
Inhibition of the Heat Shock Protein A (HSPA) Family Potentiates the Anticancer Effects of Manumycin A

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Experimental Conditions
2.2. Incubation Experiments
2.3. Cell Viability and Cytotoxicity Assays
2.4. Protein Extraction and Western Blot Analysis
2.5. Generation of Genetically Modified Cell Lines
2.6. HSF1 Functional Knockout Using the CRISPR/Cas9 Editing System
2.7. Statistical Analysis
3. Results
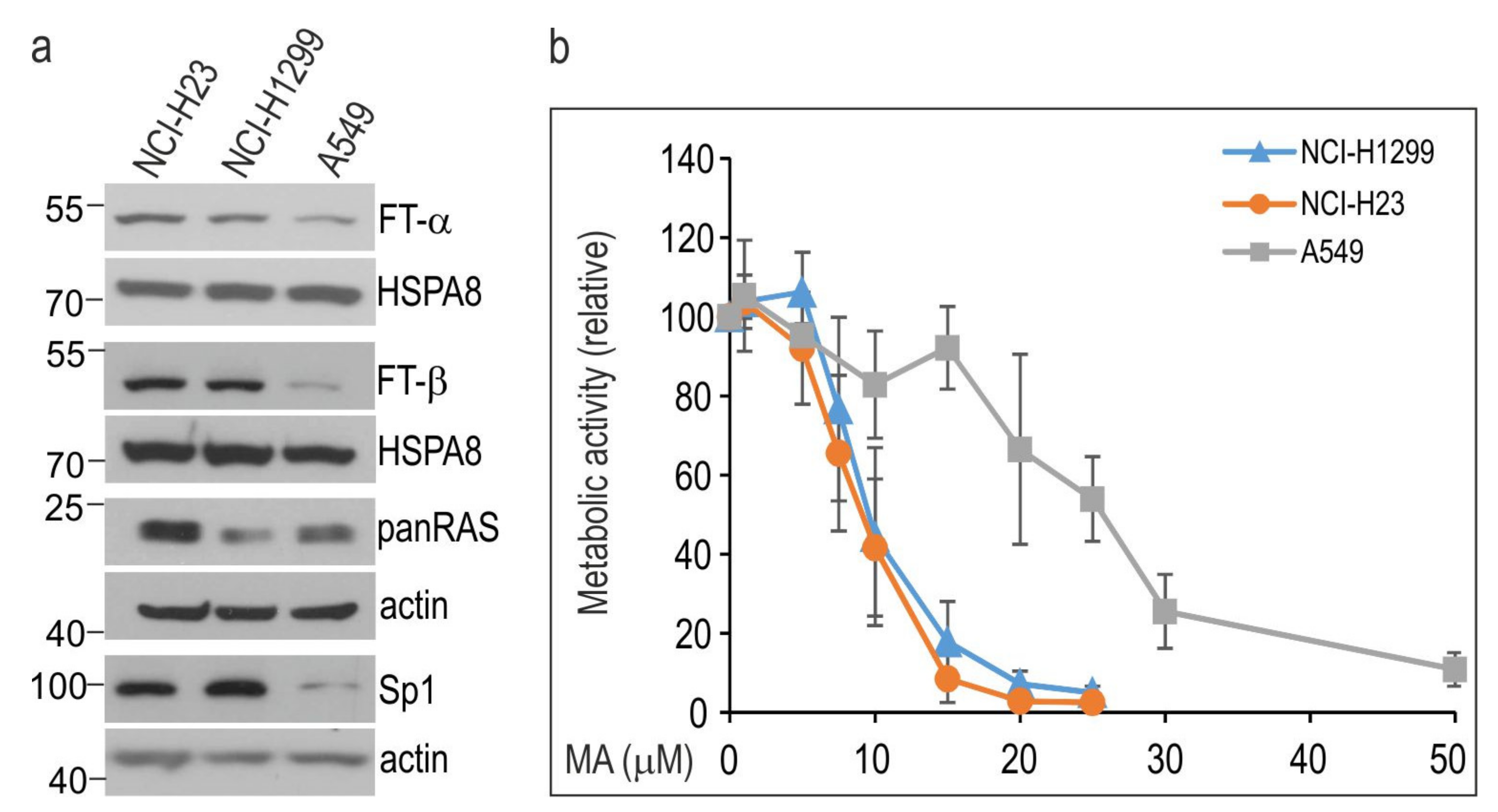
3.1. Manumycin A Inhibits the Growth of NSCLC Cells in a Cell Line-Dependent Manner
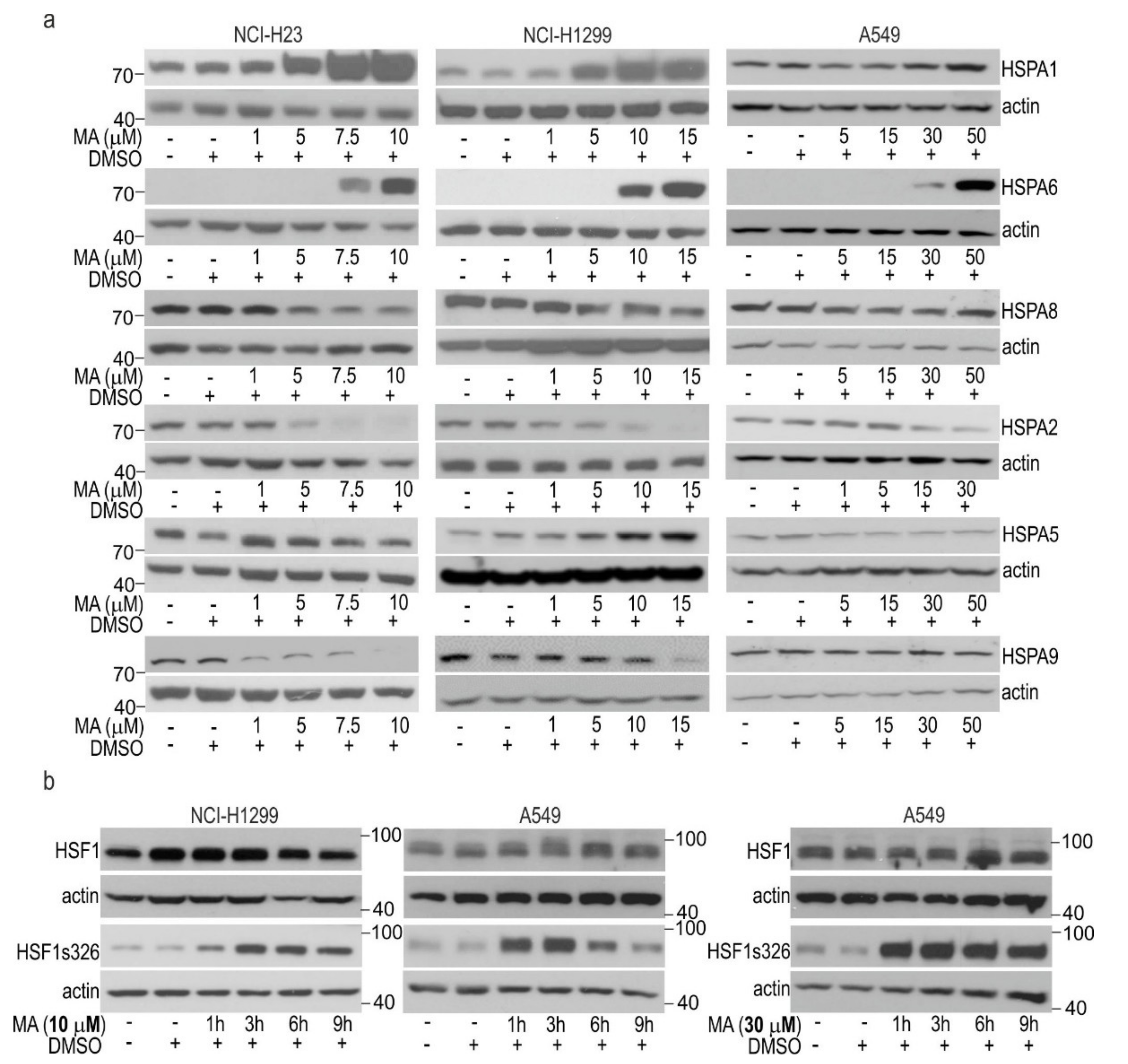
3.2. MA Modulates HSPAs Expression in Lung Cancer Cells
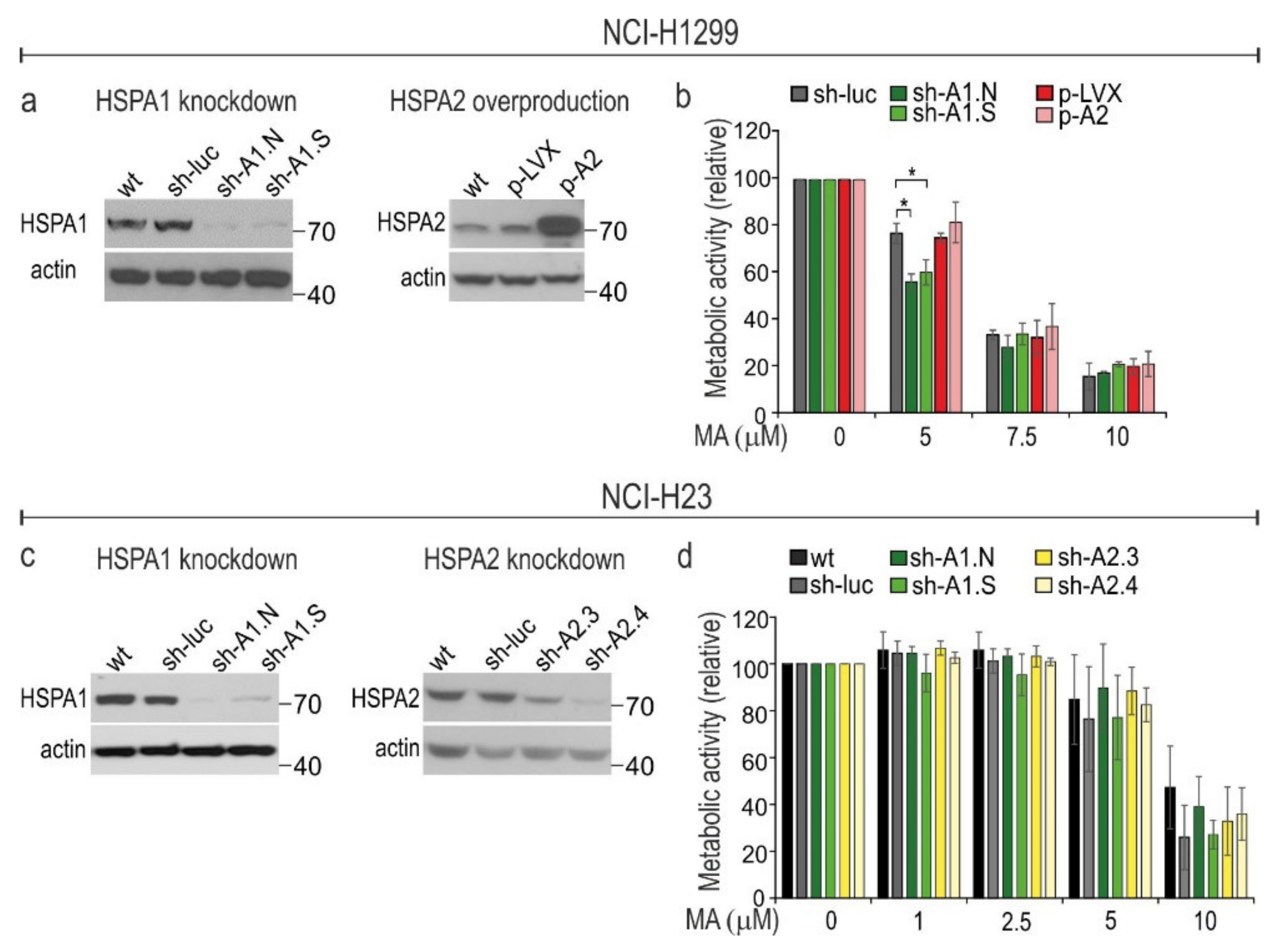
3.3. HSPA1 Exerts Very Limited, Cell Type-Dependent Protection against MA Toxicity While the Role of HSPA2 in Cytoprotection Is Negligible
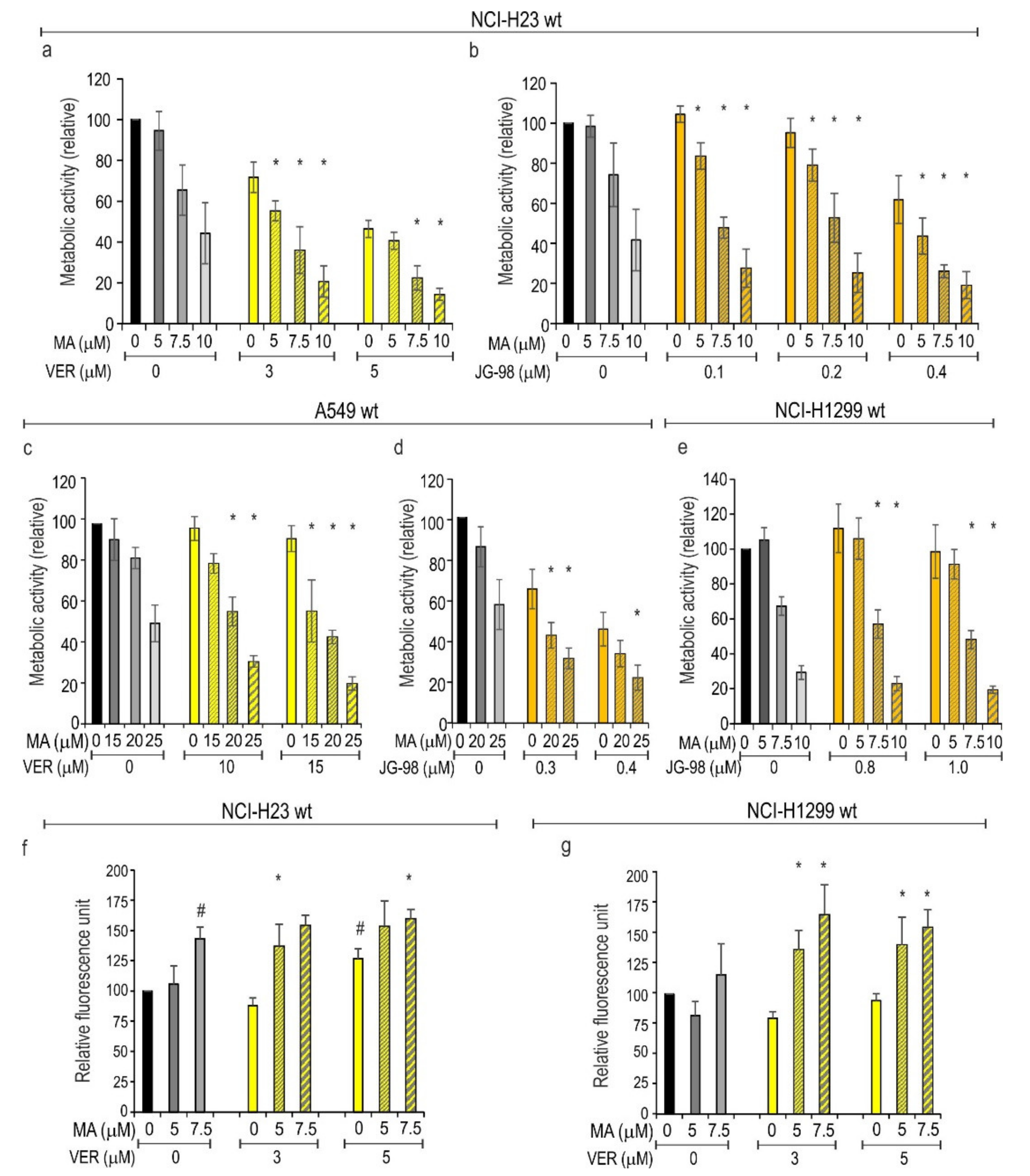
3.4. Inhibition of HSPAs Sensitize Lung Cancer Cells to MA
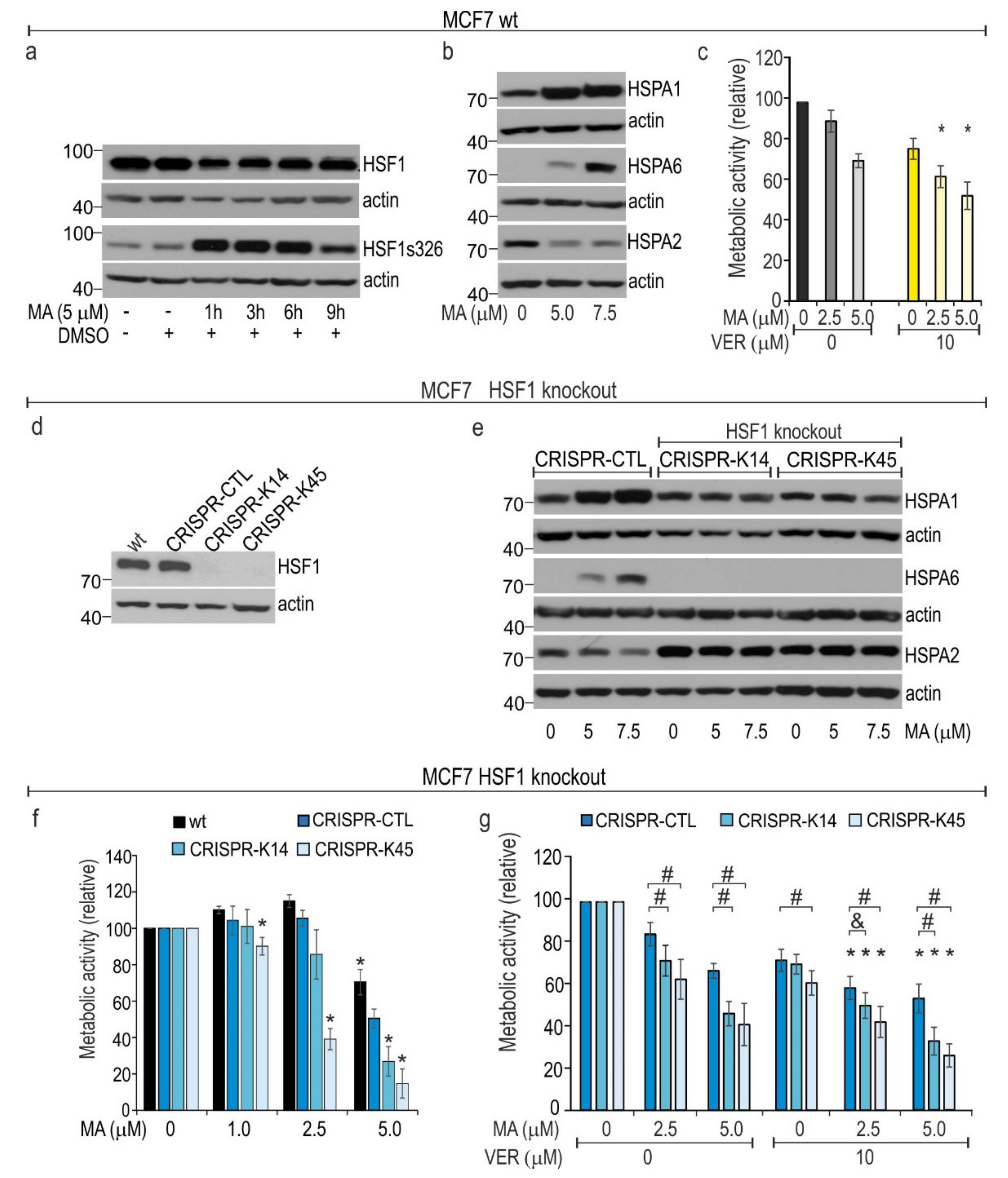
3.5. HSF1 Contributes to Cytoprotection against MA Toxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bérdy, J. Bioactive Microbial Metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Nakashima, T. Actinomycetes, an Inexhaustible Source of Naturally Occurring Antibiotics. Antibiotics 2018, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.; Akasaka, K.; Akinaga, S.; Okabe, M.; Nakano, H.; Gomez, R.; Wood, D.; Uh, M.; Tamanoi, F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. USA 1993, 90, 2281–2285. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Kawata, S.; Tamura, S.; Igura, T.; Nagase, T.; Miyagawa, J.I.; Yamazaki, E.; Ishiguro, H.; Matasuzawa, Y. Suppression of human pancreatic cancer growth in BALB/c nude mice by manumycin, a farnesyl:protein transferase inhibitor. Jpn. J. Cancer Res. 1996, 87, 113–116. [Google Scholar] [CrossRef]
- She, M.; Yang, H.; Sun, L.; Yeung, S.C. Redox control of manumycin A-induced apoptosis in anaplastic thyroid cancer cells: Involvement of the xenobiotic apoptotic pathway. Cancer Biol. Ther. 2006, 5, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Singha, P.K.; Pandeswara, S.; Venkatachalam, M.A.; Saikumar, P. Manumycin A inhibits triple-negative breast cancer growth through LC3-mediated cytoplasmic vacuolation death. Cell Death Dis. 2013, 4, e457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jiang, H.; Xie, L.; Hu, J.; Li, L.; Yang, M.; Cheng, L.; Liu, B.; Qian, X. Antitumor effect of manumycin on colorectal cancer cells by increasing the reactive oxygen species production and blocking PI3K-AKT pathway. OncoTargets Ther. 2016, 9, 2885–2895. [Google Scholar] [CrossRef] [Green Version]
- Nagase, T.; Kawata, S.; Tamura, S.; Matsuda, Y.; Inui, Y.; Yamasaki, E.; Ishiguro, H.; Ito, T.; Matsuzawa, Y. Inhibition of cell growth of human hepatoma cell line (Hep G2) by a farnesyl protein transferase inhibitor: A preferential suppression of ras farnesylation. Int. J. Cancer 1996, 65, 620–626. [Google Scholar] [CrossRef]
- Cho, J.J.; Chae, J.I.; Kim, K.H.; Cho, J.H.; Jeon, Y.-J.; Oh, H.N.; Yoon, G.; Yoon, D.Y.; Cho, Y.S.; Cho, S.-S.; et al. Manumycin A from a new Streptomyces strain induces endoplasmic reticulum stress-mediated cell death through specificity protein 1 signaling in human oral squamous cell carcinoma. Int. J. Oncol. 2015, 47, 1954–1962. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Chae, J.I.; Oh, H.; Cho, J.H.; Lee, R.-H.; Yoon, G.; Cho, S.-S.; Cho, Y.-S.; Lee, M.-H.; Liu, K.; et al. Manumycin A induces apoptosis in malignant pleural mesothelioma through regulation of Sp1 and activation of the mitochondria-related apoptotic pathway. Oncol. Rep. 2016, 36, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Isobe, Y.; Okumura, M.; McGregor, L.M.; Brittain, S.M.; Jones, M.D.; Liang, X.; White, R.; Forrester, W.; McKenna, J.M.; Tallarico, J.A.; et al. Manumycin polyketides act as molecular glues between UBR7 and P53. Nat. Chem. Biol. 2020, 16, 1189–1198. [Google Scholar] [CrossRef]
- Dixit, D.; Sharma, V.; Ghosh, S.; Koul, N.; Mishra, P.K.; Sen, E. Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radic. Biol. Med. 2009, 47, 364–374. [Google Scholar] [CrossRef]
- Tuladhar, A.; Rein, K.S. Manumycin A Is a Potent Inhibitor of Mammalian Thioredoxin Reductase-1 (TrxR-1). ACS Med. Chem. Lett. 2018, 9, 318–322. [Google Scholar] [CrossRef]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2019, 9, 1703244. [Google Scholar] [CrossRef] [Green Version]
- Akerfelt, M.; Trouillet, D.; Mezger, V.; Sistonen, L. Heat shock factors at a crossroad between stress and development. Ann. N. Y. Acad. Sci. 2007, 1113, 15–27. [Google Scholar] [CrossRef]
- Rupik, W.; Jasik, K.; Bembenek, J.; Widłak, W. The expression patterns of heat shock genes and proteins and their role during vertebrate’s development. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2011, 159, 349–366. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Scieglinska, D.; Krawczyk, Z. Expression, function, and regulation of the testis-enriched heat shock HSPA2 gene in rodents and humans. Cell Stress Chaperones 2015, 20, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Gogler-Pigłowska, A.; Klarzyńska, K.; Sojka, D.R.; Habryka, A.; Głowala-Kosińska, M.; Herok, M.; Kryj, M.; Halczok, M.; Krawczyk, Z.; Scieglinska, D. Novel role for the testis-enriched HSPA2 protein in regulating epidermal keratinocyte differentiation. J. Cell Physiol. 2018, 233, 2629–2644. [Google Scholar] [CrossRef]
- Sojka, D.R.; Gogler-Pigłowska, A.; Klarzyńska, K.; Klimczak, M.; Zylicz, A.; Głowala-Kosińska, M.; Krawczyk, Z.; Scieglinska, D. HSPA2 Chaperone Contributes to the Maintenance of Epithelial Phenotype of Human Bronchial Epithelial Cells but Has Non-Essential Role in Supporting Malignant Features of Non-Small Cell Lung Carcinoma, MCF7, and HeLa Cancer Cells. Cancers 2020, 12, 2749. [Google Scholar] [CrossRef]
- Tukaj, S. Heat Shock Protein 70 as a Double Agent Acting Inside and Outside the Cell: Insights into Autoimmunity. Int. J. Mol. Sci. 2020, 21, 5298. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Balogi, Z.; Khachatryan, W.; Gao, H.; Vígh, L.; Multhoff, G. Membrane-Associated Heat Shock Proteins in Oncology: From Basic Research to New Theranostic Targets. Cells 2020, 9, 1263. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wu, W.; Verschraegen, C.F.; Chen, L.; Mao, L.; Yeung, S.C.; Kudelka, A.P.; Freedman, R.S.; Kavanagh, J.J. Proteomic identification of heat shock protein 70 as a candidate target for enhancing apoptosis induced by farnesyl transferase inhibitor. Proteomics 2003, 3, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, Z.; Gogler-Pigłowska, A.; Sojka, D.R.; Scieglinska, D. The Role of Heat Shock Proteins in Cisplatin Resistance. Anticancer Agents Med. Chem. 2018, 18, 2093–2109. [Google Scholar] [CrossRef]
- Sojka, D.R.; Gogler-Pigłowska, A.; Vydra, N.; Cortez, A.J.; Filipczak, P.T.; Krawczyk, Z.; Scieglinska, D. Functional redundancy of HSPA1, HSPA2 and other HSPA proteins in non-small cell lung carcinoma (NSCLC); an implication for NSCLC treatment. Sci. Rep. 2019, 9, 14394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scieglinska, D.; Sojka, D.R.; Gogler-Pigłowska, A.; Chumak, V.; Krawczyk, Z. Various Anti-HSPA2 Antibodies Yield Different Results in Studies on Cancer-Related Functions of Heat Shock Protein A2. Int. J. Mol. Sci. 2020, 21, 4296. [Google Scholar] [CrossRef]
- Guettouche, T.; Boellmann, F.; Lane, W.S.; Voellmy, R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Yaglom, J.A.; Wang, Y.; Li, A.; Li, Z.; Monti, S.; Alexandrov, I.; Lu, X.; Sherman, M.Y. Cancer cell responses to Hsp70 inhibitor JG-98: Comparison with Hsp90 inhibitors and finding synergistic drug combinations. Sci. Rep. 2018, 8, 3010. [Google Scholar] [CrossRef] [Green Version]
- Schlecht, R.; Scholz, S.R.; Dahmen, H.; Wegener, A.; Sirrenberg, C.; Musil, D.; Bomke, J.; Eggenweiler, H.M.; Mayer, M.P.; Bukau, B. Functional analysis of Hsp70 inhibitors. PLoS ONE 2013, 8, e78443. [Google Scholar] [CrossRef]
- Vydra, N.; Janus, P.; Kuś, P.; Stokowy, T.; Mrowiec, K.; Toma-Jonik, A.; Krzywon, A.; Cortez, A.J.; Wojtaś, B.; Gielniewski, B.; et al. Heat Shock Factor 1 (HSF1) as a new tethering factor for ESR1 supporting its action in breast cancer. BioRXiv 2021. [Google Scholar] [CrossRef]
- Ma, L.; Sato, F.; Sato, R.; Matsubara, T.; Hirai, K.; Yamasaki, M.; Shin, T.; Shimada, T.; Nomura, T.; Mori, K.; et al. Dual targeting of heat shock proteins 90 and 70 promotes cell death and enhances the anticancer effect of chemotherapeutic agents in bladder cancer. Oncol. Rep. 2014, 31, 2482–2492. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Colvin, T.; Rauch, J.N.; Acosta-Alvear, D.; Kampmann, M.; Dunyak, B.; Hann, B.; Aftab, B.T.; Murnane, M.; Cho, M.; et al. Validation of the Hsp70-Bag3 Protein-Protein Interaction as a Potential Therapeutic Target in Cancer. Mol. Cancer Ther. 2015, 14, 642–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eugenio, A.I.P.; Fook-Alves, V.L.; de Oliveira, M.B.; Fernando, R.C.; Zanatta, D.B.; Strauss, B.E.; Silva, M.R.R.; Porcionatto, M.A.; Colleoni, G.W.B. Proteasome and heat shock protein 70 (HSP70) inhibitors as therapeutic alternative in multiple myeloma. Oncotarget 2017, 8, 114698–114709. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef]
- Barnoud, T.; Leung, J.C.; Leu, J.I.; Basu, S.; Poli, A.N.R.; Parris, J.L.D.; Indeglia, A.; Martynyuk, T.; Good, M.; Gnanapradeepan, K.; et al. A Novel Inhibitor of HSP70 Induces Mitochondrial Toxicity and Immune Cell Recruitment in Tumors. Cancer Res. 2020, 80, 5270–5281. [Google Scholar] [CrossRef]
- De Thonel, A.; Mezger, V.; Garrido, C. Implication of heat shock factors in tumorigenesis: Therapeutical potential. Cancers 2011, 3, 1158–1181. [Google Scholar] [CrossRef]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [Green Version]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S.; et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef] [Green Version]
- Xi, C.; Hu, Y.; Buckhaults, P.; Moskophidis, D.; Mivechi, N.F. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J. Biol. Chem. 2012, 287, 35646–35657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Qian, W.; Li, J.; Jiang, Z.; Cheng, L.; Yan, B.; Cao, J.; Sun, L.; Zhou, C.; Lei, M.; et al. Loss of AMPK activation promotes the invasion and metastasis of pancreatic cancer through an HSF1-dependent pathway. Mol. Oncol. 2017, 11, 1475–1492. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Liao, Y.; Zhang, J.; Huang, Q.; Luo, W.; Yu, J.; Gong, J.; Zhou, Y.; Li, X.; Tang, B.; et al. Heat shock factor 1 inhibits the mitochondrial apoptosis pathway by regulating second mitochondria-derived activator of caspase to promote pancreatic tumorigenesis. J. Exp. Clin. Cancer Res. 2017, 36, 64. [Google Scholar] [CrossRef]
- Im, C.N.; Yun, H.H.; Lee, J.H. Heat Shock Factor 1 Depletion Sensitizes A172 Glioblastoma Cells to Temozolomide via Suppression of Cancer Stem Cell-Like Properties. Int. J. Mol. Sci. 2017, 18, 468. [Google Scholar] [CrossRef]
- Nagai, N.; Nakai, A.; Nagata, K. Quercetin suppresses heat shock response by down regulation of HSF1. Biochem. Biophys. Res. Commun. 1995, 208, 1099–1105. [Google Scholar] [CrossRef]
- Au, Q.; Zhang, Y.; Barber, J.R.; Ng, S.C.; Zhang, B. Identification of inhibitors of HSF1 functional activity by high-content target-based screening. J. Biomol. Screen. 2009, 14, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.J.; Kim, J.A.; Shin, K.D.; Shin, D.S.; Han, Y.M.; Lee, Y.J.; Lee, J.S.; Kwon, B.M.; Han, D.C. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem. 2011, 286, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Widlak, W.; Vydra, N.; Malusecka, E.; Dudaladava, V.; Winiarski, B.; Scieglińska, D.; Widlak, P. Heat shock transcription factor 1 down-regulates spermatocyte-specific 70 kDa heat shock protein expression prior to the induction of apoptosis in mouse testes. Genes Cells 2007, 12, 487–499. [Google Scholar] [CrossRef]
- Yeung, S.C.; Xu, G.; Pan, J.; Christgen, M.; Bamiagis, A. Manumycin enhances the cytotoxic effect of paclitaxel on anaplastic thyroid carcinoma cells. Cancer Res. 2000, 60, 650–656. [Google Scholar]
- Yeung, S.C.; She, M.; Yang, H.; Pan, J.; Sun, L.; Chaplin, D. Combination chemotherapy including combretastatin A4 phosphate and paclitaxel is effective against anaplastic thyroid cancer in a nude mouse xenograft model. J. Clin. Endocrinol. Metab. 2007, 92, 2902–2909. [Google Scholar] [CrossRef]
- Solár, P.; Sačková, V.; Hrčková, G.; Demečková, V.; Kassayová, M.; Bojková, B.; Mudroňová, D.; Gancarčíková, S.; Jendželovský, R.; Fedoročko, P. Antitumor effect of the combination of manumycin A and Immodin is associated with antiplatelet activity and increased granulocyte tumor infiltration in a 4T1 breast tumor model. Oncol. Rep. 2017, 37, 368–378. [Google Scholar] [CrossRef]
- Xu, G.; Pan, J.; Martin, C.; Yeung, S.C. Angiogenesis inhibition in the in vivo antineoplastic effect of manumycin and paclitaxel against anaplastic thyroid carcinoma. J. Clin. Endocrinol. Metab. 2001, 86, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- She, M.; Pan, I.; Sun, L.; Yeung, S.C. Enhancement of manumycin A-induced apoptosis by methoxyamine in myeloid leukemia cells. Leukemia 2005, 19, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Goto, M.; Iwakawa, M.; Asano, T.; Kenmochi, T.; Imai, T.; Ochiai, T. Modified radiosensitivity of pancreatic cancer xenografts by farnesyl protein transferase inhibitor and MEK inhibitor. Oncol. Rep. 2003, 10, 1525–1528. [Google Scholar] [CrossRef]
- Kobayashi, H. Cancer Chemotherapy Specific to Acidic Nests. Cancers 2017, 9, 36. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sojka, D.R.; Hasterok, S.; Vydra, N.; Toma-Jonik, A.; Wieczorek, A.; Gogler-Pigłowska, A.; Scieglinska, D. Inhibition of the Heat Shock Protein A (HSPA) Family Potentiates the Anticancer Effects of Manumycin A. Cells 2021, 10, 1418. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061418
Sojka DR, Hasterok S, Vydra N, Toma-Jonik A, Wieczorek A, Gogler-Pigłowska A, Scieglinska D. Inhibition of the Heat Shock Protein A (HSPA) Family Potentiates the Anticancer Effects of Manumycin A. Cells. 2021; 10(6):1418. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061418
Chicago/Turabian StyleSojka, Damian Robert, Sylwia Hasterok, Natalia Vydra, Agnieszka Toma-Jonik, Anna Wieczorek, Agnieszka Gogler-Pigłowska, and Dorota Scieglinska. 2021. "Inhibition of the Heat Shock Protein A (HSPA) Family Potentiates the Anticancer Effects of Manumycin A" Cells 10, no. 6: 1418. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061418