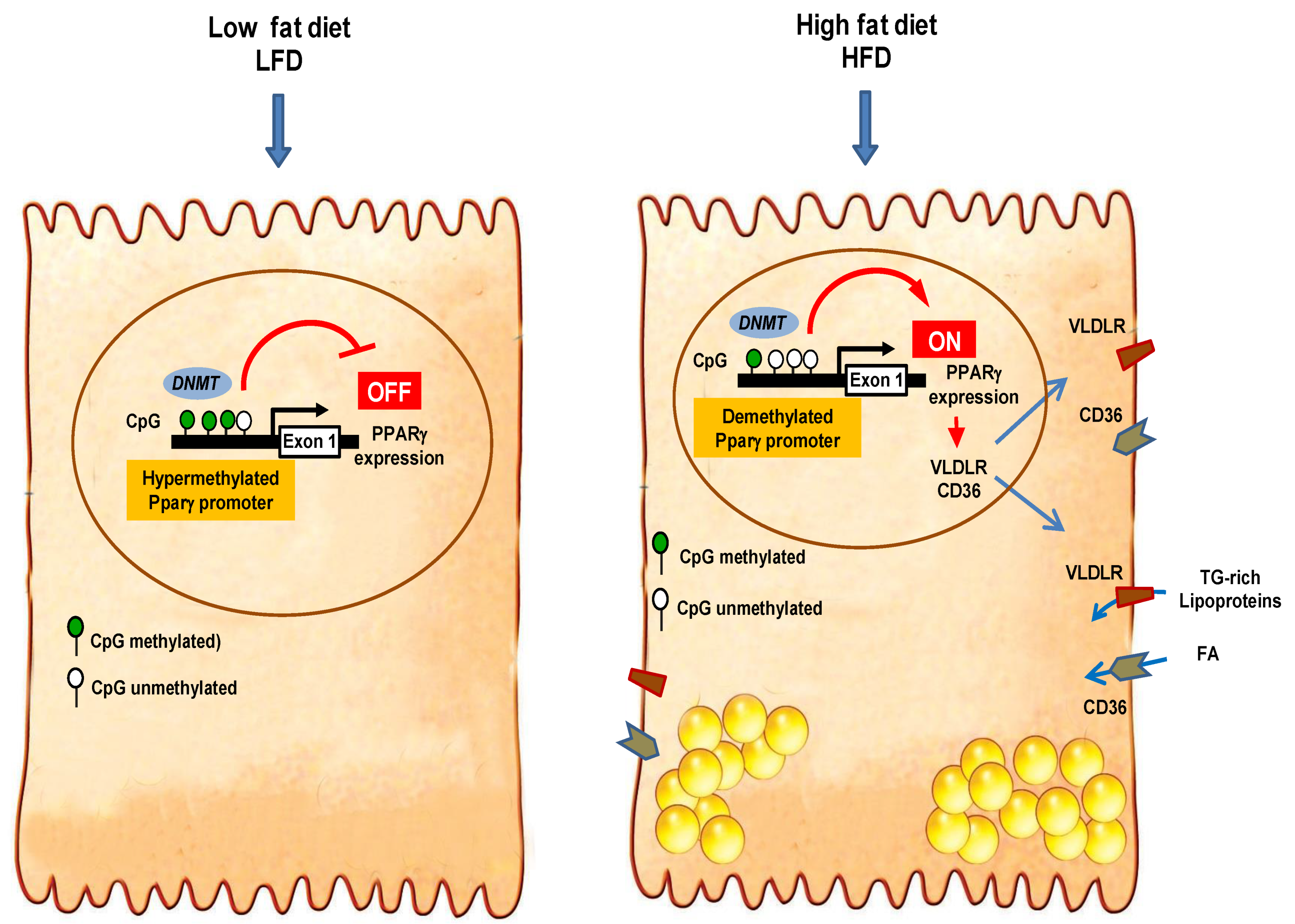
3.3. High-Fat Diet Induced Hepatic Expression of Pparγ and Its Target Genes
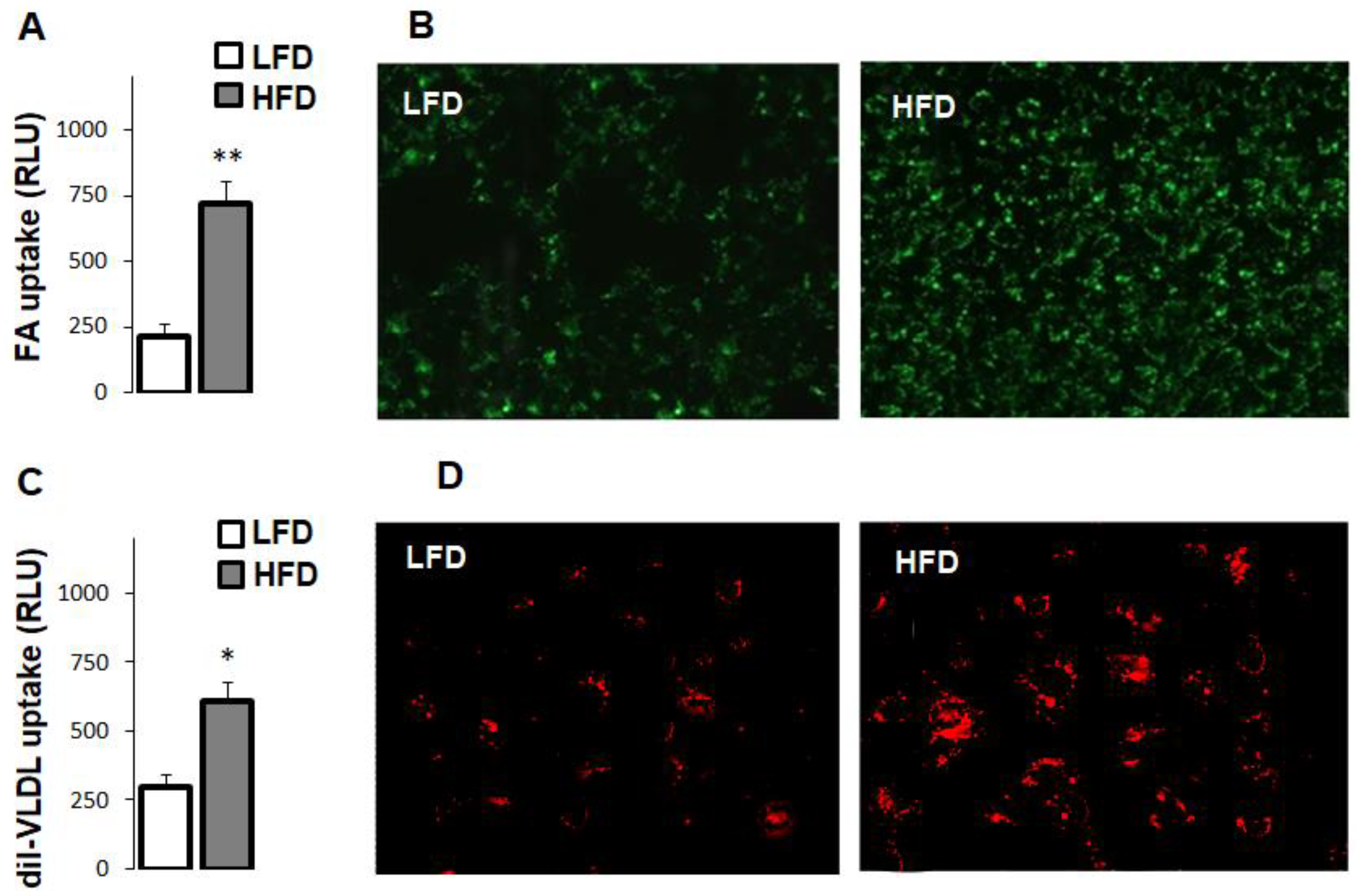
3.4. High-Fat Diet Increased Fatty Acid and Very Low Density Lipoprotein Uptake in Hepatocytes
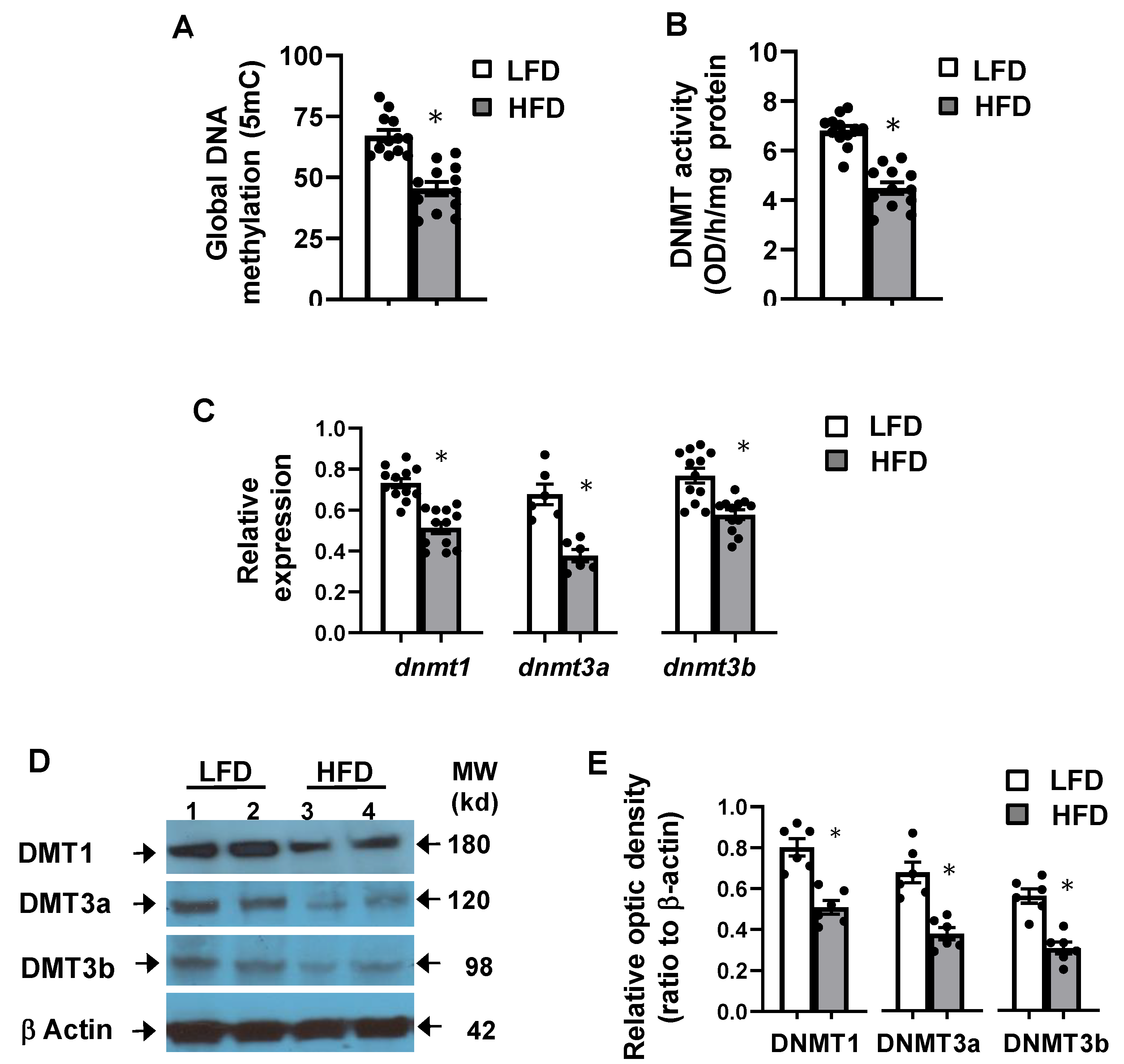
3.5. High-Fat Diet Altered Hepatic Global DNA Methylation
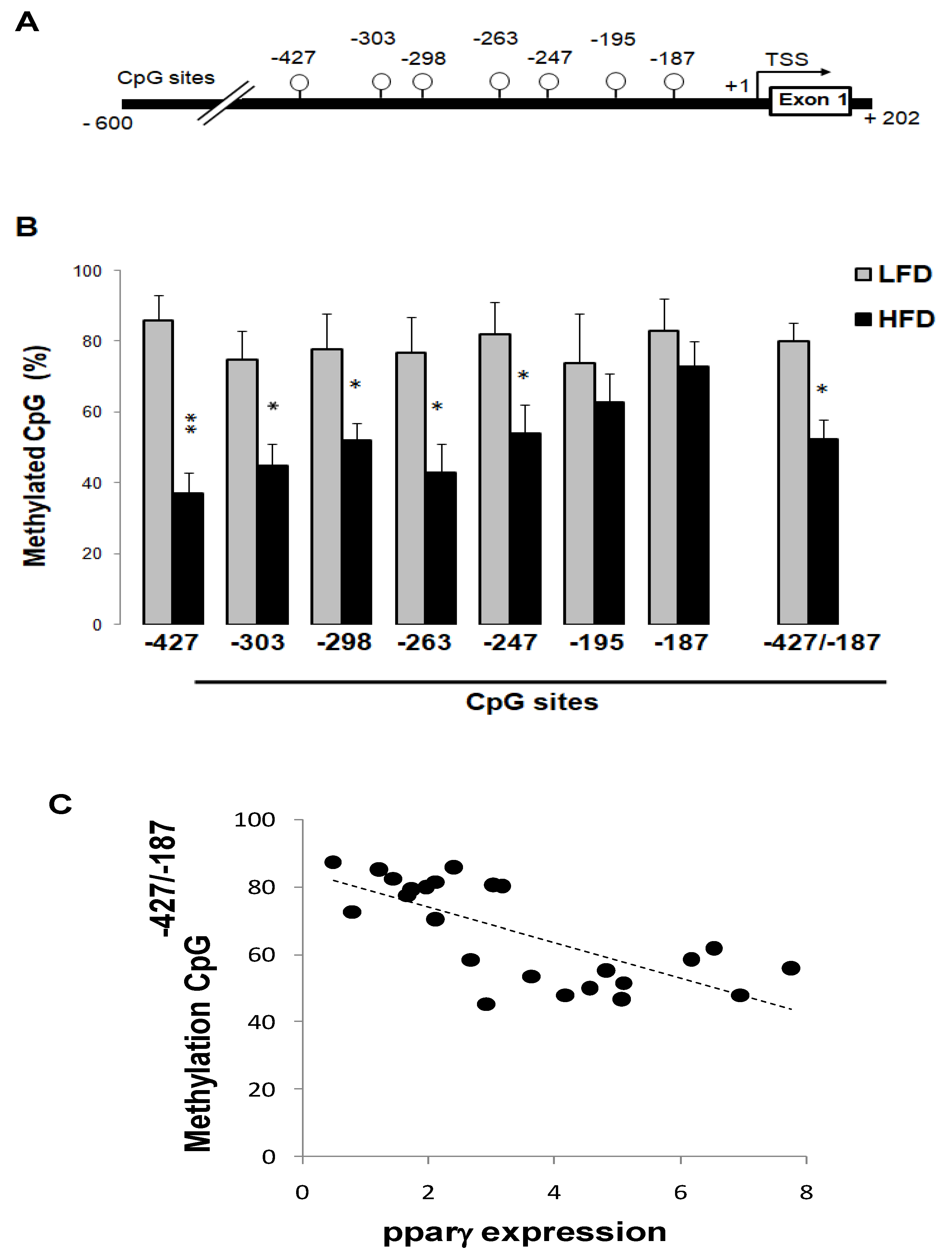
3.6. High-Fat Diet Reduced Pparγ Promoter Methylation
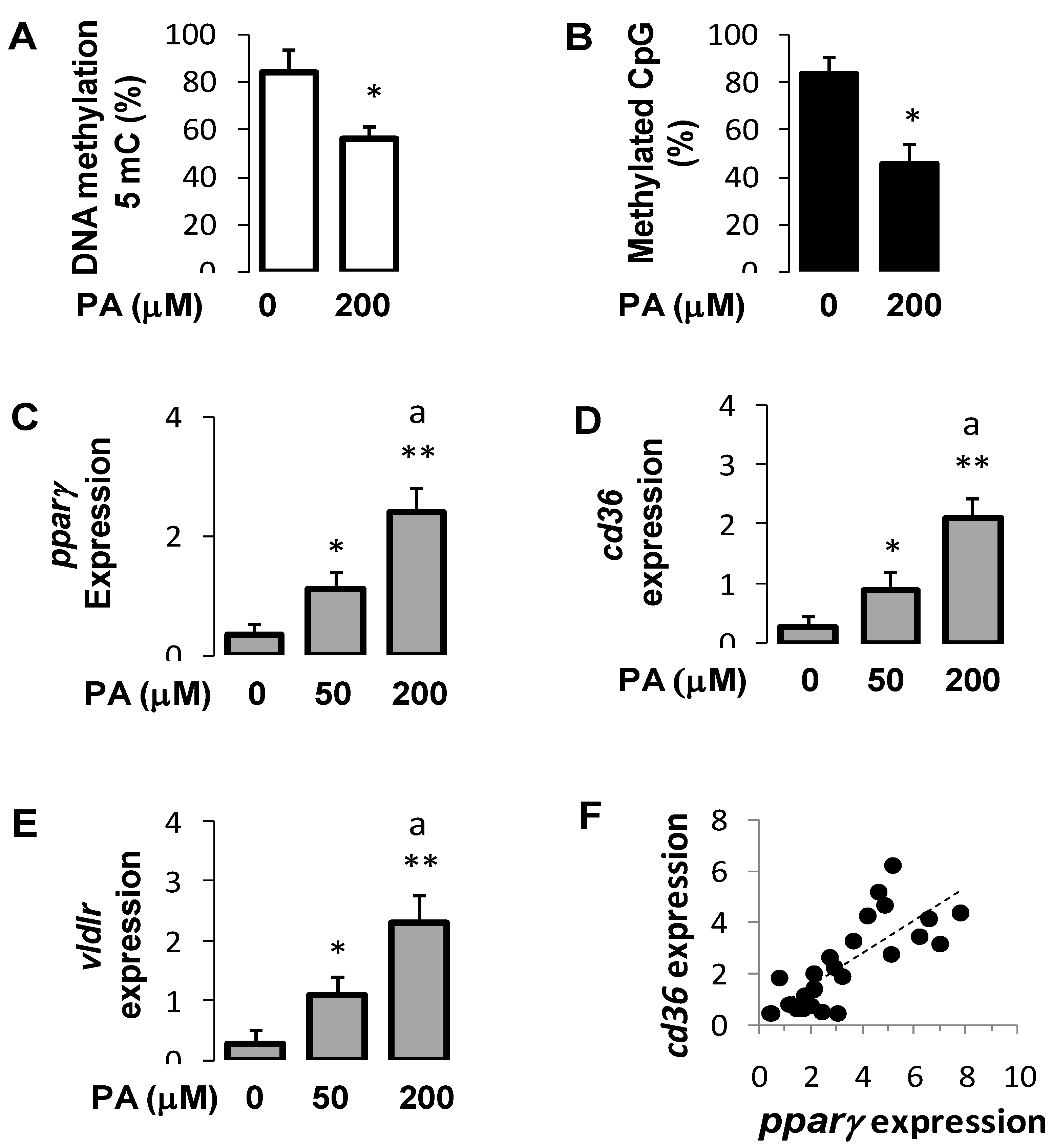
3.7. Palmitic Acid Regulated DNA Methylation and Expression of Pparγ and Its Target Genes in Hepatocytes
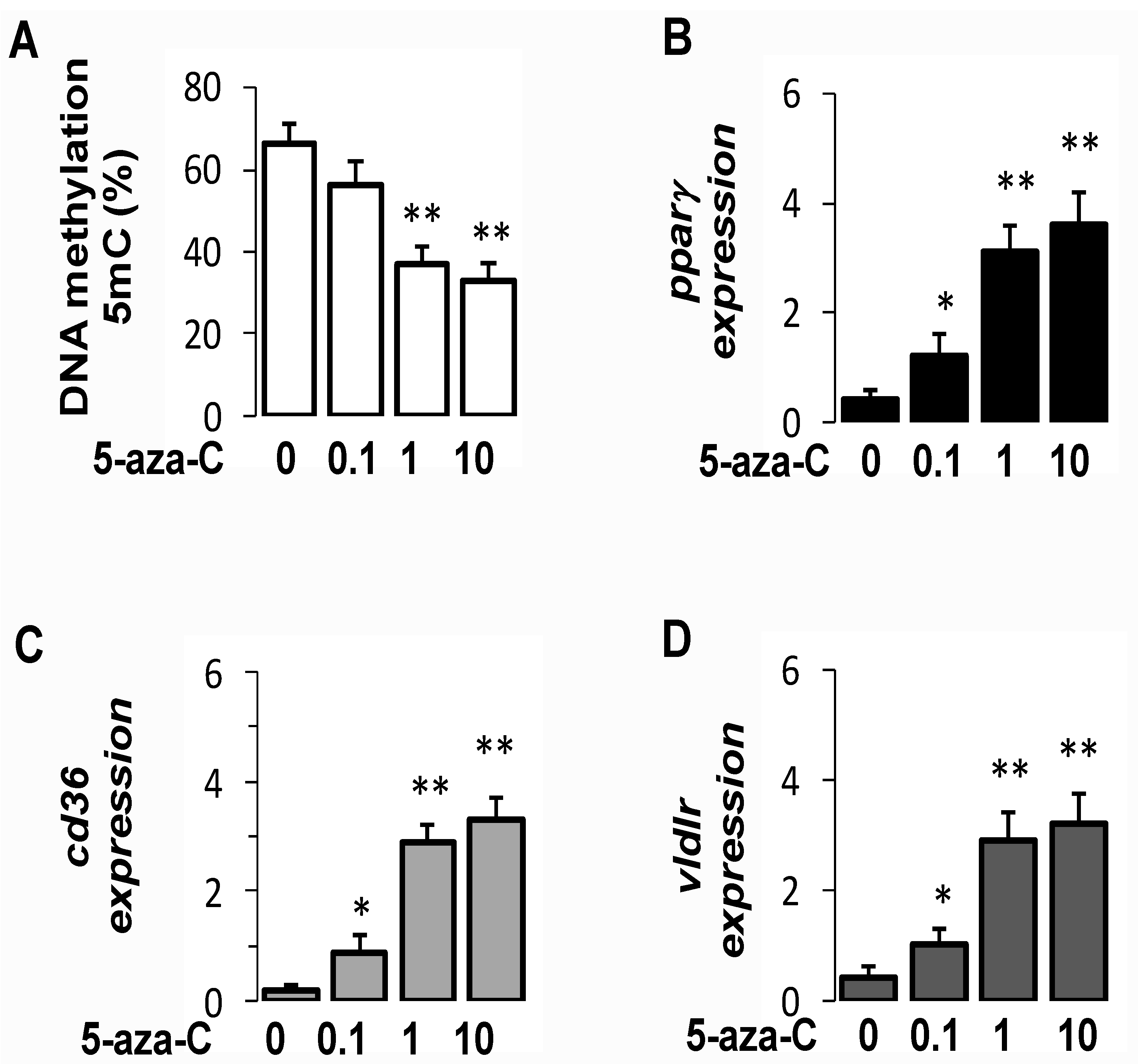
3.8. Pharmacological Inhibition of DNMT Increased PparγExpression in Hepatocytes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Asrih, M.; Jornayvaz, F.R. Diets and nonalcoholic fatty liver disease: The good and the bad. Clin. Nutr. 2014, 33, 186–190. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Dashek, R.J.; Mucinski, J.M.; Rector, R.S. A Fad too Far? Dietary Strategies for the Prevention and Treatment of NAFLD. Obesity 2020, 28, 1843–1852. [Google Scholar] [CrossRef]
- McCarthy, E.M.; Rinella, M.E. The role of diet and nutrient composition in nonalcoholic Fatty liver disease. J. Acad. Nutr. Diet 2012, 112, 401–409. [Google Scholar] [CrossRef]
- Kadayifci, F.Z.; Zheng, S.; Pan, Y.X. Molecular Mechanisms Underlying the Link between Diet and DNA Methylation. Int. J. Mol. Sci. 2018, 19, 4055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Becerra, K.; Ramos-Lopez, O.; Barron-Cabrera, E.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez-Lopez, E.; Martínez, J.A. Fatty acids, epigenetic mechanisms and chronic diseases: A systematic review. Lipids Health Dis. 2019, 18, 178. [Google Scholar] [CrossRef] [Green Version]
- Zaiou, M. Circular RNAs as Potential Biomarkers and Therapeutic Targets for Metabolic Diseases. Adv. Exp. Med. Biol. 2019, 1134, 177–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Zhou, D.; Moody, L.; Lezmi, S.; Chen, H.; Pan, Y.-X. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol. Genom. 2015, 47, 514–523. [Google Scholar] [CrossRef]
- Amaral, C.L.; Crisma, A.R.; Masi, L.N.; Martins, A.R.; Hirabara, S.M.; Curi, R. DNA Methylation Changes Induced by a High-Fat Diet and Fish Oil Supplementation in the Skeletal Muscle of Mice. J. Nutrigenet. Nutr. 2014, 7, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Zwamborn, R.A.; Slieker, R.C.; Mulder, P.C.; Zoetemelk, I.; Verschuren, L.; Suchiman, H.E.; Toet, K.H.; Droog, S.; Slagboom, P.E.; Kooistra, T.; et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 2017, 7, 43261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Martinez, G.A.; Rodriguez-Rios, D.; Alvarado-Caudillo, Y.; Vaquero, A.; Esteller, M.; Carmona, F.J.; Moran, S.; Nielsen, F.C.; Wickström-Lindholm, M.; Wrobel, K.; et al. Arachidonic and oleic acid exert distinct effects on the DNA methylome. Epigenetics 2016, 11, 321–334. [Google Scholar] [CrossRef] [Green Version]
- de la Rocha, C.; Perez-Mojica, J.E.; Leon, S.Z.; Cervantes-Paz, B.; Tristan-Flores, F.E.; Rodriguez-Rios, D.; Molina-Torres, J.; Ramírez-Chávez, E.; Alvarado-Caudillo, Y.; Carmona, F.J.; et al. Associations between whole peripheral blood fatty acids and DNA methylation in humans. Sci. Rep. 2016, 6, 25867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malodobra-Mazur, M.; Cierzniak, A.; Dobosz, T. Oleic acid influences the adipogenesis of 3T3-L1 cells via DNA Methylation and may predispose to obesity and obesity-related disorders. Lipids Health Dis. 2019, 18, 230. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Aakula, S.; Abumrad, N.N.; Hajri, T. Peroxisome proliferator-activated receptor-gamma regulates the expression and function of very-low-density lipoprotein receptor. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E68–E79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Hajri, T. Very low density lipoprotein receptor promotes adipocyte differentiation and mediates the proadipogenic effect of peroxisome proliferator-activated receptor gamma agonists. Biochem. Pharmacol. 2011, 82, 1950–1962. [Google Scholar] [CrossRef]
- Hajri, T.; Abumrad, N.A. Fatty acid transport across membranes: Relevance to nutrition and metabolic pathology. Annu. Rev. Nutr. 2002, 22, 383–415. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Kiyono, S.; Kondo, T.; Sekimoto, T.; Yokosuka, O. Palmitate-induced Regulation of PPARgamma via PGC1alpha: A Mechanism for Lipid Accumulation in the Liver in Nonalcoholic Fatty Liver Disease. Int. J. Med. Sci. 2016, 13, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Skat-Rordam, J.; Hojland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor gamma in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Hajri, T.; Khosla, P.; Pronczuk, A.; Hayes, K.C. Myristic acid-rich fat raises plasma LDL by stimulating LDL production without affecting fractional clearance in gerbils fed a cholesterol-free diet. J. Nutr. 1998, 128, 477–484. [Google Scholar] [CrossRef]
- Hajri, T.; Han, X.X.; Bonen, A.; Abumrad, N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Investig. 2002, 109, 1381–1389. [Google Scholar] [CrossRef]
- Nguyen, A.; Tao, H.; Metrione, M.; Hajri, T. Very low density lipoprotein receptor (VLDLR) expression is a determinant factor in adipose tissue inflammation and adipocyte-macrophage interaction. J. Biol. Chem. 2014, 289, 1688–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajri, T.; Tao, H.; Wattacheril, J.; Marks-Shulman, P.; Abumrad, N.N. Regulation of adiponectin production by insulin: Interactions with tumor necrosis factor-alpha and interleukin-6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E350–E360. [Google Scholar] [CrossRef]
- Gharib, M.; Tao, H.; Fungwe, T.V.; Hajri, T. Cluster Differentiating 36 (CD36) Deficiency Attenuates Obesity-Associated Oxidative Stress in the Heart. PLoS ONE 2016, 11, e0155611. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Shiota, M.; Ivanova, P.T.; Myers, D.S.; Brown, H.A.; Young, J.D. Enhanced synthesis of saturated phospholipids is associated with ER stress and lipotoxicity in palmitate treated hepatic cells. J. Lipid Res. 2014, 55, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Poirier, S.; Samami, S.; Mamarbachi, M.; Demers, A.; Chang, T.Y.; Vance, D.E.; Hatch, G.M.; Mayer, G. The epigenetic drug 5-azacytidine interferes with cholesterol and lipid metabolism. J. Biol. Chem. 2014, 289, 18736–18751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Sportsman, R.; Harris, J.; Stahl, A. Real-time quantification of fatty acid uptake using a novel fluorescence assay. J. Lipid Res. 2005, 46, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Delaney, C.; Garg, S.K.; Yung, R. Analysis of DNA Methylation by Pyrosequencing. Methods Mol. Biol. 2015, 1343, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef]
- Wang, L.J.; Zhang, H.W.; Zhou, J.Y.; Liu, Y.; Yang, Y.; Chen, X.L.; Zhu, C.H.; Zheng, R.D.; Ling, W.H.; Zhu, H.L. Betaine attenuates hepatic steatosis by reducing methylation of the MTTP promoter and elevating genomic methylation in mice fed a high-fat diet. J. Nutr. Biochem. 2014, 25, 329–336. [Google Scholar] [CrossRef]
- Fujiki, K.; Kano, F.; Shiota, K.; Murata, M. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Barchitta, M.; Maugeri, A.; Magnano San Lio, R.; Favara, G.; La Rosa, M.C.; La Mastra, C.; Quattrocchi, A.; Agodi, A. Dietary Patterns are Associated with Leukocyte LINE-1 Methylation in Women: A Cross-Sectional Study in Southern Italy. Nutrients 2019, 11, 1843. [Google Scholar] [CrossRef] [Green Version]
- Remely, M.; Ferk, F.; Sterneder, S.; Setayesh, T.; Kepcija, T.; Roth, S.; Rebhan, I.; Wagner, K.H.; Knasmüller, S.; Haslberger, A. Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients 2017, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Gracia, A.; Elcoroaristizabal, X.; Fernandez-Quintela, A.; Miranda, J.; Bediaga, N.G.M.; de Pancorbo, M.; Rimando, A.M.; Portillo, M.P. Fatty acid synthase methylation levels in adipose tissue: Effects of an obesogenic diet and phenol compounds. Genes Nutr. 2014, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Qiu, Y.; Yang, J.; Bian, S.; Chen, G.; Deng, M.; Kang, H.; Huang, L. DNMT1-PPARgamma pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci. Rep. 2016, 6, 30053. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, X.; Jia, L.; Mondal, A.K.; Diallo, A.; Hawkins, G.A.; Das, S.K.; Parks, J.S.; Yu, L.; Shi, H.; et al. Inhibiting DNA Methylation by 5-Aza-2′-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology 2014, 155, 4925–4938. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermsdorff, H.H.; Mansego, M.L.; Campion, J.; Milagro, F.I.; Zulet, M.A.; Martinez, J.A. TNF-alpha promoter methylation in peripheral white blood cells: Relationship with circulating TNFα, truncal fat and n-6 PUFA intake in young women. Cytokine 2013, 64, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Escobar, E.; Monastero, R.; Garcia-Serrano, S.; Gomez-Zumaquero, J.M.; Lago-Sampedro, A.; Rubio-Martin, E.; Colomo, N.; Rodríguez-Pacheco, F.; Soriguer, F.; Rojo-Martínez, G. Dietary fatty acids modulate adipocyte TNFa production via regulation of its DNA promoter methylation levels. J. Nutr. Biochem. 2017, 47, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Zhuo, Q.; Tseng, Y.; Wang, J.; Ma, Y.; Zhang, J.; Liu, J. TET1 promotes fatty acid oxidation and inhibits NAFLD progression by hydroxymethylation of PPARalpha promoter. Nutr. Metab. 2020, 17, 46. [Google Scholar] [CrossRef]
- Elshorbagy, A.K.; Jerneren, F.; Samocha-Bonet, D.; Refsum, H.; Heilbronn, L.K. Serum S-adenosylmethionine, but not methionine, increases in response to overfeeding in humans. Nutr. Diabetes 2016, 6, e192. [Google Scholar] [CrossRef] [Green Version]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver Peroxisome Proliferator-activated Receptorγ Contributes to Hepatic Steatosis, Triglyceride Clearance, and Regulation of Body Fat Mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MaktoobianBaharanchi, E.; Moradi Sarabi, M.; Naghibalhossaini, F. Effects of Dietary Polyunsaturated Fatty Acids on DNA Methylation and the Expression of DNMT3b and PPARalpha Genes in Rats. Avicenna J. Med. Biotechnol. 2018, 10, 214–219. [Google Scholar]
- Bozzetto, L.; Prinster, A.; Annuzzi, G.; Costagliola, L.; Mangione, A.; Vitelli, A.; Mazzarella, R.; Longobardo, M.; Mancini, M.; Vigorito, C.; et al. Liver fat is reduced by an isoenergetic MUFA diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012, 35, 1429–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.J.; James, L.; Hussey, B.; Wadley, A.J.; Lindley, M.R.; Mastana, S.S. Impact of aerobic exercise and fatty acid supplementation on global and gene-specific DNA methylation. Epigenetics 2019, 14, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | LFD(n = 16) | HFD(n = 16) |
|---|---|---|
| Body weight final (g) | 26 ± 4 | 43 ± 7 ** |
| Liver weight (g) | 2.4 ± 0.3 | 3.9 ± 0.3 *** |
| Gonadal fat weight (g) | 1.9 ± 0.1 | 3.1 ± 0.2 *** |
| TG (mg/dL) | 82 ± 5 | 134 ± 10 ** |
| Cholesterol (mg/dL) | 87.6 ± 7.6 | 113.4 ± 10.2 ** |
| FFAs (mmol/L) | 0.74 ± 0.10 | 1.09 ± 0.14 ** |
| Glucose (mg/dL) | 86 ± 6 | 106 ± 8 ** |
| Insulin (ng/mL) | 2.4 ± 0.6 | 10.1 ± 0.9 *** |
| IL-6 (ng/mL) | 13 ± 3 | 29 ± 6 ** |
| TNF-α (ng/mL) | 10 ± 4 | 19 ± 6 * |
| ALT(mU/mL) | 7.3 ± 0.9 | 19.3 ± 1.5 * |
| AST (mU/mL) | 15.6 ± 3.1 | 23.1 ± 5.2 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajri, T.; Zaiou, M.; Fungwe, T.V.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061355
Hajri T, Zaiou M, Fungwe TV, Ouguerram K, Besong S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells. 2021; 10(6):1355. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061355
Chicago/Turabian StyleHajri, Tahar, Mohamed Zaiou, Thomas V. Fungwe, Khadija Ouguerram, and Samuel Besong. 2021. "Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease" Cells 10, no. 6: 1355. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061355